

Abstract
Fusion of cellular membranes is a ubiquitous biological process requiring remodeling of two phospholipid bilayers. We believe it is very likely that merging of membranes proceeds via similar sequential intermediates. Contacting membranes form a stalk between the proximal leaflets that expands radially into an hemifusion diaphragm (HD) and subsequently open to a fusion pore. Although considered to be a key intermediate in fusion, direct experimental verification of this structure is difficult due to its transient nature. Using confocal fluorescence microscopy we have investigated the fusion of giant unilamellar vesicles (GUVs) containing phosphatidylserine and fluorescent virus derived transmembrane peptides or membrane proteins in the presence of divalent cations. Time-resolved imaging revealed that fusion was preceded by displacement of peptides and fluorescent lipid analogs from the GUV-GUV adhesion region. A detailed analysis of this area being several μm in size revealed that peptides were completely sequestered as expected for an HD. Lateral distribution of lipid analogs was consistent with formation of an HD but not with the presence of two adherent bilayers. Formation and size of the HD were dependent on lipid composition and peptide concentration.
Abbreviations used: DOPC, di-oleoyl-phosphatidylcholine; DOPE, di-oleoyl-phosphatidylethanolamine; DOPS, di-oleoyl-phosphatidylserine; GUV, giant unilamellar vesicles; HA, hemagglutinin; HD, hemifusion diaphragm; N-NBD-PE, N-(7-nitro-2,1,3-benzoxadiazol-4-yl) hexadecylphosphatidylethanolamine; SNARE, soluble NSF attachment receptor; TMD, transmembrane domain; TMR, 5-(and-6)-carboxytetramethylrhodamine ethylmaleinimid
Introduction
Fusion of biological membranes is important for intracellular vesicular trafficking, tissue genesis, fertilization, and infection by viruses. For many fusion reactions it has been shown that merging of membranes proceeds via similar sequential intermediates (1). On close approach, membranes become locally connected by formation of a stalk where proximal leaflets are fused (2). Subsequently, the stalk is thought to expand radially into an HD with the distal membrane leaflets remaining separated (3,4). Finally, opening of a fusion pore within the HD completes the fusion event. Recent studies have shown that a variety of fusion proteins mediate fusion via a hemifusion intermediate, for example viral fusion proteins and SNARE proteins mediating fusion of intracellular organelles (4–15). Although considered to be a key fusion intermediate, direct experimental verification and characterization of HDs between biological membranes turned out to be difficult (16–18).
We have used a rather simple system to study experimentally the displacement of membrane proteins from the adhesion region of approaching and fusing membranes using GUVs. The size of this vesicle type is very useful to visualize the fusion process by optical (fluorescence) microscopy as shown previously (19–22). We investigated the adhesion and fusion of GUVs containing fluorescent transmembrane domains (TMDs) of fusogenic proteins in the presence of divalent cations. To this end, we used tetramethylrhodamine (Rh)-labeled synthetic low-complexity hydrophobic model sequences (Rh-LV-Rh), that were designed to mimic the TMDs of SNARE proteins (23,24), with a 16-amino acid hydrophobic core of leucine and valine flanked by lysine triplets (Rh-LLV16-Rh: Rh-KKKKWLLVLLVLLVLLVLLVLKKKK-Rh; Rh-LV16-G8P9-Rh: Rh-KKKKWLVLVLVLGPVLVLVLVKKKK-Rh) and a 28-residue peptide (Rh-HA) corresponding to the TMD of influenza hemagglutinin (strain A/Japan/305, Rh-βA-ILAIYATVAGSLSLAIMMAGISFWMCSNKKK).
Materials and Methods
Chemicals
Phospholipids and fluorescent lipid analogs were purchased form Avanti Polar Lipids (Birmingham, AL) and used without further purification (N-NBD-PE, DOPC, DOPE, DOPS). Solvents used for vesicle preparation were of the purest available grade. Indium tin oxide (ITO)-coated glass slides were supplied by Präzisions Glas and Optik GmbH (Iserlohn, Germany).
Peptide synthesis
Rhodamine tagged LV-peptides (Rh-LV-Rh) with a 16 amino acid-long hydrophobic core of leucine and valine residues and flanked on both termini by lysins (amino acid sequences: Rh-LLV16-Rh: Rh-KKKKWLLVLLVLLVLLVLLVLKKKK-Rh; Rh-LV16-G8P9-Rh: Rh-KKKKWLVLVLVLGPVLVLVLVKKKK-Rh) were synthesized by Boc chemistry (PSL, Heidelberg, Germany). The Rh-label was added by coupling of a Lys derivative (Fmoc-Lys(Dde)-OH) to the C- and N-termini during synthesis. Reaction of the peptide with 5-(and-6)-carboxytetramethylrhodamine succinimidylester (Invitrogen, Paisley, UK) yielded Rh-labeled peptides. Rh-HA, containing 28 amino acid residues of the TMD of HA (strain Japan/305/57, H2; Rh-HA: Rh-βA-ILAIYATVAGSLSLAIMMAGISFWMCSNKKK) was synthesized using Fmoc-chemistry carried out on the TentaGel S RAM resin (0.25 mmol/g; Rapp Polymere, Tübingen, Germany) using the multiple peptide synthesizer (SYRO II; MultiSynTech GmbH, Witten, Germany). The coupling reagent benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate and N-methylmorpholine were used for activation and Fmoc-deprotection was achieved with 20% piperidine in DMF. Dye-labeling was achieved by coupling 5-(and-6)-carboxytetramethylrhodamine succinimidylester at the N-terminus via a β-alanine by benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate/N-methylmorpholine activation. The C-terminal end is flanked by three lysine residues. The presence of terminal lysines is a common approach to enhance the peptides solubility and to promote an insertion into the membrane (25). Raw products were purified by preparative high performance liquid chromatography up to >90% pureness measured by analytical high performance liquid chromatography and the peptide identity was judged by mass spectrometry.
