

Abstract
We introduce proteolytic scanning calorimetry, a modification of the differential scanning calorimetry approach to the determination of protein stability in which a proteolytic enzyme (thermolysin) is used to mimic a harsh environment. This methodology allows the straightforward calculation of the rate of irreversible denaturation as a function of temperature and concentration of proteolytic enzyme and, as a result, has the potential to probe efficiently the fundamental biophysical features of protein kinetic stability. In the particular case of Escherichia coli thioredoxin (used as an illustrative example in this article), we find that the rate of irreversible denaturation is determined by 1), the global unfolding mechanism at low thermolysin concentrations, indicating that thermodynamic stability may contribute directly to the kinetic stability of thioredoxin under moderately harsh conditions and 2), the rate of unfolding at high thermolysin concentrations, indicating that the free-energy barrier for unfolding may act as a safety mechanism that ensures significant kinetic stability, even in very harsh environments. This thioredoxin picture, however, is by no means expected to be general and different proteins may show different patterns of kinetic stabilization. Proteolytic scanning calorimetry is particularly well-suited to probe this diversity at a fundamental biophysical level.
Main Text
In vivo, proteins often face crowded or harsh conditions under which irreversible denaturation might in principle efficiently occur through unfolded or partially unfolded conformations. Hence, natural selection has endowed many proteins with kinetic stability (i.e., the native, functional protein is protected by a high free-energy barrier), a fact for which experimental evidence has been steadily accumulating (see, for instance, references 1–8).
By contrast, many in vitro denaturation experiments are performed under simple solvent conditions, involve comparatively short timescales, and use small model proteins. These in vitro experiments often lead to reversible equilibrium denaturation and emphasize thermodynamic stability (i.e., an unfolding free energy change that favors the folded state under native conditions).
Here, we explore how the environment harshness of in vitro protein denaturation experiments can be modulated and we show that fundamental information about kinetic stability can thus be derived. As a model system for this study we have chosen thioredoxin from Escherichia coli, a protein which is often taken as a paradigm of reversible denaturation and thermodynamically controlled stability (9). We use a DSC approach to the characterization of protein stability; i.e., the protein solution is heated at a constant temperature-scanning rate, heat capacity as a function of temperature is measured, and denaturation processes are revealed by transitions (heat capacity peaks) in the thermogram. Unlike conventional DSC experiments, however, we include the proteolytic enzyme thermolysin (which hydrolyzes peptide bonds at hydrophobic substrates) in the sample solution to mimic a harsh environment. The idea behind this procedure is that kinetic stability is related to a high free-energy barrier separating the folded biologically functional protein from the unfolded or partially unfolded states that may be highly susceptible to irreversible alterations and/or undesirable interactions and that such states can be probed by their susceptibility to proteolysis. Thermolysin is particularly appropriate for our modified DSC experiment because of its high denaturation temperature, broad specificificity, and robustness (10–13); although other proteases could in principle be used, provided that their denaturation temperature is higher than that for the protein under study.
Fig. 1 A shows several DSC scans of thioredodoxin plus thermolysin solutions using different concentrations of the proteolytic enzyme. The first noteworthy result is that the calorimetric transition for thioredoxin denaturation is responsive to the presence of thermolysin over several orders of magnitude of the concentration of the proteolytic enzyme. A second important result is that, although the transition temperature for thioredoxin denaturation shifts to lower temperatures upon increasing thermolysin concentration, it remains clearly above physiological temperature (37°C) even at the highest thermolysin concentrations used. This indicates (see below for further discussion) that thioredoxin is actually a kinetically stable protein, as suggested by our previous studies on the relation between mutation effects on thioredoxin unfolding rates and sequence-alignment statistics (14).
Figure 1.
