

Abstract
Blood pressure and hypertension have significant genetic underpinnings that may be age-dependent. The age-dependency, significant contributions from environmental factors such as diet and exercise, and inherent moment-to-moment variability complicate the identification of the genes contributing to the development of hypertension. While genetic abnormalities may have moderate effects, the physiological pathways involving these genes have redundant compensating mechanisms to bring the system back into equilibrium. This has the effect of reducing or completely masking the initial genetic defects, one of the hypothesized reasons for the small genetic effects found by the recent genome-wide association studies. This review article will discuss the concept of “initiators” versus “compensators” in the context of finding genes related to hypertension development. A brief review is provided of some key genes found to be associated with hypertension, including the genes identified from the nine genome-wide association studies published to date.
Keywords: Blood pressure, genetics, genome-wide association, heritability, hypertension, review, salt sensitivity
Heritability of Blood Pressure and Hypertension
Hypertension is a heritable trait with heritability estimates in the 20-40% range based on family studies and up to 60% from twin studies (1-5). For a variety of reasons, disentangling the underlying genes responsible for that heritability has been difficult. The main difficulty likely involves the small effects of any individual gene on blood pressure. As opposed to obesity which is fairly stable over months to years and cholesterol levels which remain fairly stable over time, blood pressure is highly variable from second to second, having multiple physiological pathways to maintain appropriate levels. It is also highly influenced by diet, activity, as well as obesity and lipid levels.
Age- and Time-Dependent Genetic Effects
High heritabilities for adult twin blood pressure (1, 2, 5) and low infant twin heritabilities (6) suggest that blood pressure heritability increases with age. An increasing genetic variance over time is also supported by studies that show genotype by age interactions, with the “affected” genotype having greater blood pressure increases with age than the normal genotype (7, 8). Animal models (9) and human studies (10) demonstrate that the expression of blood pressure genes varies with age and perhaps other factors. In the Hypertension Genetic Epidemiology Network (HyperGEN) study, the average age-adjusted heritability for systolic blood pressure was estimated as 0.24 in whites and 0.29 in blacks, but when the heritability was modeled as an age dependent effect, the peak heritability was 0.69 in whites and 0.68 in blacks (10). These peak heritabilities were estimated to occur at ages 74 and 59, respectively, in the two races. For diastolic blood pressure, the peak heritabilities were 0.36 and 0.46 for the two races occurring at ages 60 and 46, respectively. An additional factor to consider is that the ability to compensate for abnormal gene expression affecting blood pressure may decrease with age, allowing greater genetic effects to be measured.
Animal models also suggest that when the blood pressure system is stressed (such as with high salt intake) an initial genetic response may only be transiently expressed. Later, compensatory mechanisms counteract the abnormal genetic response to the initial stressor and may or may not reduce blood pressure back to normal levels. There also may be time windows throughout life when specific genetic factors have greater expression and contribute to the development of hypertension. Figure 1 shows a genetic cause of acute sodium retention that is subsequently hidden after compensating mechanisms increasing sodium excretion are activated, even when sodium loading continues in a microsomal prostaglandin E synthase-1 knockout mouse (11). After day 3, gene association studies would miss this association, but detection is possible between days 1 and 3. An important question raised by Figure 1 is why do the knockout mice return to the wild type levels of sodium balance after 3 days despite continued salt loading? If the sodium is merely excreted more slowly, but finally equilibrates without changes in other compensating factors, this gene would probably not be considered a hypertension gene. If compensating factors are altered and remain altered to preserve sodium balance thereby increasing blood pressure (as is the case in this model), then the microsomal prostaglandin E synthase-1 gene would be considered an initiator of hypertension even though the abnormality detected may be related to the compensatory factor. In cases such as these, genetic studies will identify genes related to defective compensating mechanisms, rather than genes initiating hypertension.
Figure 1.
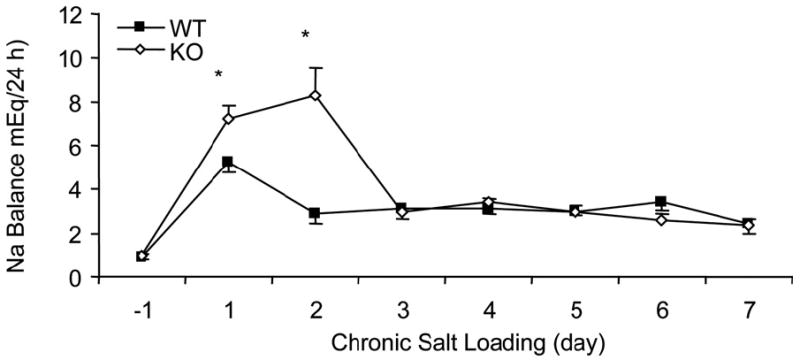
Transient acute but not chronic sodium retention in the microsomal prostaglandin E synthase-1 knockout (KO) mouse. From: Jia Z, Zhang A, Zhang H, Dong Z, Yang T. Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion. Circ Res. 2006 Nov 24;99(11):1243-51.
Figure 2 shows that delayed sodium excretion is acutely (within 1 day) unmasked going from a low to high sodium diet (but not vice versa) in an in-patient study allowing for precise quantification of sodium intake and excretion (12). This demonstrates that genetic causes of delayed sodium excretion after sodium loading in humans would be easily detectable between days 13 and 16, but undetectable after day 16 of the protocol (when on high salt or even when in balance on a low sodium diet). An earlier study found that compared to salt-resistant patients, salt-sensitive patients had slower renal responses when sodium intake was reduced (13). Therefore, whether sodium changes from low to high or high to low are best to unmask sodium excretion abnormalities remains unclear. Weinberger et al have shown that blood pressure sensitivity to such sodium challenges is reproducible, heritable, and predicts future hypertension (5, 14, 15). Heritability of salt sensitivity is also present in African Americans (16). A few studies performed predominantly in the 1970's and early 1980's investigated initial, saline-induced hormonal changes in the RAAS system (17-19) and later on the catecholamine/dopamine system (20, 21), and none tested more than a few genes for association with these changes to detect whether acute protocols such as these could unmask compensated genetic effects. An additional approach has been to use pharmacogenetics as an intervention to find genes involved with pathways specific to the medication being used for the intervention (22).
Figure 2.
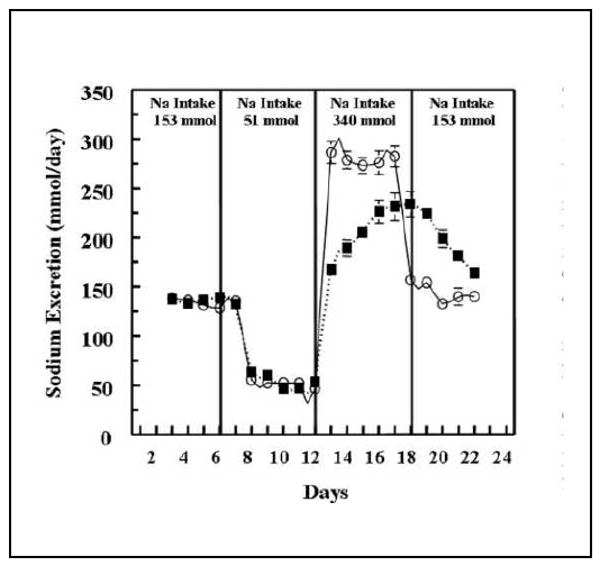
Effect of dietary sodium on sodium excretion in salt resistant (SR) (○) and salt sensitive (SS) (■) hypertensive Japanese subjects. From: Sanada H, Yatabe J, Midorikawa S, Hashimoto S, Watanabe T, Moore JH, et al. Single-nucleotide polymorphisms for diagnosis of salt-sensitive hypertension. Clin Chem. 2006 Mar;52(3):352-60.