Preparation of GUV
GUVs were prepared by the electroformation method (26). Lipid mixtures were made from stock solutions in chloroform. Finally, 100 nmol of lipids were dissolved in 30 μL chloroform along with 1 mol % of N-NBD-PE or 1 mol % of the respective peptide dissolved in trifluoroethanol. The lipid/peptide solution was spotted onto two ITO slides or two titanium plates (27) that were placed on a heater plate at 50°C to facilitate solvent evaporation, and subsequently put under high vacuum for at least 1 h for evaporation of remaining traces of solvent. Lipid-coated slides were assembled with a 1-mm Teflon spacer. The electroswelling chamber was filled with 1 mL sucrose buffer (250 mM sucrose, 15 mM NaN3, osmolarity of 280 mOsm/kg) and sealed. An alternating electrical field of 10 Hz rising from 0.02 V to 1.1 V in the first 30 min was applied for 2.5 h at room temperature followed by 30 min of 4 Hz and 1.3 V to detach the formed liposomes. Results were independent of whether GUVs were prepared on ITO slides or titanium plates.
Reconstitution of HA into GUVs
HA of influenza virus X31 was reconstituted according to the procedure of Papadopulos et al. (28). HA was labeled with TMR. See the Supporting Material for details.
Fluorescence microscopy
GUVs containing Rh-labeled peptide or N-NBD-PE were mixed and then added to glucose buffer (250 mM glucose, 11.6 mM potassium phosphate, pH 7.2) with an osmolarity of 300 mOsm/kg at a ratio of 1:1 to 1:3. The slightly hypertonic pressure allows originally spherical GUVs to undergo shape changes, e.g., those associated with adhesion, due to an altered surface to volume ratio (28). Confocal images of the equatorial plane of the GUVs were taken with an inverted confocal laser scanning microscope (FV1000; Olympus, Hamburg, Germany) with a 60× (N.A. 1.35) oil-immersion objective at room temperature. Rhodamine and NBD were excited with a 543 nm HeNe laser and the 488 nm line of an Ar-ion laser (Melles Girot, Bensheim, Germany), respectively. The emissions of rhodamine and NBD were recorded between 569 nm and 669 nm and between 500 nm and 510 nm, respectively. To image the fusion kinetics a high resolution digital B/W CCD camera (ORCA-ER, Hamamatsu, Herrsching, Germany) was used. To trigger adhesion and fusion divalent cations were added from a 100 mM stock solution by a syringe.
Fluorescence recovery after photobleaching
Fluorescence recovery after photobleaching measurements were carried out with the same confocal setup as described above. See Fig. S4 of the Supporting Material for details.
Results
GUVs were prepared from a mixture of unsaturated phospholipids DOPC/DOPE/DOPS (3:1:1, mol/mol/mol) (Material and Methods). This mixture resembles the major fraction of phospholipids in intracellular membranes, in particular of the Golgi, and in the cytoplasmic leaflet of the plasma membrane of mammalian cells, the major sites for fusion processes in the cell.
First, we studied the interaction of N-NBD-PE labeled GUVs with GUVs containing Rh-LV-Rh peptides. Aggregation of GUVs was achieved at 2 mM Ca2+ or Mg2+ (29,30). When raising the Ca2+ concentration to 6 mM, we observed the following sequence of events (Fig. 1 and Fig. S1). An area with significantly reduced fluorescence intensity emerged within the region of the initial contact between two GUVs ∼5 s after the increase of Ca2+. The first image corresponding to t = 0 refers to the last snapshot before alterations of the adhesion region between two GUVs were detected. A reduction of fluorescence in the adhesion region is essentially caused by sequestering of Rh-labeled peptides but also due to displacement of N-NBD-PE (Fig. 2). A more detailed analysis (see below) revealed that sequestering of TMDs is due to formation of an HD. Magnification of this region shows that a structure of rather high fluorescence intensity was formed at the rim of this region that may correspond to transient enrichment of sequestered molecules (Fig. 3). Finally, the diaphragm ruptures, very likely at the junction site of the three bilayers at the HD periphery and retracts to the other side (Fig. S1).
Figure 1.

Sequence of fusion between GUVs made of DOPC/DOPE/DOPS (3:1:1, mol/mol/mol), containing either 1 mol % Rh-LLV16-Rh (indicated by an arrow) or 1 mol % N-NBD-PE. Pairs of GUVs were imaged by fluorescence microscopy (rhodamine fluorescence) at 25°C. On addition of 6 mM Ca2+ fusion was monitored. The first image corresponding to t = 0 refers to the last snapshot before alterations of the adhesion region between two GUVs were observed. Magnifications of selected images are shown. Arrows indicate the dimension of the developing HD. Bright spot in the lower figure part corresponds to fluorescent aggregates inside the large GUV. In the last image the GUVs disintegrate. Scale bar = 5 μm.
