

Abstract
ClC chloride channels and transporters play major roles in cellular excitability, epithelial salt transport, volume, pH, and blood pressure regulation. One family member, ClC-ec1 from Escherichia coli, has been structurally resolved crystallographically and subjected to intensive mutagenetic, crystallographic, and electrophysiological studies. It functions as a Cl−/H+ antiporter, not a Cl− channel; however, the molecular mechanism for Cl−/H+ exchange is largely unknown. Using all-atom normal-mode analysis to explore possible mechanisms for this antiport, we propose that Cl−/H+ exchange involves a conformational cycle of alternating exposure of Cl− and H+ binding sites of both ClC pores to the two sides of the membrane. Both pores switch simultaneously from facing outward to facing inward, reminiscent of the standard alternating-access mechanism, which may have direct implications for eukaryotic Cl−/H+ transporters and Cl− channels.
Introduction
The channels and transporters of the chloride channel (ClC) gene family are crucial regulators of the resting membrane potential, cell volume, and electrical excitability of muscle cells (1). Chloride-permeable proteins play many important physiological roles (2–4). The structural resolution of bacterial ClC proteins (5,6) has made them a key biomedical research topic. They function as Cl−/H+ antiporters, not channels (7). The relation between the conformational changes in ClC channels and antiporters is elusive (8). It has been hypothesized that, like transporters, ClC channels also transport protons (9,10), an idea confirmed by a study of ClC-0 channel gating (11), which revealed the transporterlike qualities of this channel. Recently, Lísal and Maduke (12) proposed that the conformational changes in channel gating may be similar to those in the antiporter cycle, and they interpreted this as signifying that ClC-0 is a “broken” Cl−/H+ antiporter in which one of the conformational states leaks Cl−. It was suggested that the ClC-0 slow gate is a relic of antiporter gating, and that it could be one of the antiporter gates (8).
The antiporter structure (5,6) reveals a dimer with two pores, each within a unique subunit. This “double-barreled” construction is believed to be common to the entire ClC family (10). Single-channel recordings and mutagenesis studies have long suggested that a fast process independently gates each pore of ClC-0 and ClC-1, whereas the slow, or common, gating process opens and closes both pores simultaneously (4). Fast gating is thought to operate via a simple molecular mechanism where the glutamate residue located in each subunit's extracellular pore acts as a gate occluding the permeation pathway (10). There is also evidence that other protein moieties affect the fast gate (13,14). The slow gate closes both pores in ClC-0 in a period of seconds, yielding a long-lived inactivated state (4). The molecular mechanism is largely unknown, but the conformational changes are believed to be associated with a transition involving the subunits' interface (15). Both fast and slow gates are proton-modulated (16). Mutating the fast-gating glutamate in ClC-0 abolishes slow gating (9), suggesting strong coupling of the fast (protopore) and slow processes, for which there is direct experimental evidence in ClC-2, “perhaps with slow gating contributing to the operation of the pore E217 as a protopore gate” (17). Together, these results argue that the protein's overall gating is critically influenced by intersubunit structural interactions.
Matulef and Maduke (8) proposed a simple model of Cl−/H+ antiport in ClC-ec1. The protein cycles through four distinct states alternately exposing the substrate binding sites to the peri- and cytoplasm. Mutually exclusive binding and release of two Cl− and a proton on opposite sides of the membrane, i.e., a standard alternating-access transporter mechanism, induce the conformational changes (18). Miller and Nguitragool (19) proposed a different exchange mechanism to account for the available experimental observations. Rather than execute the Jardetzky (18) cycle, chloride and protons occupy the binding sites simultaneously. However, this proposal is problematic (19) in that it would necessitate ad hoc opening of the inner gate twice per transport cycle, formation of HCl (20) and an unknown proton pathway (21).
The proton transport pathway between the two proton-sensitive residues E148 and E203 is elusive (21). One possibility is that a transitory water chain is formed. Recent MD simulations (22) with explicit protons suggest that by reorientation of the E203 side chain a proton is transferred to the water chain and thus carried from the cytoplasm to the protein interior. This, in turn, delivers the excess proton to the region of the central chloride and E148 via electrostatic attraction and Grothuss shuttling. In MD simulations of the E203V mutant, proton transport was disrupted, supporting the results of recent experiments (23).
Using all-atom normal-mode analysis (NMA) (24), we identify the intrinsic large-scale motions of the ClC biomolecule. Normal-mode (NM) treatments determine and characterize the slowest motions in macromolecular systems, collective modes characteristic of a particular protein and related to its function. In a recent extensive systematic comparison of essential deformation modes for a very large set of representative proteins, it was shown that the ”important” space defined by the first, most-relevant NMA eigenvectors provide an extremely correct picture of the trace flexibility of proteins in aqueous solution” (25). In our own previous studies, we coupled all-atom NMA to our Monte Carlo normal-mode following (MC-NMF) method, identifying and predicting functional transitions in gramicidin A (gA) and KcsA (26,27). Our predictions were confirmed in detail by subsequent experiments (28,29). A target structure is usually generated from an initial minimized structure by applying single-step atomic displacement along the lowest-frequency NM eigenvector until a preset root mean-square deviation (RMSD) between the two structures, typically 2.0–3.5 Å, is attained. Our gA and KcsA studies showed that such single-step displacements do not fully elucidate the gating mechanism (see details in Miloshevsky and Jordan (26,27)), and that MC-NMF along the lowest-frequency NM is needed to completely characterize gating transitions. To our surprise, when we applied all-atom NMA to the ClC Cl−/H+ exchanger, a full transport cycle was revealed (30). Perturbing the system in either direction along the lowest-frequency NM leads to slow relative swinging of the subunits perpendicular to the membrane plane, with the intracellular interfacial domain and the two Cl− permeation pathways the regions most affected. The R and A helices execute large-scale swaying, alternately increasing and decreasing the separation of their cytoplasmic ends. This motion is in accord with observations from recent FRET experiments (31). The intracellular and extracellular permeation pathways alternately close and open in concert with the swinging motion of the subunits. When outward-facing, both extracellular pores are open and can bind Cl− ions and both intracellular pores are closed, with the proton-sensitive E203 residues deeply buried in the protein interior. When inward-facing, both extracellular pores are closed and both intracellular pores are open for release of Cl−; the E203 residues face the cytoplasm and are proton-accessible.