Most studies addressing the abnormalities that lead to elevation of blood pressure study the physiological systems in steady state. Even when interventions such as high and low salt diets are used, subjects are brought into steady state after the intervention to detect those variables that are not fully compensated and are likely to lead to increases in blood pressure. However, this design would be expected to minimize the ability to detect genes that increase the susceptibility to hypertension. The initial genetic abnormalities induced by constant over- or under-expression of the gene products would be as fully compensated by the physiological systems involved as possible. Detecting association of hypertension or blood pressure with these genes would then depend on how fully compensation is attained, even though the uncompensated effect may be large.
As shown in Figure 3, substantial individual differences in the rate of sodium excretion have long been documented. Despite recognition of marked individual differences in acute sodium excretion and early studies involving a handful of genes (14, 23-25), genetic correlates of this intermediate phenotype remain virtually unexplored.
Figure 3.

Sodium excretion (lines) in hypertensive subjects classified by rate of renin-angiotensin system suppression (solid bars). Aldosterone (not shown for clarity) was highly correlated with plasma renin activity (PRA). Subjects were in balance on 10 mEq/day Na diets and infused for 6 hours (beginning at time 0) with 0.9% NaCl at 500 ml/hr. Adapted from Tuck ML, et al. Circ Res 1976; 39:711.
Candidate Genes
Initial studies of candidate genes focused on the renin angiotensin system because of its importance in sodium and fluid volume control (26, 27). Polymorphisms in the angiotensinogen (AGT) gene have been associated with higher angiotensinogen levels (28), which would lead to constant increases in angiotensin II (A-II) levels unless compensating mechanisms were activated. Such mechanisms are normally invoked through a negative feedback loop reducing plasma renin concentrations. However, one would expect that further insults to the blood pressure control system, such as increased dietary salt intake, would make additional compensation difficult. Reducing renin further might be limited since the system is already suppressed, leading to sodium retention and increases in blood pressure. Kim et al showed that by increasing the number of copies of the AGT gene in the mouse, the amount of angiotensinogen and level of blood pressure increased, even though the compensating mechanisms in the mouse were normal (29). On the other hand, when one AGT allele was inactivated, there was a four-fold increase in the number of renin-producing cells in the afferent arterioles resulting in increased renin concentrations (30). The increased renin concentration then compensated for the lack of an AGT allele resulting in decreased angiotensinogen production.
It is hypothesized that chronic, but modest, increased angiotensinogen levels, both systemically and in the kidney, lead to increased angiotensin II production and altered capability to react to short or long term changes in dietary salt (i.e. defective compensation) (31). Angiotensinogen, though not filtered through the glomerulus, is secreted by renal tubular cells and can form local A-II, increasing proximal tubule sodium reabsorption (32). Higher angiotensin II can lead to feedback reduction of plasma renin. Distal tubule activation of a local renin angiotensin aldosterone system (RAAS) in subjects with the -6A AGT allele may also be enhanced, leading to increased sodium reabsorption that must be countered by compensatory mechanisms. Blunted renal vascular response to infused angiotensin II has been associated with this AGT polymorphism (33). Hypertensive subjects have higher urinary angiotensinogen levels that are correlated with blood pressure and reduced by RAAS blocking agents (34).
AGT-6 may not be the only site affecting angiotensinogen expression. Haplotype analysis revealed that one can divide subjects carrying AGT-6A based on a common haplotype having significant association with higher angiotensinogen levels, reduced renal plasma flow, and hypertension. Other haplotypes, even those carrying AGT-6A, had little or no association with these traits (35). This suggests that multiple genes contribute to hypertension and the presence of multiple functional polymorphisms within a single gene may have greater hypertensive effect.
In addition to the genetic and physiological implications of the AGT gene, three clinical trials have shown that dietary salt reduction significantly decreases blood pressure in those at highest genetic risk from the AGT gene for salt sensitive blood pressure (36-38). If a copy of the A allele at G-6A increases angiotensinogen levels, reduces renin, and increases salt sensitive blood pressure, limiting sodium intake would most help those having the compromised pathway since they can now compensate sufficiently to increase sodium excretion. However, salt reduction would have little effect on those not at risk for salt sensitive blood pressure due to carriage of the G allele at AGT G-6A. Table 1 summarizes the results of these studies. The Trials of Hypertension Prevention II (TOHP) enrolled subjects with borderline hypertension, the Dietary Approaches to Stop Hypertension (DASH) trial enrolled borderline or stage 1 hypertensive subjects, and the Dutch SAGA salt (SAGA) trial enrolled never-treated hypertensive subjects. All three trials showed a significant difference across AGT genotypes for change in blood pressure with the intervention. The interventions included reduced salt and weight loss in TOHP, reduced salt and increased potassium in SAGA, and an increased fruit/vegetable or full DASH diet in the DASH trial. Each showed that the AGT allele -6A, which increases risk of hypertension, was associated with greater responses to the interventions than the G allele. Therefore, the -6A subjects on average appear to be salt sensitive and respond well to appropriate interventions despite their higher baseline risk for hypertension.
Table 1.
Systolic and diastolic blood pressure reductions by angiotensinogen (AGT) genotypes after intervention in three clinical trials.
| AGT G-6A | AA | GA | GG | AA-GG difference |
|---|---|---|---|---|
| TOHP Na reduction, mmHg | -2.7/-2.2 | -1.3/-0.7 | -0.2/1.1 | -2.5/-1.5 |
| TOHP wt. loss, mmHg | -3.5/-2.4 | -0.9/-1 | -1.1/0.3 | -2.6/-1.4 |
| SAGA salt diet, mmHg | -8.6/-3.9 | -9.0/-5.2 | -5.3/-1.0 | -3.3/-2.9 |
| DASH fruit/veg. diet, mmHg | -5.2/-3.0 | -2.3/-1.2 | -1.3/0.3 | -3.9/-3.3 |
| DASH diet, mmHg | -6.9/-3.5 | -5.6/-3.2 | -2.8/0.2 | -4.1/-3.7 |
The importance of examining alterations in pathways, rather than a single variable at a time or by covariate adjustment, is illustrated by the apparent confounding of a primary AGT-initiated mechanism by obesity and lipid levels. Adipocytes are an important source of angiotensinogen, and increased angiotensinogen leads to greater angiotensin II formation inhibiting preadipocyte differentiation to mature adipocytes via the AT1 receptor (39). Therefore, the existing fat cells are not able to store excess calories, which are shunted to muscle, liver and pancreas causing insulin resistance (40, 41). Weight loss decreases the high levels of angiotensinogen, renin, aldosterone, and angiotensin converting enzyme in obese subjects (42). Similarly, higher LDL-C levels increase blood pressure responses to infused angiotensin II, presumably because LDL-C up-regulates the AT1 receptor (43, 44). Polymorphisms at the AT1 receptor also affect this physiological pathway. CC homozygotes at position A1166C of the AT1R gene had smaller blood pressure responses to infused angiotensin II than did A allele carriers (43). The effects of obesity and lipids on increased angiotensinogen and A-II levels may be as great (or greater) than any gene-specific effect, masking or decreasing the effect size of the observed genetic associations, such as those found between AGT and A-II infusion renal and adrenal responses (33).
In animal models and humans, alpha adducin is clearly related to salt sensitivity and hypertension, especially low renin hypertension (24, 45). A nonsynonymous polymorphism G460W is related to proximal tubule sodium reabsorption because it stimulates Na-K pump activity and interacts with aldosterone synthase and angiotensin converting enzyme (ACE) for hypertension risk (46). The adducin defect could be detected after both sodium depletion and sodium loading (24). More recently, acute blood pressure response to a saline load was found to be associated with variants in chloride channels (47). A-II infusions have been used as an alternative to saline infusion to directly test the relevance of the renin-angiotensin-aldosterone system in hypertension and salt sensitivity (33, 48). In addition to the effects of alpha-adducin on the Na-K ATPase transporter, the gene coding the ATP1B1 subunit of the transporter appears to be associated with hypertension (49).