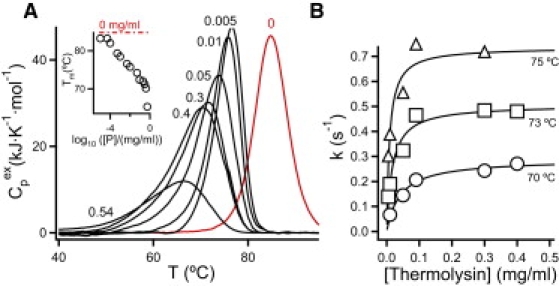
(A) Calorimetric transitions for thioredoxin denaturation in the presence of several thermolysin concentrations (shown in mg/mL as the numbers alongside the transitions). See the Supporting Material for details about calorimetric experiments and thermogram processing. (Inset) plot of transition temperature (maximum of the transition) versus log10 of thermolysin concentration. (B) Illustrative plots of rate constant for irreversible denaturation versus thermolysin concentration for the temperatures shown. Rates constant values are calculated from the DSC transitions (see text and the Supporting Material for details) and the lines are the best fits of Eq. 3.
The calorimetric transition for thioredoxin in the absence of thermolysin is to be attributed to protein unfolding and, therefore, defines the temperature range in which unfolded and partially unfolded states become significantly populated (i.e., within that temperature range, their concentration is comparable to the total protein concentration). The transitions observed at lower temperatures in the presence of thermolysin are, therefore, kinetically controlled and only involve the native protein and the final proteolysis product (F) as significantly populated states:
| (1) |
which is a phenomenological, two-state irreversible description of the denaturation process (10,15). Actually, at high thermolysin concentrations the calorimetric transitions (Fig. 1 A) show the asymmetry expected for a two-state irreversible denaturation process (10). It must be noted, however, that efficient proteolysis requires some degree of unfolding (11). Therefore, the simplest mechanism compatible with the model in Eq. 1 is
| (2) |
where X is an unfolded or partially unfolded state which, however, is not significantly populated under the conditions in which the two-state irreversible description (Eq. 1) applies (that is [X]<<[N]+[F]).
An expression for the first-order rate constant (k) for the process shown in Eq. 1 can be derived from the mechanism depicted in Eq. 2 by applying the steady-state approximation to X. The well-known result (11) can be written as
| (3) |
The meaning of the symbols in Eq. 3 is as follows: [P] is the concentration of proteolytic enzyme (thermolysin), k0 is the product K·kP, where K is the equilibrium constant for the preequilibrium N↔X and kP is the second order rate constant for the proteolysis step, X→F. Note that k ≈ k0 when the concentration of thermolysin is low and the proteolysis step becomes rate-determining (the EX2 condition).The value k1 is the rate constant for the opening step, N→X and k ≈ k1 at sufficiently high concentrations of thermolysin when the opening is rate-limiting (the EX1 condition). Equation 3 is, of course, well-known, although it is often couched in a somewhat different terminology (11).
We demonstrated in earlier work (10,15) that the rate constant k for a two-state irreversible denaturation can be calculated from the corresponding DSC thermograms using,
| (4) |
where υ is the scanning rate, Cp and <H> are the excess heat capacity, and the excess enthalpy (determined with respect to a chemical baseline) at a temperature T and ΔH is the total enthalpy of the transition. Application of Eq. 4 to the calorimetric transitions for thioredoxin denaturation (see the Supporting Material for details) leads to the value of the rate constant k, as a function of, both temperature and thermolysin concentration. Plots of k versus thermolysin concentration for given temperatures (Fig. 1 B) show the trend predicted by Eq. 3, with the transition between the EX2 and EX1 conditions clearly apparent as thermolysin concentration is increased. Excellent fits of Eq. 3 to these profiles are in fact obtained, leading to values of both k0 and k1 as a function of temperature (Fig. 2 A).
Figure 2.