Methods
All-atom NMA (24) is used to explore possible mechanisms for slow gating. Our template is the high-resolution (2.5 Å) x-ray structure of the Escherichia coli ClC-ec1 transporter (pdb entry 1OTS) (6), with four Cl− at the binding sites in the pores and 427 crystallographic waters. Protein hydrogens were added via our MCICP code (32), creating 13,524 protein atoms, and 14,809 atoms overall. The system was described with the all-atom CHARMM22 topology and parameter set (33). To remove steric clashes and to relax the system, ∼2000 vacuum minimization steps were done using steepest descent with a random step length (34); finally, the system was well tuned via conjugate gradient with guaranteed descent (35). All degrees of freedom (bond lengths, bond angles, torsion, and improper torsion angles) in the protein and waters were variable. We established system geometry with an absolute largest gradient component of <5 × 10−10 kcal mol−1 Å−1. This extremely precise minimum is needed to perform NMA of large protein structures, since even small residual derivatives can introduce serious errors in the calculated eigendirections. The Cα RMSD between the crystal and minimized ClC-ec1 structures is 1.69 Å; the RMSD for all 14,809 atoms is 2.1 Å. These RMSDs are small, indicating that minimized and crystal structures are highly similar. Standard all-mode NMA (24) was carried out in vacuum using the DSTEVR eigensolver from the LAPACK library and highly optimized BLAS routines for performing basic vector and matrix operations; the global translational and rotational NMs were removed using the conditions of Eckart (36). For the bonded and nonbonded energy terms, both gradient and Hessian were calculated analytically; for other energy terms (angle, dihedral, improper, and Urey-Bradley), a fourth-order finite-difference differentiation (37) was used.
NMA is performed in a single potential well. It provides no information on energy barriers or other stable conformational states (24). It is recognized that the large-scale deformation along the lowest-frequency (seventh) NM is energetically more favorable than that along the other NMs (38) and that NMA reliably characterizes the intrinsic directions of the cooperative displacements encoded in the protein structure (39). Backbone architecture and shape are the main factors governing allowed large-scale deformations (40). Substrate binding can trigger conformational change along the intrinsic directions encoded in the protein architecture. Substrate binding cannot induce new directionality; large-scale cooperative motion counter to NMs is physically impossible (38). Our (26,27) and other (38) NMA studies unambiguously show that the presence of ions and waters (or other substrates) leaves unaffected the intrinsic nature of large-scale deformations along the low-frequency NM. The effects of solvent and membrane environment on collective motion along NMs (38,39) indicate that these could influence the long-range gating pathways between stable states. However, enhanced NMA methods are needed to track them (26,27). NMA provides only the initial direction of the conformational changes in gating (24,38), directionality encoded in the protein's backbone architecture (39). Protein topology (40), not the side chains, ions, waters, or lipids, determines the intrinsically allowed collective motions. Large-scale intrinsic motions of the full protein or its peptide backbone are remarkably similar (38). Thus, elastic network models using only the α-carbon traces and harmonic potentials (38) very successfully predict the conformational changes that initiate gating.
Results
Low-frequency NMs of ClC-ec1 from all-atom NMA
Fig. 1 illustrates the minimized ClC-ec1 structure, with four Cl− ions (green circles) at the central and interior binding sites in the pores. Perturbation along the seventh all-atom NM in either direction (Fig. 2, B and C) leads to relative subunit swinging perpendicular to a membrane plane (Movie S1 in the Supporting Material). The swivel axis lies in a membrane plane at the subunit interface near the protein's center. Black arrows indicate domain displacement directions upon perturbation.
Figure 1.
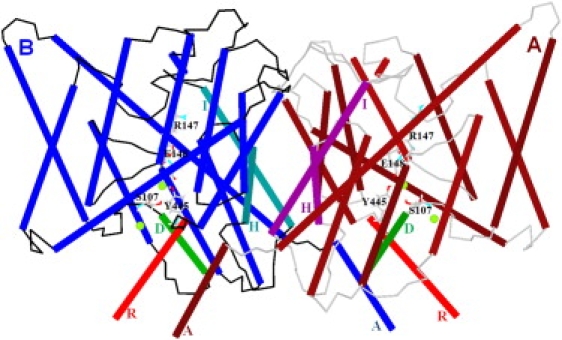
Minimized ClC-ec1 structure. View from within a membrane plane with helices in cylinder representation. Helices H and I (at the subunit interface) and helices R and D are specifically indicated for both subunits. The helices A near helices R of the other subunit are labeled. Other membrane-spanning helices are unlabeled. Polypeptide loops of subunits A and B are colored in gray and black, respectively. For clarity, crystallographic water molecules are suppressed. Four Cl− ions (intermediate-sized spheres near E148, S107, and Y 445) are shown at the central and interior binding sites of the pores. The pore lining residues S107, Y445, E148, and R147 are displayed. The E148 side-chain blocks the pore extracellularly, and side chains of S107 and Y445 constrict the pore intracellularly (30). Figs. 1–4 and 6 are generated via our MCICP code.