Sodium sensitivity of blood pressure also may result from variation in proximal tubule sodium reabsorption (50), in part relating to the proximal tubule paracrine dopamine system (20, 21). In a small study of saline infusion or dietary salt changes, urinary dopamine excretion increased by an average of 28% (20). A more recent study showed that dopamine excretion increased by an average of 53% in sodium-resistant normotensives in balance on a 40 mmol sodium/day diet in response to a 0.9% saline infusion; however, dopamine excretion did not increase in sodium-sensitive normotensive subjects (21). Differences in dopamine excretion may result from decreased production of dopamine (perhaps related to differences in the activity of dopa decarboxylase in proximal tubule cells) (51), or could result from genetic polymorphisms in the synthetic enzymes of the dopamine pathway (50). Furthermore, even if dopamine excretion is normal, decreased dopamine action can result from polymorphisms in the dopamine D1 receptor (52), primarily responsible for increasing natriuresis, the alpha subunit of Gs (GNAS1), involved in the signaling pathway of the D1 receptor (53), or defects in G-protein receptor kinase 4 (GRK4) activity (12, 54).
The catecholamine system is further implicated by studies of the tyrosine hydroxylase gene, the rate-limiting step in catecholamine synthesis. Tyrosine hydroxylase variation was associated with plasma and urine catecholamine levels, baroreflex function, and blood pressure (55). The alpha1B and beta2 adrenergic receptor and dopamine receptor type 1A genes have been implicated with blood pressure or hypertension in linkage and association studies (56-59). However, as with all candidate genes, negative studies have been reported (60).
In addition to the importance of proximal tubule reabsorption, local distal tubule and collecting duct RAAS systems exist and “fine-tune” sodium reabsorption. The epithelial sodium channel (ENAC) has recently been shown to have common polymorphisms associated with blood pressure (61, 62), in addition to the rare pseudohypoaldosteronism type 1 and Liddle syndrome mutations. A gene responsible for removal of ENAC from the cell membrane, NEDD4L has been shown associated with stressed blood pressure changes (postural change) and with hypertension (63, 64). NEDD4L is stimulated by serum glucocorticoid regulated kinase 1 (SGK1) for which polymorphisms have been associated with blood pressure (65). This suggests that if one had a combination of deleterious polymorphisms among the genes leading to ENAC activation or removal, such as NEDD4L and SGK1 or transporters on the basolateral side of the cell such as the Na-K ATPase transporter and the mineralocorticoid receptor, blood pressure could be even more significantly affected. Because so many redundant blood pressure control mechanisms exist, studying any one in isolation might miss abnormalities that become apparent when multiple deficiencies are involved.
SLC4A5 is a sodium-bicarbonate transporter regulating intracellular pH by transporting sodium bicarbonate across the cell membrane. Association of SLC4A5 with hypertension was identified through fine mapping positional candidate genes under a linkage peak from a genome-wide linkage analysis and testing for association in three of the networks in the Family Blood Pressure Program (66). The association with blood pressure was further replicated in a longitudinal study (67). The mechanisms related to increasing blood pressure remain obscure.
In elegant kidney transplantation experiments in the rat, hypertension developed in the normotensive strains when a kidney was transplanted from a hypertensive strain; and hypertension disappeared in the hypertensive strain when a kidney from the normotensive rat was transplanted (68). Based on these experiments, it is not surprising that rare monogenic hypertension and hypotension syndromes involve defects in renal electrolyte handling (69). More germane to essential hypertension are the effects of polymorphisms with more modest effect (70). Most genes significantly regulating blood pressure and hypertension involve electrolyte transport or hormonal control of electrolyte transport in the kidney.
Further complicating matters, genetic polymorphisms in one gene may act differently based upon the tissue in which the gene is expressed. Experiments using the transplantation model reveal that functioning AT1 receptors in the kidney were required for development of hypertension but not the AT1 receptors in the extrarenal vasculature (even though the extrarenal receptors help control blood pressure) (71, 72). Therefore, measurement of local intrarenal hormones such as angiotensinogen, aldosterone, dopamine, etc may be critical to elucidate important genetic mechanisms of hypertension involving kidney function.
All hypertension genes are not expressed in kidney. Other pathways likely contribute to hypertension, particularly in 50-60% of individuals who do not appear to have salt sensitive blood pressure. A positional candidate gene study following a linkage signal identified genes that may affect the vascular endothelium (49). Genes such as endothelin may make both systemic and renal contributions to blood pressure control (73). The drug metabolizing gene CYP3A5 may have one allele that is associated with higher blood pressures, and another allele that is associated with improved treatment responses (25). Many of the genes detected through genome-wide association analysis have no clear renal expression or function.
A number of studies suggest that high salt diets can lead to adverse cardiovascular changes independent of blood pressure. Salt-related end-organ damage includes cardiac hypertrophy, fibrosis, impaired vasodilatory response, increased endothelial activation, and renal damage (74-79). All of these conditions would increase the likelihood of hypertension regardless of whether or not a salt-sensitive genetic background is present. Both the Framingham and Bogalusa studies detected gene associations with arterial stiffness (80, 81).
Genome-wide Linkage Studies
The NHLBI Family Blood Pressure Program (FBPP), consisting of 4 large networks of collaborating universities, was designed to find hypertension genes in over 16,000 subjects. Genome-wide linkage scans for hypertension have been published for each of the four FBPP networks, including a meta-analysis of the combined dataset (82-86). Multiple linkage scans have also been published for related phenotypes including blood pressure, lipids, and various biochemical variables. From the best linkage region on chromosome 2 for hypertension, one FBPP network narrowed down associated SNPs to one gene, SLC4A5 (a renal sodium bicarbonate/chloride transporter), and replicated the findings among the other networks (66). Subsequently, typing the variants in the Utah Pedigree Study verified an association with SBP and DBP at baseline and 25-year follow-up and showed an association with baseline stressed mental math and isometric handgrip blood pressures (67). Three other blood pressure genes (ATP1B1, SELE, RGS5) under a linkage peak on chromosome 1 were found in the FBPP to be associated with blood pressure (49). Novel associations have been also found for ADRB2 (56), NEDD4L (87), HUT2 (88), PTPN1 (89), ADRB1 (90), and calcineurin (91). The BRIGHT study in Britain is similar in design to the FBPP and has published multiple linkage and association studies (92, 93), including contributing to a recent large genome-wide association study (94).
Genome-wide Association Studies
Initial genome-wide association (GWA) studies failed to detect genes related to hypertension at the genome-wide significance level in contrast to the highly successful detection of regions related to diabetes, obesity, lipids, uric acid, and noncardiovascular disease phenotypes. Of the 9 genome-wide scans listed in Table 2, only the 9p21 locus in the Amish study (95), two SNPs in the Global BPgen study (94), 2 chromosomal regions in the CHARGE consortium (96), and 6 regions in the African American sample from Washington D.C. (97) had genome-wide significance. After combining results with the replication samples in each study (many of which overlap with each other), additional regions were detected. Nevertheless, based on the “primary” scans, there were only two loci found in more than one study (and then only in two of the studies). These loci contained the genes SH2B3 and CACNB2. Ten total loci were identified between the CHARGE and Global BPgen studies when the total replication samples were combined with the main studies and then compared between the two large meta-analyses (96).
Table 2.