Figure 2.
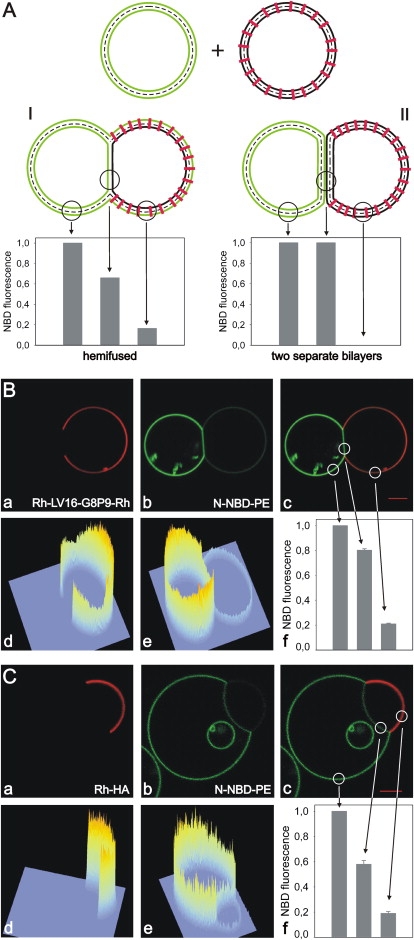
Fluorescence intensity of fluorescent lipid analogs in the contact region. (A) Expected fluorescence intensity of N-NBD-PE in membranes of adherent GUVs (left GUV labeled with N-NBD-PE (green); right GUV with inserted peptide (red)). Intensity is shown for two possible different structures of the adhesion region: (I) HD; (II) two separate adherent bilayers. Although no N-NBD-PE is found in the peptide-containing GUV for II, the outer leaflet of the peptide-containing GUV becomes labeled by the lipid analog for I. However, NBD intensity is reduced by ∼50% due to FRET from NBD to Rh-labeled peptides. (B and C) GUVs containing the peptide Rh-LV16-G8P9-Rh (B) or Rh-HA (C) and N-NBD-PE labeled GUVs were mixed. A total of 2 mM Ca2+ or Mg2+ were added to trigger adhesion of GUVs. Distribution of (a) Rh-labeled peptide; (b) distribution of N-NBD-PE; (c) overlay of a and b. Fluorescence intensity profiles of (d) rhodamine and (e) NBD. N-NBD-PE fluorescence intensity in three different bilayer regions is given in (f). Region of the NBD-labeled GUV outside the HD (intensity was set to 100%), HD, and region of the peptide-containing GUV outside the HD. Differences between B and C with respect to the relative intensities are due to the different sizes of GUVs.
Figure 3.
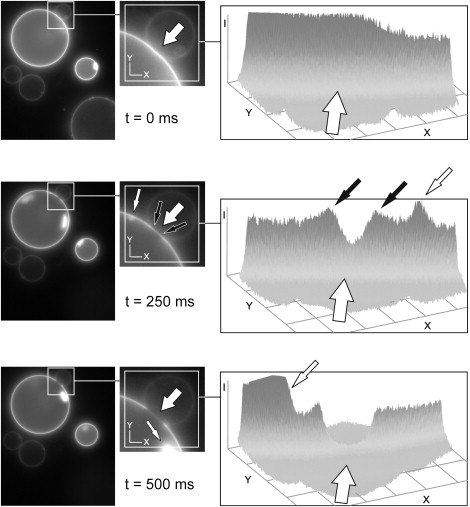
Temporary enrichment of TMDs at the rim of the forming HD. CCD camera images of the fusion kinetic of Fig. 1 are presented in an intensity plot showing the forming HD and its rim. On formation of the HD (see fluorescence decrease in the forming HD (large open arrow)) there is a temporary local fluorescence increase at the rim of the forming HD (small solid arrows) as the TMD gets sequestered. The small open arrow marks structures in the GUV not related to fusion. Note the large open arrow in the intensity plots indicates also the direction of view (from back to front).
At 2 mM Ca2+ or Mg2+ GUVs attached but did not fuse immediately or even remained unfused. Using these conditions we could visualize and quantify the distribution of N-NBD-PE and Rh-LV-Rh within the area of contact that was stable on a timescale of seconds to minutes (Fig. 2). To mimic the TMD of a native fusogenic protein, we also studied the peptide Rh-HA. For both types of peptides we observed sequestering from the adhesion region. For Rh-HA the peptide was sequestered in 77 of 91 cases (85%). Because both LV- and HA-TMDs were sequestered, displacement seems to be typical for TMD peptides and not related to a specific sequence. In the remaining cases, we found Rh-HA was not or only partially sequestered (Fig. S2). When both contacting GUVs contained peptides we also found contact regions with sequestered peptides (see below and Fig. S3), but less frequently.