Figure 2.
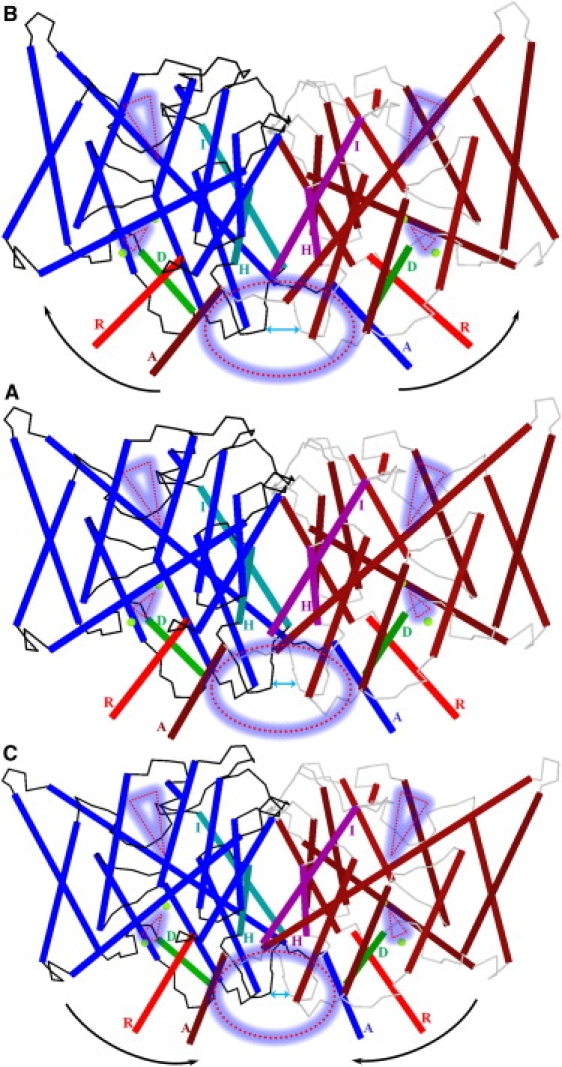
Perturbation of the ClC-ec1 system along the lowest-frequency NM, viewed from within a membrane plane. The feature identification scheme of Fig. 1 is used. (A) The minimized system. (B and C) “Positive” and “negative” displacement along the seventh all-atom NM. Pore lining residues are suppressed. The oval and triangles highlight the protein regions most affected during perturbation.
The intracellular part of the subunit interface (Fig. 2, dotted red circle) is the region most affected. Both protein halves separate (Fig. 2 B) and then approach closely (Fig. 2 C). The intracellular ends of the H and I helices and their connecting polypeptide loops separate and approach each other (Fig. 2, double-headed cyan arrow). The R and A helices undergo large-scale swaying, increasing and decreasing the distance between their cytoplasmic ends. Conversely, backbone geometries of the pores and their immediately surrounding helices are nearly unaffected. They move as whole units. As the subunits separate, the intracellular pore tilt with respect to the membrane plane changes substantially. Thus, the pore region between the two Cl− ions constricts when perturbed “negatively” (Fig. 2 C) and expands when perturbed “positively” (Fig. 2 B). This constriction and expansion affects the flexibility of side chains of S107 and Y445 in the regions between the central and inner Cl− ions (small dotted red triangles).
In contrast, the extracellular regions and the majority of the subunit interface are much less affected, although there are some important changes. The extracellular regions (Fig. 2, large dotted red triangles) structurally affected by the slow swinging subunit motion are localized near the extracellular Cl− pathway. When perturbed “positively” (Fig. 2 B), these domains compress (note rearrangements of the extracellular parts of peripheral helices at triangle marks relative to the I helices), possibly shutting the extracellular pores. When perturbed “negatively” (Fig. 2 C), the extracellular regions near the conduction pathways relax, possibly opening the extracellular pores. Thus, the dotted oval and triangles highlight the regions that mainly contribute to the seventh NM, domains that have been implicated in the control of slow gating (11,4). Extracellular interfacial H+- and Cl−-dependent functionally relevant conformational changes were observed during the ClC-ec1 transport cycle via solution-state 19F NMR (41). However, by themselves these data provide no information on the nature or magnitude of the changes.
Perturbing along the eighth all-atom NM in either direction leads to relative subunit rotational twisting in the membrane plane (Fig. S1). Subunits twist oppositely about a point at their interface and near the center of the protein. The entire interface is affected, but both intra- and extracellular pores are nearly unperturbed. However, the separation of the R helices is affected. The seventh and eighth all-atom NMs describe nearly rigid-body relative global motion (swinging and rotation) of the subunits, whereas perturbation along higher-frequency NMs leads to relative motions and rotations of large protein blocks connected by flexible hinges.
Extracellular pores of ClC-ec1 perturbed along the seventh NM
Our data suggest that structural changes near the extracellular Cl− pathway, arising from the slow subunit motions, could possibly block the periplasmic pores (Fig. 2). The affected residues are identified in Fig. 3. In “positive” perturbation to an RMSD of 3.5 Å along the seventh NM, the R147, F317, and V236 side chains sterically block the outer pores (Fig. 3 B), making nearly direct contact, with further occlusion by the Q61, N62, N318, L319, and E235 side chains (Fig. 3 B). In “negative” perturbation, the outer pore opens (Fig. 3 C). The R147 side chain, its backbone segment effectively hinged to its strictly conserved neighbor, G146, alternately nears and separates from the opposing side chains, F317 and V236 (Fig. 3, B and C, and Movie S2). F317 rotates slightly. Although the backbone segment of E148 is also hinged to a strictly conserved side chain, the adjacent G149, it is far less affected: its side chain remains hydrogen-bonded to the backbone HNs and backbone HAs of G146 and G355 (32). The side chains of Q61, N62, and M65 undergo large-scale motion, blocking and unblocking the outer mouths (Fig. 3, B and C). In this extracellular region, there are numerous glycines, G146, G149, G315, G316, and G66, which could promote substantial protein flexibility.