Primary* genome-wide study results for blood pressure or hypertension.
| Study or Location | Chromosome – Position (Mb) | ||
|---|---|---|---|
| WTCCC (98) (Affymetrix 500k) No signals met genome-wide significance. P-values of listed SNP regions p<5.8 × 10-5. |
SBP | DBP | HBP |
| 1 - 235.7 | |||
| 8 - 140.2 | |||
| 12 - 24.9 | |||
| 12 - 100.5 | |||
| 13 - 67.0 | |||
| 15 - 94.6 | |||
| Framingham (81) (Affymetrix 100k) No signals met genome-wide significance. P-values of listed SNP regions p<10-5. |
1 – 63.3 | 3 – 2.6 CNTN4 | |
| 5 – 110.8 CAMK4 | |||
| 6 – 88.7 | |||
| 8 – 73.3 | |||
| 14 – 75.7 C14orf118 | |||
| Finland (102) (Illumina 370k) No blood pressure signals met genome-wide significance. Interactions with environmental factors were identified. |
|||
| Amish (95) (Affymetrix 100k) Only chromosome 9 SNP genome-wide significant. Chromosome 2 at p<10-5. |
2 - 168.7 STK39 | ||
| 9 – 24.7 | |||
| Global BPgen (94) (Both Affymetrix and Illumina platforms of varying sizes were used in this meta analysis) Only two SNPs genome-wide significant. Others have p-values <10-5. |
1 – 11.8 MTHFR | 4 – 81.5 FGF5 | All BP loci were also associated with HBP |
| 10 – 104.8 CYP17A1 | 10 – 63.2 C10orf107 | ||
| 17 – 40.6 PLCD3 | 12 – 110.5 SH2B3 | ||
| 15 – 72.9 CYP1A2 | |||
| 17 – 44.8 ZNF652 | |||
| KORA (103) (Affymetrix 500k) No signals met genome-wide significance. P-values of listed SNP regions p<5 × 10-6. |
2 – 21.3 APOB | 5 – 121.3 SRFBP1 | |
| 5 – 162.6 CCNG1 | 16 – 81.2 CDH13 | ||
| 10 – 28.7 MMP7 | 18 – 68.9 NETO1 | ||
| 10 – 71.2 COL13A1 | |||
| 12 – 65.4 GRIP1 | |||
| 17 – 17.3 MED9 | |||
| CHARGE (96) (Both Affymetrix and Illumina platforms of varying sizes were used in this meta analysis) p< 10-6. Chromosome region 12 was genome-wide significant for SBP, DBP and HBP, as was chromosome 18 for SBP. |
1 – 10.7 CASZ1 | 2 – 190.6 MSTN | 3 – 37.6 ITGA9 |
| 2 – 190.5 PMS1 | 3 – 41.9 ULK4 | 10 – 18.7 CACNB2 | |
| 11 – 16.8 PLEKHA7 | 10 – 18.7 CACNB2 | 12 – 88.5 ATP2B1 | |
| 12 – 88.5 ATP2B1 | 11 – 16.9 PLEKHA7 | ||
| 12 – 110.4 SH2B3 | 12 – 88.5 ATP2B1 | ||
| 18 – 13.4 C18orf1 | 12 – 110.4 SH2B3 and ATXN2 | ||
| 12 – 111.1 TRAFD1, C12orf30, C12orf51 | |||
| 12 – 113.8 TBX3-TBX5 | |||
| 15 – 72.9 CYP1A2 | |||
| Taiwan (104) (Affymetrix 100k) Young onset hypertension. No single SNP hypertension signals met genome-wide significance. Significant 3-SNP haplotypes and SNP-SNP interactions were found. |
|||
| African-Americans (97) (Affymetrix 1M) |
2 – 190.4 PMS1 | ||
| 6 – 16.0 AL3652652 3 | |||
| 8 – 102.0 YWHAZ | |||
| 11 – 9.4 IPO7 | |||
| 14 – 91.9 SLC24A4 | |||
| 16 – 1.2 CACNA1H | |||
SBP: systolic blood pressure; DBP: diastolic blood pressure; HBP: hypertension.
Main study sample only. Does not include regions that are genome-wide significant after combination with the replication samples. P-value cutoff for listing SNPs varies somewhat by study.
It should be noted that the Wellcome Trust Case Control Consortium WTCCC study controls did not have blood pressure data available, so that 20-30% of subjects would be expected to have hypertension, thereby diminishing the statistical power for this trait(98). In addition, many of the SNPs with p-values that did not meet genome-wide significance levels may have overlapped with those loci listed in Table 2, if they had been added to the table. The Bonferonni correction may be too stringent, eliminating true positive signals. This is suggested by a study of atrial natriuretic peptides A and B (99). In this study, there was highly significant association of one or more SNPs in the two genes (10-70 and 10-68, respectively) coding for these peptides and an accompanying association with decreased blood pressure in the 10-5 range. If this had been a genome-wide study, these SNPs would have only been borderline significant for blood pressure after correction, yet appear to indicate true associations because of the additional intermediate phenotype results.
An unknown number of positive results listed in Table 2 may prove to be false positives or have such small effect that most studies would fail to detect them. In 11,433 subjects, the NHLBI Family Blood Pressure Program tested the top 6 SNPs reported in the WTCCC study as suggestive. Only one SNP was replicated, and that SNP was associated in opposite directions in two of the racial groups and not associated in another race (100). The Utah Pedigree study including more than 2000 pedigree members followed for 25 years failed to reveal a blood pressure association with 7 SNPs typed that were suggestive from the Framingham 100k project results (81), nor with 10 SNPs that were significant or suggestive in the CHARGE or Global BPgen studies (94, 96) (unpublished results). Power is certainly an issue with the Utah study and other similarly sized studies, as the effect sizes detected in the large consortia studies are less than 1 mmHg and required more than 20,000 subjects. Therefore, it is not surprising that there are negative studies published for replicated genes with small effect.
Furthermore, none of the genome-wide studies have yet found significant results for SNPs covering the physiological candidate genes that have fairly consistently been implicated in blood pressure control and hypertension development. The Framingham 100k study found AGT and the mineralocorticoid receptor (NR3C2) associated with blood pressure, but not at genome-wide significance levels (81). Many of the studies in Table 2 used the Affymetrix 100k or 500k chips, which did not cover the genes as well as Illumina or the Affymetrix 1M chips, and may explain the lack of association with these candidate genes. A study of SNPs from the Affymetrix 500k chip across 160 candidate genes for blood pressure has shown that the associations with the candidate genes are stronger when the SNPs on the chip tag over 50% of the HapMap SNPs (101). Even when the SNPs on the chip tag the majority of the HapMap SNPs, many of the candidate genes were not replicated in a second cohort. This suggests that the relevant SNPs are still not captured and/or the candidate genes have small effects just like the novel genes suggested in the genome-wide studies.
Summary
Why do genetic effect sizes appear to be small and explain relatively little of the variation in blood pressure? As hypothesized at the beginning of this article, genetic polymorphisms in or near genes related to the initial cause of increasing blood pressure may be more reliably detected with larger effect sizes if the compensating mechanisms were not in operation or the genetic signals could be uncovered by acute stresses on the pathway involved with the gene. We may often be studying the genetic effects of lack of compensation, which are also likely to be small. Note in Table 1 that the mean blood pressure differences among AGT genotypes after an intervention were in the range of 1.4-4.1 mmHg, much larger than the gene effect sizes in the genome-wide studies of resting blood pressure. On top of these genetic considerations, diet, exercise, stress and other environmental effects on blood pressure have not been comprehensively examined for confounding or interaction with the genes identified to date. The good news is that potential blood pressure and hypertension genes are being suggested, many of which are likely to be further replicated. However, we are left to conclude that most the detectable initiating and compensating genetic effect sizes are small. Thus, systems pathway analysis may be necessary to better delineate how small perturbations in the blood pressure control system are integrated into an age-related increase in blood pressure leading to hypertension.