A contact region devoid of TMDs could be indicative of an HD. To unravel the membrane organization in this region, we quantified NBD fluorescence intensity. As illustrated in Fig. 2 A, NBD fluorescence allows us to distinguish between adhered, yet unfused bilayers and an HD. For hemifusion we can identify two criteria. First, N-NBD-PE is expected to redistribute to the outer leaflet of the peptide-containing GUV. Note that the intensity of N-NBD-PE in the outer leaflet of the peptide-containing GUV is decreased by Förster resonance energy transfer (FRET) to rhodamine (acceptor). Second, as a consequence of lipid analog redistribution in the outer but not in the inner leaflets between GUVs the NBD fluorescence intensity in an HD should be about two-thirds of that found outside of this region in the GUV labeled originally with N-NBD-PE. On the other hand, if the contact region still consists of two separate bilayers, the NBD fluorescence in and outside this region would be similar for the N-NBD-PE labeled GUV and N-NBD-PE would not redistribute to the peptide-containing GUV (cf. Fig. S2 A). The NBD intensity pattern of images in Fig. 2, B and C, indeed suggests that an HD has been formed. Further, peptide did not redistribute to the N-NBD-PE labeled GUV, whereas we found N-NBD-PE labeling of the GUV containing the peptide (N-NBD-PE fluorescence is reduced by FRET). The latter would not be expected if two bilayers would form an adhesion region.
Further evidence for the formation of an HD was obtained by studying the contact region between Rh-HA containing GUVs and nonlabeled GUVs (no N-NBD-PE present). To GUVs forming a contact region with sequestered peptides we added the short-chain lipid analog C6-NBD-PC that is known to insert rapidly into the exposed, outer membrane leaflet (Fig. 4 A, drawing). We found rapid labeling of both GUV membranes except for the contact region (Fig. 4 B). The intensity profile of the analog outside this region shows that insertion of the analog was completed within ∼1 min (Fig. 4 Bc). Again, the NBD fluorescence in GUVs containing the TMD peptides is lower due to FRET (Fig. 4 Bc). This labeling pattern supports the existence of an HD because an adhesion region formed by two separate bilayers would be rapidly labeled by lateral diffusion of analogs. Assuming a typical lateral lipid diffusion rate of ∼1 μm2/s, an analog would diffuse ∼2 μm/s, or migrate into a 10-μm wide contact region within 5 s. However, weak NBD fluorescence was detected only after ∼130 s in the HD. We surmise that slow labeling of the HD is due to redistribution of the short-chain analogs from the outer to the inner leaflet of GUVs caused by peptide mediated perturbations of the bilayer and/or by the membrane structure at the junction site of three bilayers at the HD periphery. The fluorescence in the HD slowly increased to a level comparable to that of the NBD intensity in the peptide-free GUV outside this region. The latter observation argues also for the formation of an HD. If this region would consist of two intact bilayers with only the outer leaflets labeled, the final fluorescence intensity would be twice as much as that observed outside this region. To verify that labeling of the HD is due to redistribution of analogs to the inner leaflet and not due to restricted diffusion of analogs between two adhered intact bilayers we carried out fluorescence recovery after photobleaching measurements in the equatorial plane of the GUVs after a constant fluorescence of C6-NBD-PC in the HD has been reached. We found the same recovery pattern in the HD and outside of this region (Fig. S4) that would not be expected in the case of restricted diffusion between two bilayers. Hence, neither the slow labeling kinetics nor the final fluorescence intensity and the lateral diffusion of analogs are compatible with the presence of two intact separated bilayers in the adhesion region.
Figure 4.
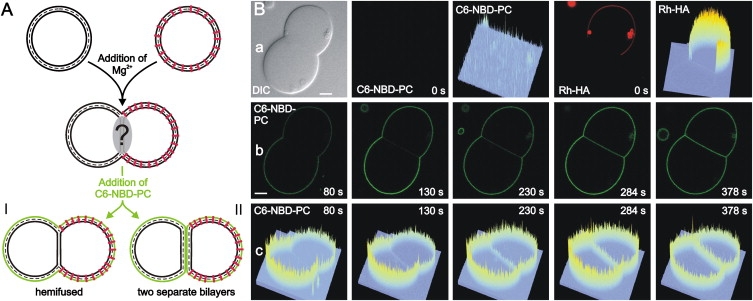
Lipids in the outer leaflet cannot enter the HD. C6-NBD-PC was added to pairs of GUVs with sequestered Rh-HA peptides. After insertion of the lipid analog in the outer leaflet, labeling of the contact region was studied by following the lateral distribution of the NBD fluorescence. (A) Sketch of C6-NBD-PC localization. In case of HD formation no redistribution of the lipid analog to the HD is observed (I) whereas the adhesion region becomes labeled when it is formed by two separated bilayers (II). (B) Lateral distribution of C6-NBD-PC observed by confocal fluorescence microscopy. (a) Images of a GUV pair before addition of C6-NBD-PC (t = 0). From left to right: Differential interference contrast; distribution of C6-NBD-PC (green); intensity profile of NBD fluorescence; distribution of Rh-labeled peptide (red); intensity profile of rhodamine fluorescence; (b) distribution of C6-NBD-PC and (c) corresponding intensity profile at various times after addition of C6-NBD-PC. Scale bar = 5 μm.