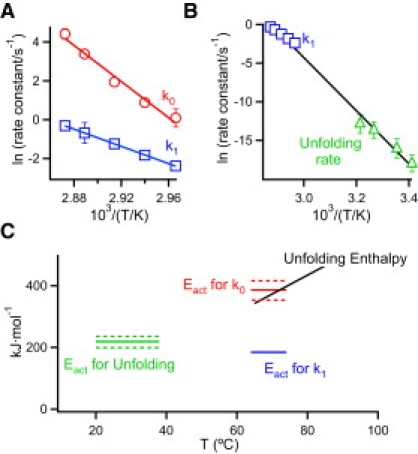
(A) Arrhenius plots for irreversible denaturation of thioredoxin in the presence of thermolysin under EX2 and EX1 conditions (rate constants k0 and k1 respectively, derived from the fittings illustrated in Fig. 1B). (B) Arrhenius plot for the indicated rate constants. Those labeled unfolding rates were determined from guanidine-induced denaturation experiments by extrapolation to zero denaturant concentration (see the Supporting Material for details). Error bars are the associated uncertainties calculated from the fitting. Errors bars are not shown when they are smaller than the size of the symbol. (C) Thioredoxin unfolding enthalpy (data from Georgescu et al. (9) and activation energy values for the rates of irreversible denaturation (k0 and k1) and unfolding. Dashed lines indicate the uncertainties from the Arrhenius fittings.
The Arrhenius plot for k0 leads to an activation energy of 386 kJ/mol, very close to the total unfolding enthalpy for thioredoxin in the temperature range under consideration (Fig. 2 C). Since k0 is the product K·kP, it appears reasonable to attribute its very large temperature dependence to K (the equilibrium constant for the N ⇔ X step) and to assume that X is in fact the unfolded state of the protein. Accordingly, proteolysis under the EX2 condition occurs through the global unfolding mechanism in this case.
The Arrhenius plot for k1 indicates an activation energy of 184 kJ/mol, significantly lower than that determined for k0 (Fig. 2 C). For a two-state irreversible denaturation (Eq. 1) there is an inverse relationship between the activation energy value and the width of the calorimetric transition (see Appendix in Costas et al. (8). The decrease in activation energy as thermolysin concentration is increased (and the mechanism changes from EX2 to EX1) is, in fact, visually apparent in the calorimetric transitions of Fig. 1 A. The most interesting implication of the calculated k1 values is, however, that they agree reasonably well with the unfolding rates calculated from guanidine-induced denaturation by extrapolation to zero denaturant concentration (Fig. 2 B). Accordingly, proteolysis under EX1 conditions is determined by the rate of unfolding.
Overall, the above results delineate fundamental features of how kinetic stability is implemented in the thioredoxin molecule. At low concentrations of thermolysin, irreversible denaturation occurs through the global unfolding mechanism and, therefore, thermodynamic stability (implying a low concentration of unfolded state in equilibrium with the folded protein) contributes directly to kinetic stability. At high thermolysin concentrations (very harsh environment), the rate of irreversible denaturation is determined by the rate of unfolding; therefore, as we demonstrated several years ago in an earlier study (1), the free-energy barrier for unfolding may act as a kind of safety device, since irreversible denaturation that occurs through unfolded or partially unfolded states cannot be faster than the crossing of this barrier.
Proteolytic scanning calorimetry provides, therefore, a particularly clear picture of thioredoxin kinetic stability and its relation with thermodynamic stability. This thioredoxin picture, however, is by no means expected to be general. Different proteins may show different degrees of kinetic stabilization, different types of relation between the kinetic and thermodynamic stabilities, and different mechanisms of irreversible denaturation (involving, for instance, local unfolding processes and partially unfolded forms). Proteolytic scanning calorimetry seems well-suited to probe this diversity at a fundamental biophysical level and, in this sense, it should be complementary to the recently developed high-throughput methodologies (6,7) for the determination of kinetic stability.
Acknowledgments
This work was supported by Feder funds and grant BIO2006-07332 (Spanish Minsitry of Science and Education) to J.M.S.R. and grant CVI 1668 (Junta de Andalucia) to B.I.M.