Acknowledgments
Supported by NIH grants AG18734 and HL090668.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feinleib M, Garrison RJ, Fabsitz R, Christian JC, Hrubec Z, Borhani NO, et al. The NHLBI twin study of cardiovascular disease risk factors: methodology and summary of results. Am J Epidemiol. 1977;106:284–95. doi: 10.1093/oxfordjournals.aje.a112464. [DOI] [PubMed] [Google Scholar]
- 2.Hunt SC, Hasstedt SJ, Kuida H, Stults BM, Hopkins PH, Williams RR. Genetic heritability and common environmental components of resting and stressed blood pressures, lipids, and body mass index in Utah pedigrees and twins. Am J Epidemiol. 1989;129:625–38. doi: 10.1093/oxfordjournals.aje.a115175. [DOI] [PubMed] [Google Scholar]
- 3.Tambs K, Moum T, Holmen J, Eaves LJ, Neale MC, Lund-Larsen G, et al. Genetic and environmental effects on blood pressure in a Norwegian sample. Genet Epidemiol. 1992;9(1):11–26. doi: 10.1002/gepi.1370090104. [DOI] [PubMed] [Google Scholar]
- 4.Robinson WM, Borges-Osório MR, Callegari-Jacques SM, Achutti AC, de Silveira LG, Klein CH, et al. Genetic and nongenetic determinants of blood pressure in a southern Brazilian sample. Genet Epidemiol. 1991;8:55–67. doi: 10.1002/gepi.1370080106. [DOI] [PubMed] [Google Scholar]
- 5.Miller JZ, Weinberger MH, Christian JC, Daugherty SA. Familial resemblance in the blood pressure response to sodium restriction. Am J Epidemiol. 1987;126:822–30. doi: 10.1093/oxfordjournals.aje.a114719. [DOI] [PubMed] [Google Scholar]
- 6.Levine RS, Hennekens CH, Perry A, Cassady J, Gelband H, Jesse MJ. Genetic variance of blood pressure levels in infant twins. Am J Epidemiol. 1982;116:759–64. doi: 10.1093/oxfordjournals.aje.a113465. [DOI] [PubMed] [Google Scholar]
- 7.Pérusse L, Moll PP, Sing CF. Evidence that a single gene with gender- and age-dependent effects influences systolic blood pressure determination in a population-based sample. Am J Hum Genet. 1991;49:94–105. [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng LSC, Carmelli D, Hunt SC, Williams RR. Evidence for a major gene influencing 7-year increases in diastolic blood pressure with age. Am J Hum Genet. 1995;57:1169–77. [PMC free article] [PubMed] [Google Scholar]
- 9.Vaughn TT, Pletscher LS, Peripato A, King-Ellison K, Adams E, Erikson C, et al. Mapping quantitative trait loci for murine growth: a closer look at genetic architecture. Genet Res. 1999 Dec;74(3):313–22. doi: 10.1017/s0016672399004103. [DOI] [PubMed] [Google Scholar]
- 10.Shi G, Gu CC, Kraja AT, Arnett DK, Myers RH, Pankow JS, et al. Genetic effect on blood pressure is modulated by age: the Hypertension Genetic Epidemiology Network Study. Hypertension. 2009 Jan;53(1):35–41. doi: 10.1161/HYPERTENSIONAHA.108.120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia Z, Zhang A, Zhang H, Dong Z, Yang T. Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion. Circ Res. 2006 Nov 24;99(11):1243–51. doi: 10.1161/01.RES.0000251306.40546.08. [DOI] [PubMed] [Google Scholar]
- 12.Sanada H, Yatabe J, Midorikawa S, Hashimoto S, Watanabe T, Moore JH, et al. Single-nucleotide polymorphisms for diagnosis of salt-sensitive hypertension. Clin Chem. 2006 Mar;52(3):352–60. doi: 10.1373/clinchem.2005.059139. [DOI] [PubMed] [Google Scholar]
- 13.Weinberger MH, Stegner JE, Fineberg NS. A comparison of two tests for the assessment of blood pressure responses to sodium. Am J Hypertens. 1993 Mar;6(3 Pt 1):179–84. [PubMed] [Google Scholar]
- 14.Weinberger MH, Miller JZ, Luft FC, Fineberg NS, Grim CE, Christian JC. Genetic aspects of the blood pressure control system. Clin Exp Hypertens. 1981;3(4):569–81. doi: 10.3109/10641968109033684. [DOI] [PubMed] [Google Scholar]
- 15.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996 Mar;27(3 Pt 2):481–90. doi: 10.1161/01.hyp.27.3.481. [DOI] [PubMed] [Google Scholar]
- 16.Svetkey LP, McKeown SP, Wilson AF. Heritability of Salt Sensitivity in Black Americans. Hypertension. 1996 November 1;28(5):854–8. doi: 10.1161/01.hyp.28.5.854. [DOI] [PubMed] [Google Scholar]
- 17.Tuck ML, Williams GH, Dluhy RG, Greenfield M, Moore TJ. A delayed suppression of the renin-aldosterone axis following saline infusion in human hypertension. Circ Res. 1976 Nov;39(5):711–7. doi: 10.1161/01.res.39.5.711. [DOI] [PubMed] [Google Scholar]
- 18.Tuck ML, Dluhy RG, Williams GH. Sequential responses of the renin-angiotensin-aldosterone axis to acute postural change: effect of dietary sodium. J Lab Clin Med. 1975 Nov;86(5):754–63. [PubMed] [Google Scholar]
- 19.Weinberger MH, Miller JZ, Luft FC, Grim CE, Fineberg NS. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986 Jun;8(6 Pt 2):II127–34. doi: 10.1161/01.hyp.8.6_pt_2.ii127. [DOI] [PubMed] [Google Scholar]
- 20.Alexander RW, Gill JR, Jr, Yamabe H, Lovenberg W, Keiser HR. Effects of dietary sodium and of acute saline infusion on the interrelationship between dopamine excretion and adrenergic activity in man. J Clin Invest. 1974 Jul;54(1):194–200. doi: 10.1172/JCI107743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damasceno A, Santos A, Serrao P, Caupers P, Soares-da-Silva P, Polonia J. Deficiency of renal dopaminergic-dependent natriuretic response to acute sodium load in black salt-sensitive subjects in contrast to salt-resistant subjects. J Hypertens. 1999 Dec;17(12 Pt 2):1995–2001. doi: 10.1097/00004872-199917121-00033. [DOI] [PubMed] [Google Scholar]
- 22.Turner ST, Boerwinkle E. Genetics of blood pressure, hypertensive complications, and antihypertensive drug responses. Pharmacogenomics. 2003 Jan;4(1):53–65. doi: 10.1517/phgs.4.1.53.22587. [DOI] [PubMed] [Google Scholar]
- 23.Weinberger MH, Miller JZ, Fineberg NS, Luft FC, Grim CE, Christian JC. Association of haptoglobin with sodium sensitivity and resistance of blood pressure. Hypertension. 1987;10:443–6. doi: 10.1161/01.hyp.10.4.443. [DOI] [PubMed] [Google Scholar]
- 24.Manunta P, Cusi D, Barlassina C, Righetti M, Lanzani C, D'Amico M, et al. Alpha-adducin polymorphisms and renal sodium handling in essential hypertensive patients. Kidney Int. 1998 Jun;53(6):1471–8. doi: 10.1046/j.1523-1755.1998.00931.x. [DOI] [PubMed] [Google Scholar]
- 25.Ho H, Pinto A, Hall SD, Flockhart DA, Li L, Skaar TC, et al. Association Between the CYP3A5 Genotype and Blood Pressure. Hypertension. 2005 February 1;45(2):294–8. doi: 10.1161/01.HYP.0000151361.31736.96. [DOI] [PubMed] [Google Scholar]
- 26.Jeunemaitre X, Rigat B, Charru A, Houot AM, Soubrier F, Corvol P. Sib pair linkage analysis of renin gene haplotypes in human essential hypertension. Hum Genet. 1992;88:301–6. doi: 10.1007/BF00197264. [DOI] [PubMed] [Google Scholar]