In another approach, we labeled the outer leaflet of Rh-HA peptide-containing GUVs with C6-NBD-PC before allowing them to adhere. In the contact region of those GUVs both the peptide as well as the lipid analog were displaced (see Fig. S3). Again, the latter would not have been observed if this region would consist of two intact bilayers. Only after longer incubation we observed labeling of the HD by C6-NBD-PC that very likely is due to redistribution of analogs to the inner leaflet (see above). Both approaches gave the same results for GUVs without peptide (not shown). Based on these various observations, we conclude that the contact region with sequestered peptides corresponds to an HD.
Growth of an HD is expected to decrease the total membrane area, accompanied by a reduction of membrane tension. This reduction of tension is observable as an increase of the contact angle between GUV and coverslip (31). Indeed, we found from Z-stack images (1 μm slices) that the GUV-coverslip contact angle for hemifused GUVs (83 ± 9)° was much larger than for nonhemifused GUVs (35 ± 14)°.
The size of the HD was dependent on the surface area of GUVs. We found an almost linear increase of the surface area of HD with that of the GUV pair (Fig. 5). Notably, reduction of phosphatidylserine (PS) from 20 to 10 mol % did not affect the linear dependence. Only in case that the size of the two hemifused GUVs was very different we found shallower dependence of HD size from that of GUVs (Fig. 5 and Table S1). For a more detailed analysis see Discussion.
Figure 5.
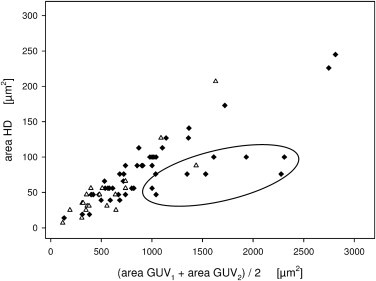
HD area versus GUV surface area. HD area plotted against the mean surface area of the two hemifused GUVs. (Solid symbols) GUVs containing 20 mol % PS lipids. (Open symbols) GUVs with 10 mol % PS. A shallower dependence was observed in case the size of the two hemifused GUVs was very different (encircled, ratio of GUV diameters >4).
We observed a dependence of the HD size on TMD peptide concentration, i.e., for increasing peptide concentration we found a decreasing HD size (Fig. S6). When the peptide concentration was raised above 1 mol % we found only occasionally formation of HD not sufficient for statistics. At 5 mol % we never observed HD formation (see Supporting Material and Fig. S2 D). We surmise that the increased amount of TMD in the membrane outside the HD produces a 2D osmotic pressure pressing on the HD boundary thus resisting HD growth. When GUVs were prepared without DOPE we did not find any hemifused GUVs.
To address whether full length HA is also sequestered from contact regions, we reconstituted HA into DOPC/DOPE/DOPS (3:1:1, mol/mol/mol) GUVs containing 1 mol % N-NBD-PE. We rarely observed sequestered HA on addition of 2 mM Ca2+ (Fig. 6). Unfortunately, at low pH conditions known to trigger a conformational change of HA leading to membrane fusion, we could not study the formation of HD because GUVs became instable.
Figure 6.
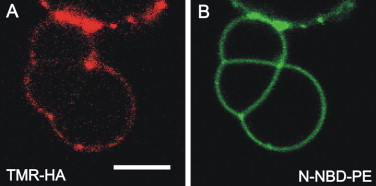
Sequestering of full length HA from contact regions. HA was labeled with TMR and reconstituted into GUVs made of DOPC/DOPE/DOPS (3:1:1, mol/mol/mol), containing 1 mol % N-NBD-PE. On addition of 2 mM Ca2+ adhesion of GUVs and formation of regions depleted of HA could be observed. (A) TMR-HA and (B) N-NBD-PE fluorescence. See the Supporting Material for details. Scale bar = 5 μm.
Discussion
Due to their size GUVs are very useful to follow the fusion process between membranes by light (fluorescence) microscopy. Recently, fusion between GUVs triggered either by fusogenic substances or by electroporation has been studied by using a high time resolution camera (32). The opening kinetics of the fusion necks between GUVs was very fast with an expansion velocity of centimeters per seconds. In this study, we have investigated the organization of the contact region between TMD peptide-containing GUVs preceding divalent cation induced fusion. We observed that this region can be formed by a microscopic visible structure for which a sequestering of peptides as well as a significant reduction of fluorescent lipid analogs was typical. Although the structure was short-lived and followed by full membrane fusion, at lower divalent cation concentration it was stable and allowed us to investigate its organization by fluorescence microscopy.
Displacement of TMDs from the contact region and (re)distribution of lipid analogs between the contact region and the remaining membrane provided strong evidence for the formation of an HD. For GUVs labeled on both leaflets with N-NBD-PE, a comparison of the fluorescence intensity of lipid analogs between the contact region and the membrane outside this region was consistent with hemifusion but not with adhering nonhemifused GUVs (Fig. 2). Obviously, lipid analogs of the outer leaflet, but not those of the inner leaflet were sequestered from this region. This was confirmed when membranes were labeled on the outer leaflet with C6-NBD-PC after preparation of GUVs. On adhesion, lipid analogs were sequestered from the contact region (Fig. S3). Furthermore, we found that lipid analogs externally inserted into the outer leaflet could not rapidly enter the contact region as it would be expected if this region would consist of two adhered bilayers (Fig. 4).