Figure 3.
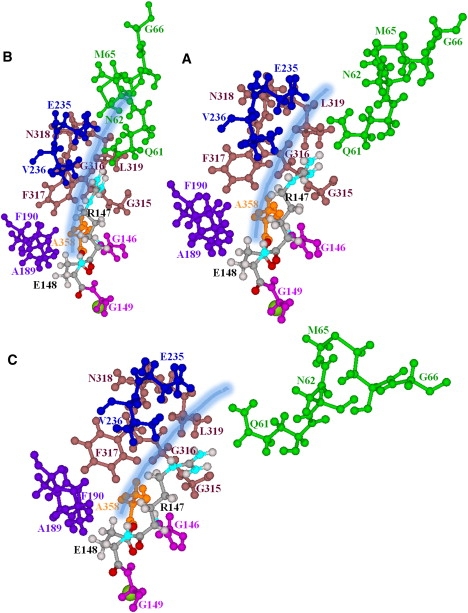
Residues forming the extracellular pores, viewed from within the membrane. (A) Minimized ClC-ec1. (B and C) “Positive” and “negative” displacement along the seventh all-atom NM. Only the subunit A pore is illustrated. Near the central binding site, it is bordered by G146, R147, A358, F190, and E148 (32), which block it. The side chains bounding the pore's outer mouth are G315, G316, F317, and V236, and the guanidinium group of R147 (32). R147 and E148 are explicitly shown; their backbone segments are effectively hinged to their adjacent strictly conserved residues (G146 and G149). The other pore lining groups explicitly illustrated are A358, A189, F190, G315, G316, F317, N318, L319, E235, V236, Q61, N62, M65, and G66. The pore is indicated by the thick shaded line.
Intracellular pores of ClC-ec1 perturbed along the seventh NM
Our data also suggest that the pore region between the central and inner Cl− ions may be affected by the slow motion of the ClC subunits (Fig. 2). The molecular details are illustrated in Fig. 4. Unperturbed (Fig. 4 A), the S107 and Y445 side chains constrict the inner pore. The S107 side chain protrudes into the pore, blocking intersite Cl− movement. An earlier work (32) identified steric contacts with various heavy atoms of S107 and Y445, with a constriction ∼1.84 Å in diameter, suggesting a putative anion trajectory that follows the contour of the S107 side chain. Here, in “positively” perturbing along the seventh NM to a 3.5-Å RMSD, the S107 and Y445 side chains separate and the pore diameter increases to ∼3.4 Å (Fig. 4 B), roughly comparable to the vdW diameter of chloride (∼3.6 Å), thus effectively eliminating the constriction and nearly opening the intracellular pore (Movie S3). The S107 backbone is hinged to its strictly conserved neighbors, G106 and G108, and its side chain rotates slightly in the course of subunit swinging. In “negative” perturbation, the S107, Y445, and F348 side chains sterically block the intracellular pores (Fig. 4 C), making tight contacts that completely occlude the intracellular pathways. Fig. S2 illustrates alternating closure and opening of the intra- and extracellular pathways.
Figure 4.
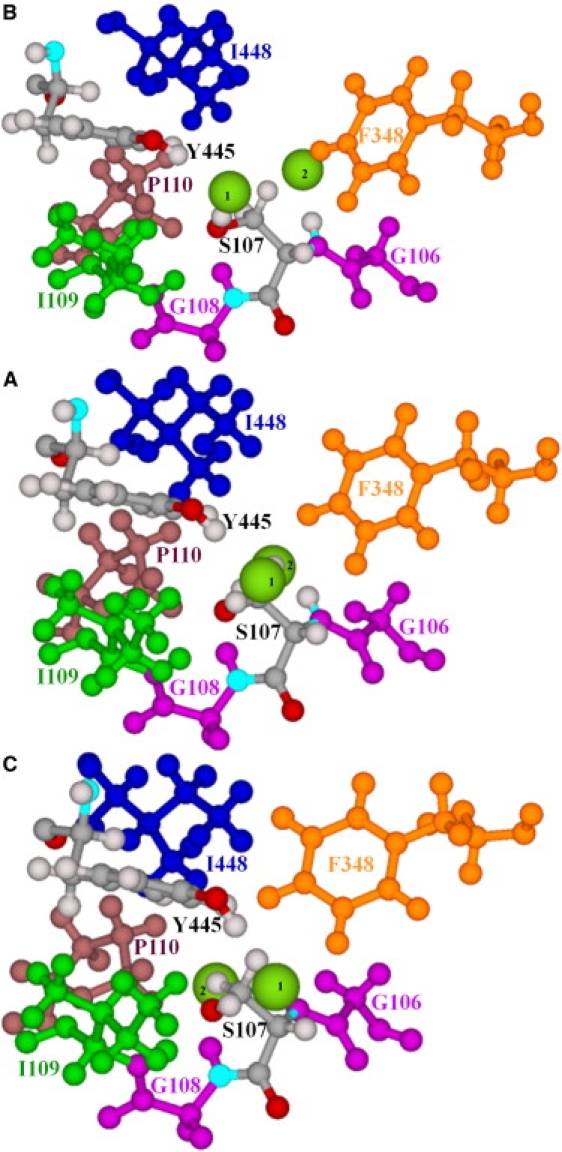
Residues forming the intracellular pores, viewed from the central binding site along a line nearly connecting the central and inner binding sites. (A) Minimized ClC-ec1. (B and C) “Positive” and “negative” displacement along the seventh all-atom NM. Only the subunit A pore is illustrated. S107 and Y445 are explicitly shown. The backbone segment of S107 is hinged to adjacent strictly conserved glycines, G106 and G108. Other groups shown are F348, I109, P110, and I448. Two Cl− ions (intermediate-sized spheres), one at the central binding site (1) and the other at the inner binding site (2), are shown. In A, the S107 side chain protrudes into the pore between the Cl− ions blocking the pathway.