- 27.Jeunemaitre X, Lifton RP, Hunt SC, Williams RR, Lalouel JM. Absence of linkage between the angiotensin converting enzyme locus and human essential hypertension. Nature Genetics. 1992;1:72–5. doi: 10.1038/ng0492-72. [DOI] [PubMed] [Google Scholar]
- 28.Jeunemaitre X, Soubrier F, Kotelevtsev Y, Lifton RP, Williams CS, Charru A, et al. Molecular basis of human hypertension: Role of angiotensinogen. Cell. 1992;71:169–80. doi: 10.1016/0092-8674(92)90275-h. [DOI] [PubMed] [Google Scholar]
- 29.Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, et al. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci. 1995;92:2735–9. doi: 10.1073/pnas.92.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim HS, Maeda N, Oh GT, Fernandez LG, Gomez RA, Smithies O. Homeostasis in mice with genetically decreased angiotensinogen is primarily by an increased number of renin-producing cells. J Biol Chem. 1999;274(20):14210–7. doi: 10.1074/jbc.274.20.14210. [DOI] [PubMed] [Google Scholar]
- 31.Lalouel JM, Rohrwasser A. Genetic susceptibility to essential hypertension: insight from angiotensinogen. Hypertension. 2007 Mar;49(3):597–603. doi: 10.1161/01.HYP.0000257145.20363.9c. [DOI] [PubMed] [Google Scholar]
- 32.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, et al. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension. 1999 Dec;34(6):1265–74. doi: 10.1161/01.hyp.34.6.1265. [DOI] [PubMed] [Google Scholar]
- 33.Hopkins PN, Hunt SC, Jeunemaitre X, Smith B, Solorio D, Fisher ND, et al. Angiotensinogen genotype affects renal and adrenal responses to angiotensin II in essential hypertension. Circulation. 2002 Apr 23;105(16):1921–7. doi: 10.1161/01.cir.0000014684.75359.68. [DOI] [PubMed] [Google Scholar]
- 34.Kobori H, Alper AB, Jr, Shenava R, Katsurada A, Saito T, Ohashi N, et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension. 2009 Feb;53(2):344–50. doi: 10.1161/HYPERTENSIONAHA.108.123802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watkins WS, Hunt SC, Williams GH, Tolpinrud W, Jeunemaitre X, Lalouel JM, et al. Genotype-phenotype analysis of angiotensinogen polymorphisms and essential hypertension: the importance of haplotypes. J Hypertens. 2009 Sep 16; doi: 10.1097/HJH.0b013e328332031a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunt SC, Cook NR, Oberman A, Cutler JA, Hennekens CH, Allender PS, et al. Angiotensinogen genotype, sodium reduction, weight loss, and prevention of hypertension: trials of hypertension prevention, phase II. Hypertension. 1998;32(3):393–401. doi: 10.1161/01.hyp.32.3.393. [DOI] [PubMed] [Google Scholar]
- 37.Svetkey LP, Moore TJ, Simons-Morton DG, Appel LJ, Bray GA, Sacks FM, et al. Angiotensinogen genotype and blood pressure response in the Dietary Approaches to Stop Hypertension (DASH) study. J Hypertens. 2001 Nov;19(11):1949–56. doi: 10.1097/00004872-200111000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Hunt SC, Geleijnse JM, Wu LL, Witteman JCM, Williams RR, Grobbee DE. Enhanced blood pressure response to mild sodium reduction in subjects with the 235T variant of the angiotensinogen gene. Am J Hypertens. 1999;12:460–6. doi: 10.1016/s0895-7061(99)00014-x. [DOI] [PubMed] [Google Scholar]
- 39.Janke J, Engeli S, Gorzelniak K, Luft FC, Sharma AM. Mature adipocytes inhibit in vitro differentiation of human preadipocytes via angiotensin type 1 receptors. Diabetes. 2002 Jun;51(6):1699–707. doi: 10.2337/diabetes.51.6.1699. [DOI] [PubMed] [Google Scholar]
- 40.Danforth E., Jr Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000 Sep;26(1):13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 41.Sharma AM, Janke J, Gorzelniak K, Engeli S, Luft FC. Angiotensin blockade prevents type 2 diabetes by formation of fat cells. Hypertension. 2002 Nov;40(5):609–11. doi: 10.1161/01.hyp.0000036448.44066.53. [DOI] [PubMed] [Google Scholar]
- 42.Engeli S, Bohnke J, Gorzelniak K, Janke J, Schling P, Bader M, et al. Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005 Mar;45(3):356–62. doi: 10.1161/01.HYP.0000154361.47683.d3. [DOI] [PubMed] [Google Scholar]
- 43.Vuagnat A, Giacche M, Hopkins PN, Azizi M, Hunt SC, Vedie B, et al. Blood pressure response to angiotensin II, low-density lipoprotein cholesterol and polymorphisms of the angiotensin II type 1 receptor gene in hypertensive sibling pairs. J Mol Med. 2001 May;79(4):175–83. doi: 10.1007/s001090100205. [DOI] [PubMed] [Google Scholar]
- 44.Nickenig G, Sachinidis A, Michaelsen F, Bohm M, Seewald S, Vetter H. Upregulation of vascular angiotensin II receptor gene expression by low-density lipoprotein in vascular smooth muscle cells. Circulation. 1997 Jan 21;95(2):473–8. doi: 10.1161/01.cir.95.2.473. [DOI] [PubMed] [Google Scholar]
- 45.Bianchi G, Tripodi G, Casari G, Salardi S, Barber B, Garcia R, et al. Two point mutations within the adducin genes are involved in blood pressure variation. Proc Natl Acad Sci. 1994;91:3999–4003. doi: 10.1073/pnas.91.9.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staessen JA, Wang JG, Brand E, Barlassina C, Birkenhager WH, Herrmann SM, et al. Effects of three candidate genes on prevalence and incidence of hypertension in a Caucasian population. J Hypertens. 2001 Aug;19(8):1349–58. doi: 10.1097/00004872-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Barlassina C, Dal Fiume C, Lanzani C, Manunta P, Guffanti G, Ruello A, et al. Common genetic variants and haplotypes in renal CLCNKA gene are associated to salt-sensitive hypertension. Hum Mol Genet. 2007 Jul 1;16(13):1630–8. doi: 10.1093/hmg/ddm112. [DOI] [PubMed] [Google Scholar]
- 48.Williams GH, Dluhy RG, Lifton RP, Moore TJ, Gleason R, Williams R, et al. Non-modulation as an intermediate phenotype in essential hypertension. Hypertension. 1992;20:788–96. doi: 10.1161/01.hyp.20.6.788. [DOI] [PubMed] [Google Scholar]
- 49.Chang YPC, Liu X, Kim JDO, Ikeda MA, Layton MR, Weder AB, et al. Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am J Hum Genet. 2007 Feb;80(2):253–64. doi: 10.1086/510918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiolero A, Maillard M, Nussberger J, Brunner HR, Burnier M. Proximal sodium reabsorption: An independent determinant of blood pressure response to salt. Hypertension. 2000 Oct;36(4):631–7. doi: 10.1161/01.hyp.36.4.631. [DOI] [PubMed] [Google Scholar]
- 51.Carey RM. Theodore Cooper Lecture: Renal dopamine system: paracrine regulator of sodium homeostasis and blood pressure. Hypertension. 2001 Sep;38(3):297–302. doi: 10.1161/hy0901.096422. [DOI] [PubMed] [Google Scholar]
- 52.Sato M, Soma M, Nakayama T, Kanmatsuse K. Dopamine D1 receptor gene polymorphism is associated with essential hypertension. Hypertension. 2000 Aug;36(2):183–6. doi: 10.1161/01.hyp.36.2.183. [DOI] [PubMed] [Google Scholar]