Based on these observations, we conclude that the contact region with sequestered peptides correspond to an HD. In our model system, sequestering of peptides was independent of their amino acid sequence as well as secondary structure. Whereas Rh-LLV-16-Rh display ∼80% α-helical and ∼20% β-sheet structure, Rh-LV16-G8P9-Rh consist of ∼20% α-helical and ∼80% β-sheet structure in membranes (33). The Rh-HA peptide is essentially of α-helical structure (J. Nikolaus and A. Herrmann, unpublished results).
The formation of such large HDs is remarkable. Whether a stalk can expand to an HD has been the focus of many theoretical studies. HD growth increases the length of its rim where monolayer curvature is large (2,4,34–36). This is energetically unfavorable unless the lipid spontaneous curvature is sufficiently negative to favor and drive HD growth (2,4,36). This is consistent with our report here and previous observations (21) that HD formation requires negatively curved DOPE.
HD formation can also be driven by an external force pulling on the diaphragm rim. Although this may be achieved by specialized membrane proteins, it is not obvious that TMD peptides could develop such a pulling force. Indeed, we observed such large HDs also in the absence of peptides. We surmise that in our system HDs are formed by the following reasons. First, the interaction of negatively charged phospholipids with divalent cations crosslinks GUVs leading to adhesion (21). Binding of Ca2+ and Mg2+ to PS causes a shielding of the negatively charge and the dehydration of headgroups (37,38) and, hence, reduces repulsion between headgroups. Second, monolayer studies revealed a 7.4% decrease of the DOPS surface area on addition of Ca2+ (38). A similar observation has been made for PC bilayers on addition of Ca2+ although surface area reduction (5%) was less pronounced in comparison to PS (39). Addition of Ca2+ to PS bilayers also lead to a phase change from the fluid to crystalline state and condensation of the surface area (19,29,38,40). Because divalent cations can only interact with the outer leaflet surface but not with the luminal leaflet, surface area reduction is asymmetric (41) and the surface area difference between both leaflets has to be compensated to preserve stability of GUVs. This could be achieved by formation of an HD. Taking into account the molar fractions of phospholipids in GUVs (DOPC/DOPE/DOPS (3:1:1, mol/mol/mol)) and the decrease of their molecular area in the presence of Ca2+ (38,39), the condensation of the outer monolayer should be ∼5.5% of total membrane surface (because no data on DOPE were available we assume the same reduction as for PC because PE and PC are both zwitterionic lipids). The dependence of the HD size on cation condensation of lipids would predict that the HD size should increase with increasing surface area of GUVs that was indeed the case. That PS is not the sole contributor to surface condensation is sustained by the observation that reduction of PS by 10 mol % did not affect within the error of measurement the area of the HD (Fig. 5). Third, additionally to the cationic component bilayer tension also drives hemifusion and fusion (42). Our measurements show that the relative surface area of HDs is ∼8.7% of the mean surface area of GUV pairs (Fig. S5 and Table S1). This is in good agreement with the predicted reduction of the outer leaflet by cation adsorption (5.5%) plus a contribution of membrane tension driving HD growth (presumably in the range of the remainders; J. M. Warner and B. O'Shaughnessy, Columbia University, New York, personal communication, 2009). Tension in our experiments results from the addition of cations (43,44), from the adhesion of vesicle bilayers that flatten against each other with their volume remaining constant (45) and from adhesion with the substrate (31). Membrane tension may strongly affect the fusion pathway. Dissipative particle dynamics simulations for fusion events of a vesicle with a planar membrane by Grafmuller et al. (46) predict a variation of the adhesion time depending strongly on tension (large tension, fast fusion; small tension, large contact area and long adhesion times). Consistent with this for variation of cation concentration and thus also a variation of tension (43) we find either rapid full fusion (Fig. 1 and Fig. S1) for high Ca2+ concentration or stable adhesion (Fig. S2 A) and hemifusion (Fig. 2 and Fig. 4) for low concentration of Mg2+ or Ca2+. Fourth, another mechanism that can compensate for the excess of lipids in the outer leaflet could be the formation of the bleb-like structures we have observed close to the rim of the diaphragm.
Why are μm-sized HDs not observed commonly in vivo? Although we observed also microscopic HDs with sequestered full length HA reconstituted into GUVs, the situation is different to viruses and cellular membranes. Membrane proteins in biological membranes are much more densely packed than in our model system. Merging of the contacting leaflets requires sequestering even of those proteins that are not involved in fusion. Sequestering might be energetically unfavorable and interfere with expansion and even stability of an HD. Indeed, we found that the presence of peptides at higher concentration or in both attached GUVs significantly reduced formation of HDs. Another factor could be the interaction of membrane proteins with the membrane cytoskeleton. Finally, an important conclusion of our work is that formation of large HDs requires efficient mechanisms to deal with outer leaflet lipids by for example reducing outer leaflet surface area. As yet it is not known if such mechanisms are available to cells.
Acknowledgments
We are indebted to Ben O'Shaughnessy and Jason M. Warner (Columbia University, New York) for very fruitful discussions and to Thomas Pomorski (Humboldt University Berlin) for critical reading.
This work was supported by the Leibniz Graduate School of Molecular Biophysics (fellowship to J.N.) and the Deutsche Forschungsgemeinschaft DFG (grants SFB 740 and 449 to A.H. and R.V).