Perturbation of ClC-ec1 along the seventh NM affects water access to E203
Neutralizing E203 abolishes H+/Cl− coupling in ClC-ec1 (21). This residue is located near the cytoplasm side and is partially buried in the protein interior. In the crystal structure (6), two water molecules are located between its side chain and the Y445 backbone. In the absence of perturbation, cytoplasmic water can access E203, since, in addition to the waters near E203, crystal waters are found in the cytoplasmic protein interior. How do large-scale conformational changes affect this site? To identify a putative water path from E203 to the cytoplasm, we inserted hard spheres with a radius of 1.4 Å (the “hard-core” radius of water) within a cylinder of radius 10 Å centered on the CD atom of E203. Another cylinder of the same size was centered on the CD atom of E148 and hard cores 1.4 Å in radius were inserted to identify water occupation of the extracellular mouth. Crystal waters were removed and protein vdW radii were reduced (by a factor of 0.8) to mimic hard spheres. We made 2 × 108 random MC trials, finding continuous water-filled pockets near the E203 and E148 side chains. The blue trace in Fig. 5 A shows the E203 side chain and a putative water pathway to it from the cytoplasm. Water access to E203 is interrupted. The extracellular R147 residue is hydrated, but E148 is inaccessible. In “positive” displacement along the seventh NM, the intracellular pores significantly tilt and open and the E203 residues move toward the cytoplasm (Fig. 5 B). Due to significant protein expansion, these side chains are completely exposed to the cytoplasm and easily water-accessible. A continuous water chain forms. On the extracellular side, water is pushed out of the mouth due to pore closure. In “negative” perturbation, the inner pores are sealed and an array of densely packed protein forms on the intracellular side between the A and R helices (Fig. 5 C). The E203 side chains move toward the periplasm and are buried in the protein interior. For this configuration, MC attempts at finding a continuous water-filled route from the cytoplasm to the E203 side chain failed. Water access to E203 is completely blocked. The extracellular pore opens and E148 side chains become water-accessible (Fig. 5 C). In a recent MD simulations including explicit protons, using a special method to explicitly treat proton transfer from side chains to water, it was shown that E203 residues can deliver protons from the intracellular solution to the protein core via side-chain rotation (22).
Figure 5.
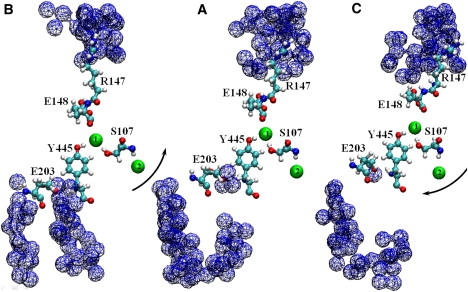
Water access to the side chains of E203 and E148 upon perturbation of ClC-ec1 along the seventh NM, viewed from within the membrane. The side chains of E203, Y445, S107, E148, and R147 are explicitly shown. Two Cl− ions (1 and 2) are shown in the central and inner binding sites as intermediate-sized spheres. Traces of overlapping cross-hatched spheres illustrate water access from the cytoplasm to the E203 and from the periplasm to the E148. (A) Minimized ClC-ec1 corresponding to the crystal structure. (B and C) “Positive” and “negative” perturbation along the seventh all-atom NM. Black arrows indicate the direction of the intracellular pore tilt. The figure is drawn using VMD.
Discussion
Our data show that in perturbing along the seventh NM, the intracellular portion of the interface between the ClC subunits and the intracellular and extracellular Cl− permeation pathways (Fig. 2) are the regions most affected. The exchanger shows an alternate conformational cycle: an outward-facing conformation, with both pores simultaneously open to the periplasm (E148 is protonated and dislodged (Fig. 3 C)) and sealed off on the interior side at the S107/Y445 locus (Fig. 4 C); and an inward-facing conformation, with both pores simultaneously open to the cytoplasm (Fig. 4 B) and blocked on the periplasmic side (Fig. 3 B). There is also a transitional (unperturbed) conformation (Figs. 3 A and 4 A), with the Cl− permeation pathways blocked on both sides, resulting in a locked state. Both E148 and R147 side chains, with backbone segments hinged to adjacent strictly conserved glycines, can swing during gating. The R147 side chain, along with those of Q61, N62, and M65 can sterically block the extracellular pore and mouth. Thus, the extracellular gate requires a complex structural change, not just simple movement of the E148 side chain. Experimental evidence supports the theory that unexpected domains are involved in the conformational changes of the outer pores (4). Attaching fluorescent labels to helix R showed that structural changes extend beyond E148 and propagate to the intracellular region (14). Here, the S107 side chain directly blocks the chloride's intersite motion (Fig. 4 A). This residue is less often studied than Y445, which is generally felt to be the intracellular gate (19). Our data show that the S107 side chain, its backbone segment hinged to its strictly conserved neighbors, G106 and G108, swings during rearrangement. Again, the intracellular gate involves more than S107 movement. The intracellular pores tilt significantly and the side chains of S107, Y445, and F348 tightly block (Fig. 4 C) and unblock (Fig. 4 B) the permeation pathways. The proton-sensing residues E203 are also significantly affected (Fig. 5). With the inner pores tightly blocked, these residues are cytoplasmically inaccessible (Fig. 5 C). Such large-scale rearrangements, occurring on both sides of the protein in regions far from the binding sites, are characteristic of transporters like LacY (42), which operate via alternating access (18). Once the extracellular gates have closed, two Cl− ions occupy the central and outer binding sites, and are the transported substrate. Each subunit alternates about a two-site substrate-binding region, and both subunits operate in unison. However, the molecular details for subunits A and B differ. The displacement of the R147-E148 hairpin is smaller in B than in A. There are also small differences in the behavior of the intracellular pores and their effects on the E203 sites. Relative Cα displacements of two cross-bridge pairs at opposite edges of the subunit interface also differ (see below). This may reflect the protein's antiparallel, pseudosymmetric architecture. In perturbing along other NMs, there is relative subunit rotation, which significantly affects the entire interface, but not the permeation pathways (eighth NM (Fig. S1)) or relative motion of large protein domains (higher-frequency NMs).