- 53.Jia H, Hingorani AD, Sharma P, Hopper R, Dickerson C, Trutwein D, et al. Association of the G(s)alpha gene with essential hypertension and response to beta-blockade. Hypertension. 1999 Jul;34(1):8–14. doi: 10.1161/01.hyp.34.1.8. [DOI] [PubMed] [Google Scholar]
- 54.Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, et al. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci U S A. 2002 Mar 19;99(6):3872–7. doi: 10.1073/pnas.062694599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao F, Zhang L, Wessel J, Zhang K, Wen G, Kennedy BP, et al. Tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis: discovery of common human genetic variants governing transcription, autonomic activity, and blood pressure in vivo. Circulation. 2007 Aug 28;116(9):993–1006. doi: 10.1161/CIRCULATIONAHA.106.682302. [DOI] [PubMed] [Google Scholar]
- 56.Krushkal J, Xiong M, Ferrell R, Sing CF, Turner ST, Boerwinkle E. Linkage and association of adrenergic and dopamine receptor genes in the distal portion of the long arm of chromosome 5 with systolic blood pressure variation. Hum Mol Genet. 1998;7(9):1379–83. doi: 10.1093/hmg/7.9.1379. [DOI] [PubMed] [Google Scholar]
- 57.Ranade K, Shue WH, Hung YJ, Hsuing CA, Chiang FT, Pesich R, et al. The glycine allele of a glycine/arginine polymorphism in the beta2-adrenergic receptor gene is associated with essential hypertension in a population of Chinese origin. Am J Hypertens. 2001 Dec;14(12):1196–200. doi: 10.1016/s0895-7061(01)02213-0. [DOI] [PubMed] [Google Scholar]
- 58.Wallerstedt SM, Eriksson AL, Ohlsson C, Hedner T. Haplotype association analysis of the polymorphisms Arg16Gly and Gln27Glu of the adrenergic beta2 receptor in a Swedish hypertensive population. J Hum Hypertens. 2005 Sep;19(9):705–8. doi: 10.1038/sj.jhh.1001897. [DOI] [PubMed] [Google Scholar]
- 59.Wu H, Tang W, Li H, Zhou X, Yang Y, Yu H, et al. Association of the beta2-adrenergic receptor gene with essential hypertension in the non-Han Chinese Yi minority human population. J Hypertens. 2006 Jun;24(6):1041–7. doi: 10.1097/01.hjh.0000226193.21311.e1. [DOI] [PubMed] [Google Scholar]
- 60.Tomaszewski M, Brain NJ, Charchar FJ, Wang WY, Lacka B, Padmanabahn S, et al. Essential hypertension and beta2-adrenergic receptor gene: linkage and association analysis. Hypertension. 2002 Sep;40(3):286–91. doi: 10.1161/01.hyp.0000029105.21202.fe. [DOI] [PubMed] [Google Scholar]
- 61.Busst CJ, Scurrah KJ, Ellis JA, Harrap SB. Selective genotyping reveals association between the epithelial sodium channel gamma-subunit and systolic blood pressure. Hypertension. 2007 Oct;50(4):672–8. doi: 10.1161/HYPERTENSIONAHA.107.089128. [DOI] [PubMed] [Google Scholar]
- 62.Büsst CJ, Scurrah KJ, Ellis JA, Xin Y, Brinton EA, Hopkins PN, et al. Epithelial sodium channel gamma-subunit polymorphisms are associated with 25-year follow-up blood pressures in Utah pedigrees. Hypertension. 2007 abstract-O92, Council for High Blood Pressure Research. [Google Scholar]
- 63.Pankow JS, Dunn DM, Hunt SC, Leppert MF, Miller MB, Rao DC, et al. Further evidence of a quantitative trait locus on chromosome 18q21 influencing postural change in systolic blood pressure: The HyperGEN study. Am J Hypertens. 2005;18:672–8. doi: 10.1016/j.amjhyper.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 64.Russo CJ, Melista E, Cui J, DeStefano AL, Bakris GL, Manolis AJ, et al. Association of NEDD4L ubiquitin ligase with essential hypertension. Hypertension. 2005 Sep;46(3):488–91. doi: 10.1161/01.HYP.0000178594.63193.c0. [DOI] [PubMed] [Google Scholar]
- 65.Busjahn A, Aydin A, Uhlmann R, Krasko C, Bahring S, Szelestei T, et al. Serum- and glucocorticoid-regulated kinase (SGK1) gene and blood pressure. Hypertension. 2002 Sep;40(3):256–60. doi: 10.1161/01.hyp.0000030153.19366.26. [DOI] [PubMed] [Google Scholar]
- 66.Barkley RA, Chakravarti A, Cooper RS, Ellison RC, Hunt SC, Province MA, et al. Positional identification of hypertension susceptibility genes on chromosome 2. Hypertension. 2004 Feb;43(2):477–82. doi: 10.1161/01.HYP.0000111585.76299.f7. [DOI] [PubMed] [Google Scholar]
- 67.Hunt SC, Xin Y, Wu LL, Cawthon RM, Coon H, Hasstedt SJ, et al. Sodium bicarbonate cotransporter polymorphisms are associated with baseline and 10-year follow-up blood pressures. Hypertension. 2006 Mar;47(3):532–6. doi: 10.1161/01.HYP.0000196949.26088.3c. [DOI] [PubMed] [Google Scholar]
- 68.Dahl LK, Heine M, Thompson K. Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ Res. 1974 Jan;40(4):94–101. doi: 10.1161/01.res.40.4.94. [DOI] [PubMed] [Google Scholar]
- 69.Lifton RP. Genetic dissection of human blood pressure variation: common pathways from rare phenotypes. Harvey Lect. 2004;100:71–101. [PubMed] [Google Scholar]
- 70.Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005 Apr;85(2):679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 71.Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, et al. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest. 2005 Apr;115(4):1092–9. doi: 10.1172/JCI200523378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006 Nov 21;103(47):17985–90. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ge Y, Bagnall A, Stricklett PK, Webb D, Kotelevtsev Y, Kohan DE. Combined knockout of collecting duct endothelin A and B receptors causes hypertension and sodium retention. Am J Physiol Renal Physiol. 2008 Dec;295(6):F1635–40. doi: 10.1152/ajprenal.90279.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Wardener HE, MacGregor GA. Sodium and blood pressure. Curr Opin Cardiol. 2002 Jul;17(4):360–7. doi: 10.1097/00001573-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 75.Frohlich ED, Varagic J. Sodium directly impairs target organ function in hypertension. Curr Opin Cardiol. 2005 Sep;20(5):424–9. doi: 10.1097/01.hco.0000175519.34933.a5. [DOI] [PubMed] [Google Scholar]
- 76.Frohlich ED. The role of salt in hypertension: the complexity seems to become clearer. Nat Clin Pract Cardiovasc Med. 2008 Jan;5(1):2–3. doi: 10.1038/ncpcardio1087. [DOI] [PubMed] [Google Scholar]
- 77.Varagic J, Frohlich ED, Susic D, Ahn J, Matavelli L, Lopez B, et al. AT1 receptor antagonism attenuates target organ effects of salt excess in SHRs without affecting pressure. Am J Physiol Heart Circ Physiol. 2008 Feb;294(2):H853–8. doi: 10.1152/ajpheart.00737.2007. [DOI] [PubMed] [Google Scholar]
- 78.Lombard JH, Sylvester FA, Phillips SA, Frisbee JC. High-salt diet impairs vascular relaxation mechanisms in rat middle cerebral arteries. Am J Physiol Heart Circ Physiol. 2003 Apr;284(4):H1124–33. doi: 10.1152/ajpheart.00835.2002. [DOI] [PubMed] [Google Scholar]