Supporting Material
References
- 1.Chernomordik L.V., Zimmerberg J., Kozlov M.M. Membranes of the world unite! J. Cell Biol. 2006;175:201–207. doi: 10.1083/jcb.200607083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kozlov M.M., Markin V.S. Possible mechanism of membrane fusion. Biofizika. 1983;28:242–247. [PubMed] [Google Scholar]
- 3.Chernomordik L.V., Melikyan G.B., Chizmadzhev Y.A. Biomembrane fusion: a new concept derived from model studies using two interacting planar lipid bilayers. Biochim. Biophys. Acta. 1987;906:309–352. doi: 10.1016/0304-4157(87)90016-5. [DOI] [PubMed] [Google Scholar]
- 4.Chernomordik L.V., Kozlov M.M. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 2003;72:175–207. doi: 10.1146/annurev.biochem.72.121801.161504. [DOI] [PubMed] [Google Scholar]
- 5.Chernomordik L.V., Frolov V.A., Zimmerberg J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998;140:1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong R.T., Kushnir A.S., White J.M. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 2000;151:425–437. doi: 10.1083/jcb.151.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kemble G.W., Danieli T., White J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- 8.Melikyan G.B., Lin S., Cohen F.S. Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol. Biol. Cell. 1999;10:1821–1836. doi: 10.1091/mbc.10.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nüssler F., Clague M.J., Herrmann A. Meta-stability of the hemifusion intermediate induced by glycosylphosphatidylinositol-anchored influenza hemagglutinin. Biophys. J. 1997;73:2280–2291. doi: 10.1016/S0006-3495(97)78260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cleverley D.Z., Lenard J. The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. USA. 1998;95:3425–3430. doi: 10.1073/pnas.95.7.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu T., Wang T., Weisshaar J.C. Productive hemifusion intermediates in fast vesicle fusion driven by neuronal SNAREs. Biophys. J. 2008;94:1303–1314. doi: 10.1529/biophysj.107.107896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y., Zhang F., Shin Y.K. Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 2005;12:417–422. doi: 10.1038/nsmb921. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann M.W., Peplowska K., Langosch D. Self-interaction of a SNARE transmembrane domain promotes the hemifusion-to-fusion transition. J. Mol. Biol. 2006;364:1048–1060. doi: 10.1016/j.jmb.2006.09.077. [DOI] [PubMed] [Google Scholar]
- 14.Giraudo C.G., Hu C., Rothman J.E. SNAREs can promote complete fusion and hemifusion as alternative outcomes. J. Cell Biol. 2005;170:249–260. doi: 10.1083/jcb.200501093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jahn R., Scheller R.H. SNAREs-engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 16.Kozlovsky Y., Chernomordik L.V., Kozlov M.M. Lipid intermediates in membrane fusion: formation, structure, and decay of hemifusion diaphragm. Biophys. J. 2002;83:2634–2651. doi: 10.1016/S0006-3495(02)75274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melikyan G.B., White J.M., Cohen F.S. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J. Cell Biol. 1995;131:679–691. doi: 10.1083/jcb.131.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frolov V.A., Cho M.S., Zimmerberg J. Multiple local contact sites are induced by GPI-linked influenza hemagglutinin during hemifusion and flickering pore formation. Traffic. 2000;1:622–630. doi: 10.1034/j.1600-0854.2000.010806.x. [DOI] [PubMed] [Google Scholar]
- 19.Lei G., MacDonald R.C. Lipid bilayer vesicle fusion: intermediates captured by high-speed microfluorescence spectroscopy. Biophys. J. 2003;85:1585–1599. doi: 10.1016/S0006-3495(03)74590-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lei G., MacDonald R.C. Effects on interactions of oppositely charged phospholipid vesicles of covalent attachment of polyethylene glycol oligomers to their surfaces: adhesion, hemifusion, full fusion and “endocytosis”. J. Membr. Biol. 2008;221:97–106. doi: 10.1007/s00232-007-9089-x. [DOI] [PubMed] [Google Scholar]
- 21.Pantazatos D.P., MacDonald R.C. Directly observed membrane fusion between oppositely charged phospholipid bilayers. J. Membr. Biol. 1999;170:27–38. doi: 10.1007/s002329900535. [DOI] [PubMed] [Google Scholar]
- 22.Heuvingh J., Pincet F., Cribier S. Hemifusion and fusion of giant vesicles induced by reduction of inter-membrane distance. Eur. Phys. J. E. Soft Matter. 2004;14:269–276. doi: 10.1140/epje/i2003-10151-2. [DOI] [PubMed] [Google Scholar]