The bacterial ClC-ec1 homolog has been successfully used to predict and rationalize electrophysiological behavior of eukaryotic ClC channels ((43); reviewed briefly in Miller (10)). Many studies strongly suggest that there are overall structural similarities among the ClC channels and antiporters (8). Thus, prokaryote structures provide a sound template for understanding the eukaryotes. Our data reveal relative subunit swinging, where the C-termini approach and separate, motion that is directly seen in FRET experiments on ClC-0 (31) and which indicates that C-termini have a functional role in slow gating. It is known that a common or slow gate involving both subunits can simultaneously shut both pores of ClC-0. Slow gate closure accompanies physical separation of the C-termini of the subunits (31). In opening, separation of C-termini decreases. Our results agree with these FRET data (31). We find that when the C-termini separate, side chains of R147, together with Q61, N62, and M65, sterically block the extracellular pore and mouth. When C-termini approach each other, extracellular regions near the ion conduction pathways relax, opening the outer pores.
Although the serine and tyrosine residues are conserved in ClC-0, we hypothesize that the intracellular gate is absent in ClC-0 but present in ClC-ec1. This hypothesis is based on evidence that ClC-0 is a “degraded” (10) or “broken” (11) transporter, in which the intracellular conformational changes, in contrast to those of ClC-ec1, cannot prevent Cl− ion leak. Thus, the conformational change involved in Cl−/H+ exchange by ClC-ec1 could be closely related to the conformational change of coupled fast and slow gating in ClC-0. Our results suggest a direct interaction between fast and slow gates in ClC-0, as both gates are localized in the same spatial domain in ClC-ec1. Indeed, E166A and E166D mutations of ClC-0 eliminated slow gating, causing the slow gate to be permanently locked open (9).
ClC-ec1 exchanges two Cl− ions/proton in its transport cycle (7) with a wild-type (WT) turnover rate of ∼2000 s−1/subunit (44), roughly the maximum characteristic transporter rate (1 × 103 s−1) (45). In mutants lacking both inner and outer gates, transport was only 10-fold faster than in the WT (45). The doubly ungated E148A/Y445S mutant was just threefold faster (∼36 × 103 s−1) than the fastest known transporter, but ∼20-fold slower than the ClC-1 channel. In contrast, Jayaram et al. (45) noted that somewhere in the conformational cycle, the two eukaryotic ion-transport proteins, CFTR and the Na+/K+ pump, can operate in a leaky regime with both gates simultaneously open, with an ion turnover rate of ∼106 s−1, substantially higher than the conduction rate in this degated ClC-ec1. For the Na+/K+ pump, the leak requires the presence of a toxin. Although it has been suggested that slow ion electrodiffusion reflects unusual narrowness of the pore of ClC-ec1 (narrower than the vdW diameter of Cl−), the cycle of conformational changes revealed in our study could account for the low turnover.
Concatemeric channels coupling a ClC-0 subunit with one from either ClC-1 or ClC-2 exhibited conductance levels characteristic of the individual subunits, demonstrating functional and structural separation of the individual pores (46). Individual-pore fast gating was observed, but the joint slow gating property of ClC-0 was lost. Thus, the channel properties of the ClC-0 monomer are altered by interaction with other subunits. Recent concatemer studies of the antiporter hClC-5, coupling WT and nonconducting subunits, suggest that each subunit can effect Cl−/H+ exchange individually (47). However, as these studies do not identify the associated conformational change, they actually do not speak to our NMA data, since the swinging motion of ClC subunits is a consequence of protein architecture, not ion or protein transport. A subunit can be nonconducting but still effect large-scale motion. Moreover, recent NMR data indicate that ClC-ec1 mutants in which H+ transport is lost or impaired still exhibit H+-dependent conformational changes (41). Experiments designed to measure single-channel conductance from ClC-ec1 were unsuccessful (48), indicating that ClC-ec1 conductance might be small or its transport mechanism fundamentally different from that of the eukaryotes, ClC-0, ClC-1, and ClC-2. In support of the latter possibility, the NMR studies also showed that the ClC-ec1 channel-like mutant did not exhibit the same substrate-dependent conformational changes that occurred in the ClC-ec1 transporters (41).