- 79.Ferri C, Bellini C, Desideri G, Giuliani E, De Siati L, Cicogna S, et al. Clustering of endothelial markers of vascular damage in human salt-sensitive hypertension: influence of dietary sodium load and depletion. Hypertension. 1998 Nov;32(5):862–8. doi: 10.1161/01.hyp.32.5.862. [DOI] [PubMed] [Google Scholar]
- 80.Chen W, Srinivasan SR, Boerwinkle E, Berenson GS. Beta-adrenergic receptor genes are associated with arterial stiffness in black and white adults: the Bogalusa Heart Study. Am J Hypertens. 2007 Dec;20(12):1251–7. doi: 10.1016/j.amjhyper.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 81.Levy D, Larson MG, Benjamin EJ, Newton-Cheh C, Wang TJ, Hwang SJ, et al. Framingham Heart Study 100K Project: genome-wide associations for blood pressure and arterial stiffness. BMC Med Genet. 2007;8 1:S3. doi: 10.1186/1471-2350-8-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rao DC, Province MA, Leppert MF, Oberman A, Heiss G, Ellison RC, et al. A genome-wide affected sibpair linkage analysis of hypertension: the HyperGEN network. Am J Hypertens. 2003 Feb;16(2):148–50. doi: 10.1016/s0895-7061(02)03247-8. [DOI] [PubMed] [Google Scholar]
- 83.Kardia SL, Rozek LS, Krushkal J, Ferrell RE, Turner ST, Hutchinson R, et al. Genome-wide linkage analyses for hypertension genes in two ethnically and geographically diverse populations. Am J Hypertens. 2003 Feb;16(2):154–7. doi: 10.1016/s0895-7061(02)03249-1. [DOI] [PubMed] [Google Scholar]
- 84.Thiel BA, Chakravarti A, Cooper RS, Luke A, Lewis S, Lynn A, et al. A genome-wide linkage analysis investigating the determinants of blood pressure in whites and african americans. Am J Hypertens. 2003 Feb;16(2):151–3. doi: 10.1016/s0895-7061(02)03246-6. [DOI] [PubMed] [Google Scholar]
- 85.Ranade K, Hinds D, Hsiung CA, Chuang LM, Chang MS, Chen YT, et al. A genome scan for hypertension susceptibility loci in populations of Chinese and Japanese origins. Am J Hypertens. 2003 Feb;16(2):158–62. doi: 10.1016/s0895-7061(02)03245-4. [DOI] [PubMed] [Google Scholar]
- 86.Province MA, Kardia SL, Ranade K, Rao DC, Thiel BA, Cooper RS, et al. A meta-analysis of genome-wide linkage scans for hypertension:The National Heart, Lung and Blood Institute Family Blood Pressure Program. Am J Hypertens. 2003 Feb;16(2):144–7. doi: 10.1016/s0895-7061(02)03248-x. [DOI] [PubMed] [Google Scholar]
- 87.Dunn DM, Ishigami T, Pankow J, Von Niederhausern A, Alder J, Hunt SC, et al. Common variant of human NEDD4L activates a cryptic splice site to form a frameshifted transcript. J Hum Genet. 2002;47(12):665–76. doi: 10.1007/s100380200102. [DOI] [PubMed] [Google Scholar]
- 88.Ranade K, Wu KD, Hwu CM, Ting CT, Pei D, Pesich R, et al. Genetic variation in the human urea transporter-2 is associated with variation in blood pressure. Hum Mol Genet. 2001 Sep 15;10(19):2157–64. doi: 10.1093/hmg/10.19.2157. [DOI] [PubMed] [Google Scholar]
- 89.Olivier M, Hsiung CA, Chuang LM, Ho LT, Ting CT, Bustos VI, et al. Single nucleotide polymorphisms in protein tyrosine phosphatase 1beta (PTPN1) are associated with essential hypertension and obesity. Hum Mol Genet. 2004 Sep 1;13(17):1885–92. doi: 10.1093/hmg/ddh196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ranade K, Jorgenson E, Sheu WH, Pei D, Hsiung CA, Chiang FT, et al. A polymorphism in the beta1 adrenergic receptor is associated with resting heart rate. Am J Hum Genet. 2002 Apr;70(4):935–42. doi: 10.1086/339621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tang W, Arnett DK, Devereux RB, Panagiotou D, Province MA, Miller MB, et al. Identification of a novel 5-base pair deletion in calcineurin B (PPP3R1) promoter region and its association with left ventricular hypertrophy. Am Heart J. 2005 Oct;150(4):845–51. doi: 10.1016/j.ahj.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 92.Caulfield M, Munroe P, Pembroke J, Samani N, Dominiczak A, Brown M, et al. Genome-wide mapping of human loci for essential hypertension. Lancet. 2003 Jun 21;361(9375):2118–23. doi: 10.1016/S0140-6736(03)13722-1. [DOI] [PubMed] [Google Scholar]
- 93.Newhouse SJ, Wallace C, Dobson R, Mein C, Pembroke J, Farrall M, et al. Haplotypes of the WNK1 gene associate with blood pressure variation in a severely hypertensive population from the British Genetics of Hypertension study. Hum Mol Genet. 2005 Jul 1;14(13):1805–14. doi: 10.1093/hmg/ddi187. [DOI] [PubMed] [Google Scholar]
- 94.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009 May 10; doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, O'Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, et al. Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci U S A. 2009 Jan 6;106(1):226–31. doi: 10.1073/pnas.0808358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009 May 10; doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Adeyemo A, Gerry N, Chen G, Herbert A, Doumatey A, Huang H, et al. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009 Jul;5(7):e1000564. doi: 10.1371/journal.pgen.1000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007 Jun 7;447(7145):661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet. 2009 Mar;41(3):348–53. doi: 10.1038/ng.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ehret GB, Morrison AC, O'Connor AA, Grove ML, Baird L, Schwander K, et al. Replication of the Wellcome Trust genome-wide association study of essential hypertension: the Family Blood Pressure Program. Eur J Hum Genet. 2008 Jun 4;102 doi: 10.1038/ejhg.2008.102. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sober S, Org E, Kepp K, Juhanson P, Eyheramendy S, Gieger C, et al. Targeting 160 candidate genes for blood pressure regulation with a genome-wide genotyping array. PLoS One. 2009;4(6):e6034. doi: 10.1371/journal.pone.0006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sabatti C, Service SK, Hartikainen AL, Pouta A, Ripatti S, Brodsky J, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet. 2009 Jan;41(1):35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Org E, Eyheramendy S, Juhanson P, Gieger C, Lichtner P, Klopp N, et al. Genome-wide scan identifies CDH13 as a novel susceptibility locus contributing to blood pressure determination in two European populations. Hum Mol Genet. 2009 Jun 15;18(12):2288–96. doi: 10.1093/hmg/ddp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang HC, Liang YJ, Wu YL, Chung CM, Chiang KM, Ho HY, et al. Genome-wide association study of young-onset hypertension in the Han Chinese population of Taiwan. PLoS One. 2009;4(5):e5459. doi: 10.1371/journal.pone.0005459. [DOI] [PMC free article] [PubMed] [Google Scholar]