Supporting Material
References and Footnotes
- 1.Plaza del Pino I.M., Ibarra-Molero B., Sanchez-Ruiz J.M. Lower kinetic limit to protein thermal stability: a proposal regarding protein stability in vivo and its relation with misfolding diseases. Proteins. 2000;40:58–70. doi: 10.1002/(sici)1097-0134(20000701)40:1<58::aid-prot80>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 2.Jaswal S.S., Sohl J.L., Agard D.A. Energetic landscape of alpha-lytic protease optimizes longevity through kinetic stability. Nature. 2002;415:343–346. doi: 10.1038/415343a. [DOI] [PubMed] [Google Scholar]
- 3.Hammarström P., Wiseman R.L., Kelly J.W. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 4.Jayaraman S., Gantz D., Gursky O. Structural basis for thermal stability of human low-density lipoprotein. Biochemistry. 2005;44:3965–3971. doi: 10.1021/bi047493v. [DOI] [PubMed] [Google Scholar]
- 5.Flaugh S.L., Mills I.A., King J. Glutamine deamidation destabilizes human gammaD-crystallin and lowers the kinetic barrier to unfolding. J. Biol. Chem. 2006;281:30782–30793. doi: 10.1074/jbc.M603882200. [DOI] [PubMed] [Google Scholar]
- 6.Park C., Zhou S., Marqusee S. Energetics-based protein profiling on a proteomic scale: identification of proteins resistant to proteolysis. J. Mol. Biol. 2007;368:1426–1437. doi: 10.1016/j.jmb.2007.02.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia K., Manning M., Colon W. Identifying the subproteome of kinetically stable proteins via diagonal 2D SDS/PAGE. Proc. Natl. Acad. Sci. USA. 2007;104:6043–6049. doi: 10.1073/pnas.0705417104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costas M., Rodríguez-Larrea D., Sanchez-Ruiz J.M. Between-species variation in the kinetic stability of TIM proteins linked to solvation-barrier free energies. J. Mol. Biol. 2009;385:924–937. doi: 10.1016/j.jmb.2008.10.056. [DOI] [PubMed] [Google Scholar]
- 9.Georgescu R.E., Garcia-Mira M.M., Sanchez-Ruiz J.M. Heat capacity analysis of oxidized Escherichia coli thioredoxin fragments (1--73, 74--108) and their noncovalent complex. Evidence for the burial of apolar surface in protein unfolded states. Eur. J. Biochem. 2001;268:1477–1485. doi: 10.1046/j.1432-1327.2001.02014.x. [DOI] [PubMed] [Google Scholar]
- 10.Sánchez-Ruiz J.M., López-Lacomba J.L., Mateo P.L. Differential scanning calorimetry of the irreversible thermal denaturation of thermolysin. Biochemistry. 1988;27:1648–1652. doi: 10.1021/bi00405a039. [DOI] [PubMed] [Google Scholar]
- 11.Park C., Marqusee S. Probing the high energy states in proteins by proteolysis. J. Mol. Biol. 2004;343:1467–1476. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 12.Park C., Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat. Methods. 2005;2:207–212. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 13.Thermolysin, in fact, has been recently used in the development of the pulse-proteolysis method for the high-throughput screening of protein thermodynamic stability (12). In pulse proteolysis, thermolysin is allowed to act during a comparatively short time on a slow-relaxing equilibrium mixture of native and unfolded protein, thus cleaving only the unfolded protein and probing the position of the folding/unfolding equilibrium. Our proteolysis scanning calorimetry experiment is, on the other hand, meant to probe kinetic stability at a fundamental biophysical level and, therefore, thermolysin is not pulsed, but is present during the whole temperature range of the experiment.
- 14.Godoy-Ruiz R., Ariza F., Sanchez-Ruiz J.M. Natural selection for kinetic stability is a likely origin of correlations between mutational effects on protein energetics and frequencies of amino acid occurrences in sequence alignments. J. Mol. Biol. 2006;362:966–978. doi: 10.1016/j.jmb.2006.07.065. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez-Ruiz J.M. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry. Biophys. J. 1992;61:921–935. doi: 10.1016/S0006-3495(92)81899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.