It was recently suggested that individual Cl−/H+ exchange assemblies are contained in each subunit, and that large structural rearrangements, such as those known to occur in ClC-0, are not necessary for the Cl−/H+ transport cycle in ClC-ec1 (49). The subunits were straitjacketed by many interfacial cross-links near the protein edges to prevent their relative movement. The basic assumption of that study is that common gating requires conformational changes at the dimer interface, and the authors concluded that conformational cross talk between subunits in the transport cycle is weak. Our results demonstrate that the subunit interface remains almost intact except for the intracellular region between the polypeptide loops connecting helices H and I (Fig. 2). We examined relative motion of Cαs of cross-bridge pairs studied experimentally (49). “Positive” and “negative” perturbation along the seventh NM to RMSDs of 3.5 Å causes the Cαs of the 207/207 pair of residues to separate and approach by 1.81 Å and 1.79 Å, respectively. This bridge is located on the cytoplasmic side of the interface linking the polypeptide loops of helices H and I. The Cαs of the two 230/249 pairs, located at opposite corners of the extracellular edge of the interface, move by 0.61/0.51 and 0.20/0.17 Å, respectively. Finally, the Cαs of the two 216/433 cross-links, located near the opposite edges of the intracellular subunit interface, move by 0.19/0.19 and 0.41/0.39 Å, respectively. These results show that in perturbation along the seventh NM (the putative slow gate initiator), relative subunit movements are small. The 207/207 residue pair is separated by ∼15.7 Å; when cross-linked, the residues move from their original WT positions, distorting the I-H loops. This cross-linking, and the accompanying distortion, could prevent the intracellular gate from opening fully, thus reducing transport rates to about half (49) the WT turnover rate. We used ElNémo (50), the web-interface to the Elastic Network Model, to identify and characterize the seventh NM of the straightjacketed ClC-ec1 mutant (PDB code 2R9H) (49) in the rotation-translation-block (RTB) approximation (51). Perturbation in either direction revealed the symmetric swinging of the subunits relative to each other, results discussed in the Supporting Material (Fig. S3), with animation directly captured from the ElNémo web page in Movie S4. ElNémo RTB analysis of other WT and mutant ClC structures (1OTS (WT), 1OTT (E148A), 1OTU (E148Q), 3DET (E148A and Y445A), 2HTL (Y445F), 2HT3 (Y445L), 2HLF (Y445E), and 2HT2 (Y445H)) all showed similar behavior. The 3DET mutant has an empty pore. The central and inner binding sites in 2HT3 and 2HTL are occupied by Br−. These studies and experimental investigations of helix involvement at the dimer interface during slow gating in ClC-1 (52) demonstrate that slow gating processes are not confined to the dimer interface.
Our data suggest a possible transport cycle for ClC-ec1. The two glutamates, E148 and E203, located ∼15–20 Å apart, participate directly in proton transport (21). E148 also controls Cl− permeation, blocking or unblocking the extracellular pathway, depending on its protonation state. The intermediate proton-binding sites between these are currently unknown. There is evidence that the central Cl− ion and the Y445 side chain somehow contribute to proton transport. A schematic, speculative Cl−/H+ antiport model for ClC-ec1 is illustrated in Fig. 6. It is an extension of an antiport model proposed previously (8) and associates conformational states with the individual steps. The transport cycle (Step 1) starts with the protein facing outward (Fig. 3 C) and the pores pinched shut at the S107-Y445 locus. The extracellular pores are open, with E148 protonated and dislodged. The side chain of E148 would be accessible from the periplasm, and protonation is favored. The central and outer binding sites are empty. In E148A this “external-open” conformation, with empty central and outer binding sites, was observed crystallographically when Cl− was replaced by small nonhalide anions (53). In WT protein, the central binding site was empty, but the outer binding site was blocked by E148. We speculate that the outer binding site is not occupied when E148 is protonated. The intracellular gates are tightly locked (Figs. 4 C and 5 C). The E203 side chains are proton-inaccessible (Fig. 5 C). In Step 2, two Cl− ions bind to the central and outer sites, triggering a transition to the inward-facing conformation, which could correspond to the E148Q mutant with Cl− ions at the central and outer binding sites and the Q148 side chain displaced (6). The extracellular pores start to close with the structural rearrangements shown in Figs. 3 A and 4 B. Before the extracellular gates close, E148 loses its proton to the periplasm. Due to gate closure, the E148 side chain becomes buried in the protein interior, depressing protonation. As illustrated, E148 is protonated during Step 2, since it is unclear at what point during the closure process deprotonation occurs. At this point in the cycle (Step 3), the paths to the periplasm are closed, the intracellular pores open significantly (Fig. 4 B), and E203 is cytoplasmically accessible (Fig. 5 B). A proton binds to E203 on the intracellular side and can move toward E148 along a pathway suggested on the basis of the recent MD simulations (22). The driving force for proton transport in the protein interior could be electrostatic, due to two Cl− and a deprotonated E148, possibly assisted by the transmembrane pH gradient. Simultaneously, two Cl− exit to the cytoplasm, expelled by the deprotonated side chain of E148 (and also by the Cl− concentration gradient), and at some point, E148 captures an intracellular proton. Binding of a proton to E203, subsequent protonation of E148, and removal of Cl− ions (Step 4) trigger a transition to the outward-facing conformation. The intracellular pores close, E203 residues become buried in the protein interior, the extracellular pores open, and two Cl− ions can enter from the extracellular solution. The protein returns to Step 1 with E148 protonated and two binding sites awaiting Cl− ions. The cycle is repeated, moving two Cl− ions and one proton in opposite directions.
Figure 6.