- 23.Langosch D., Crane J.M., Reed J. Peptide mimics of SNARE transmembrane segments drive membrane fusion depending on their conformational plasticity. J. Mol. Biol. 2001;311:709–721. doi: 10.1006/jmbi.2001.4889. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann M.W., Weise K., Langosch D. De novo design of conformationally flexible transmembrane peptides driving membrane fusion. Proc. Natl. Acad. Sci. USA. 2004;101:14776–14781. doi: 10.1073/pnas.0405175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hesselink R.W., Koehorst R.B., Hemminga M.A. Membrane-bound peptides mimicking transmembrane Vph1p helix 7 of yeast V-ATPase: a spectroscopic and polarity mismatch study. Biochim. Biophys. Acta. 2005;1716:137–145. doi: 10.1016/j.bbamem.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Angelova M., Soléau S., Bothorel P. Preparation of giant vesicles by external AC electric fields: kinetics and application. Prog. Colloid Polym. Sci. 1992;89:127–131. [Google Scholar]
- 27.Ayuyan A.G., Cohen F.S. Lipid peroxides promote large rafts: effects of excitation of probes in fluorescence microscopy and electrochemical reactions during vesicle formation. Biophys. J. 2006;91:2172–2183. doi: 10.1529/biophysj.106.087387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papadopulos A., Vehring S., Herrmann A. Flippase activity detected with unlabeled lipids by shape changes of giant unilamellar vesicles. J. Biol. Chem. 2007;282:15559–15568. doi: 10.1074/jbc.M604740200. [DOI] [PubMed] [Google Scholar]
- 29.Papahadjopoulos D., Vail W.J., Lazo R. Studies on membrane fusion. III. The role of calcium-induced phase changes. Biochim. Biophys. Acta. 1977;465:579–598. doi: 10.1016/0005-2736(77)90275-9. [DOI] [PubMed] [Google Scholar]
- 30.Papahadjopoulos D., Nir S., Düzgünes N. Molecular mechanisms of calcium-induced membrane fusion. J. Bioenerg. Biomembr. 1990;22:157–179. doi: 10.1007/BF00762944. [DOI] [PubMed] [Google Scholar]
- 31.Rädler J.O., Feder T.J., Sackmann E. Fluctuation analysis of tension-controlled undulation forces between giant vesicles and solid substrates. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 1995;51:4526–4536. doi: 10.1103/physreve.51.4526. [DOI] [PubMed] [Google Scholar]
- 32.Haluska C.K., Riske K.A., Dimova R. Time scales of membrane fusion revealed by direct imaging of vesicle fusion with high temporal resolution. Proc. Natl. Acad. Sci. USA. 2006;103:15841–15846. doi: 10.1073/pnas.0602766103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ollesch J., Poschner B.C., Langosch D. Secondary structure and distribution of fusogenic LV-peptides in lipid membranes. Eur. Biophys. J. 2008;37:435–445. doi: 10.1007/s00249-007-0233-4. [DOI] [PubMed] [Google Scholar]
- 34.Kozlovsky Y., Kozlov M.M. Stalk model of membrane fusion: solution of energy crisis. Biophys. J. 2002;82:882–895. doi: 10.1016/S0006-3495(02)75450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.May S. Structure and energy of fusion stalks: the role of membrane edges. Biophys. J. 2002;83:2969–2980. doi: 10.1016/S0006-3495(02)75303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozlovsky Y., Efrat A., Kozlov M.M. Stalk phase formation: effects of dehydration and saddle splay modulus. Biophys. J. 2004;87:2508–2521. doi: 10.1529/biophysj.103.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feigenson G.W. On the nature of calcium ion binding between phosphatidylserine lamellae. Biochemistry. 1986;25:5819–5825. doi: 10.1021/bi00367a071. [DOI] [PubMed] [Google Scholar]
- 38.Mattai J., Hauser H., Shipley G.G. Interactions of metal ions with phosphatidylserine bilayer membranes: effect of hydrocarbon chain unsaturation. Biochemistry. 1989;28:2322–2330. doi: 10.1021/bi00431a051. [DOI] [PubMed] [Google Scholar]
- 39.Uhríková D., Kucerka N., Balgavý P. Structural changes in dipalmitoylphosphatidylcholine bilayer promoted by Ca2+ ions: a small-angle neutron scattering study. Chem. Phys. Lipids. 2008;155:80–89. doi: 10.1016/j.chemphyslip.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Kozlov M.M., Markin V.S. On the theory of membrane fusion. The adhesion-condensation mechanism. Gen. Physiol. Biophys. 1984;3:379–402. [PubMed] [Google Scholar]
- 41.Chanturiya A., Scaria P., Woodle M.C. The role of membrane lateral tension in calcium-induced membrane fusion. J. Membr. Biol. 2000;176:67–75. doi: 10.1007/s00232001076. [DOI] [PubMed] [Google Scholar]
- 42.Shillcock J.C., Lipowsky R. Tension-induced fusion of bilayer membranes and vesicles. Nat. Mater. 2005;4:225–228. doi: 10.1038/nmat1333. [DOI] [PubMed] [Google Scholar]
- 43.Ohki S. A mechanism of divalent ion-induced phosphatidylserine membrane fusion. Biochim. Biophys. Acta. 1982;689:1–11. doi: 10.1016/0005-2736(82)90182-1. [DOI] [PubMed] [Google Scholar]
- 44.Sinn C.G., Antonietti M., Dimova R. Binding of calcium to phosphatidylcholine-phosphatidylserine membranes. Colloids Surf. A. Physicochem. Eng. Asp. 2006;282–283:410–419. [Google Scholar]
- 45.Kachar B., Fuller N., Rand R.P. Morphological responses to calcium-induced interaction of phosphatidylserine-containing vesicles. Biophys. J. 1986;50:779–788. doi: 10.1016/S0006-3495(86)83518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grafmüller A., Shillcock J., Lipowsky R. The fusion of membranes and vesicles: pathway and energy barriers from dissipative particle dynamics. Biophys. J. 2009;96:2658–2675. doi: 10.1016/j.bpj.2008.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.