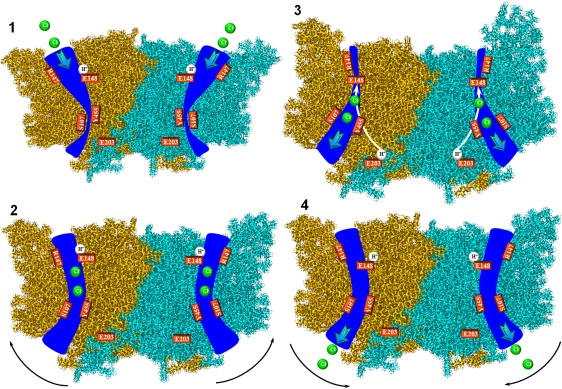
Schematic model of the possible chloride-proton antiport cycle in ClC-ec1. ClC-ec1 subunits are shown. Chloride ions (spheres in permeation pathway) and protons (spheres neighboring E148 (Steps 1, 2, and 4) or E203 (Step 3)) and coordinating key residues (labeled rectangles) are depicted in a structural model of ClC-ec1. The Cl− pathways are shown as wide pores. Step 1 corresponds to the outward-facing conformation with blocked intracellular pores. Step 2 is the transient state leading to the inward-facing conformation. Step 3 is the inward-facing conformation. Solid arrows in Step 3 represent putative proton pathways. Step 4 is the transition to the outward-facing conformation. For Steps 2 and 4, black arrows indicate the direction of subunit swing.
Our findings suggest how large-scale structural changes may be alternately transduced to the outer and inner pores affecting the gates, and they suggest directions for new experiments. Site-directed fluorescence labeling experiments have been performed on liposomes containing equilibrated ClC-ec1 proteins of both orientations, and have been used to study conformational changes in the equilibrium populations of states in the transport cycle (14); the fluorescence signal was a weighted average of contributions from multiple conformational states. In analogy to the FRET studies of ClC-0 (31), fluorophores attached to different helices on both intracellular and extracellular sides of ClC-ec1 can be used to determine whether the relative motion of these helices corresponds to the motion revealed here. The diffracted x-ray tracking method (28), involving gold nanocrystals attached to a protein and irradiated with white x-rays, might be tried. This method can trace the diffraction spot from a nanocrystal at video rate and directly report on conformational changes. Crystallization of some mutants (similar to the C154G LacY mutant), capturing ClC-ec1 in transient conformations, might help characterize the transport cycle. Mutating strictly conserved glycine pairs, G106/G108 and G146/G149, might significantly affect the transport cycle, locking the gates. Finally, synthesizing a single functional monomer could help determine whether transport is carried out independently by each subunit (49).
All-atom NMA only describes the onset of large-scale transitions. It cannot identify other distinct, stable conformation(s) where the protein might be trapped for long periods. Such state(s) could be found using MC-NMF to track the low-frequency NM(s) (27). Standard NMA leaves open the questions of how Cl− permeates to the central and outer binding sites, what is the interior proton pathway; how E148 protonates/deprotonates and dislodges/blocks the pore; how and why the binding of two Cl− to the extracellular pore or their release to the intracellular solution, and/or the binding of protons to E203, works as triggers; and what happens first—protonation of E203 or release of two Cl− ions into the cytoplasm. Addressing these issues requires further atomistic simulations using approaches other than NMA. Recent MD simulations that describe explicit proton transport (22) suggest the presence of a proton transport mechanism between the E203 and E148 sites.
Acknowledgments
We are indebted to Chris Miller and Joe Mindell for valuable comments and suggestions. We thank Merritt Maduke for sharing with us preliminary 19F NMR results.
This work was supported by a grant from the National Institutes of Health, GM-28643, and Purdue University.
Supporting Material
References
- 1.Jentsch T.J. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit. Rev. Biochem. Mol. Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- 2.Chen T.-Y. Structure and function of ClC channels. Annu. Rev. Physiol. 2005;67:809–839. doi: 10.1146/annurev.physiol.67.032003.153012. [DOI] [PubMed] [Google Scholar]
- 3.Zifarelli G., Pusch M. CLC chloride channels and transporters: a biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol. 2007;158:23–76. doi: 10.1007/112_2006_0605. [DOI] [PubMed] [Google Scholar]
- 4.Chen T.-Y., Hwang T.-C. CLC-0 and CFTR: chloride channels evolved from transporters. Physiol. Rev. 2008;88:351–387. doi: 10.1152/physrev.00058.2006. [DOI] [PubMed] [Google Scholar]
- 5.Dutzler R., Campbell E.B., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 6.Dutzler R., Campbell E.B., MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 7.Accardi A., Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- 8.Matulef K., Maduke M. The CLC “chloride channel” family: revelations from prokaryotes. Mol. Membr. Biol. 2007;24:342–350. doi: 10.1080/09687680701413874. [DOI] [PubMed] [Google Scholar]
- 9.Traverso S., Zifarelli G., Pusch M. Proton sensing of CLC-0 mutant E166D. J. Gen. Physiol. 2006;127:51–65. doi: 10.1085/jgp.200509340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440:484–489. doi: 10.1038/nature04713. [DOI] [PubMed] [Google Scholar]
- 11.Lísal J., Maduke M. The ClC-0 chloride channel is a “broken” Cl−/H+ antiporter. Nat. Struct. Mol. Biol. 2008;15:805–810. doi: 10.1038/nsmb.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lísal J., Maduke M. Review. Proton-coupled gating in chloride channels. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:181–187. doi: 10.1098/rstb.2008.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He L., Denton J., Strange K. Carboxy terminus splice variation alters ClC channel gating and extracellular cysteine reactivity. Biophys. J. 2006;90:3570–3581. doi: 10.1529/biophysj.105.078295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell S.P., Curran P.K., Mindell J.A. Site-directed fluorescence studies of a prokaryotic ClC antiporter. Biochemistry. 2006;45:6773–6782. doi: 10.1021/bi0523815. [DOI] [PubMed] [Google Scholar]
- 15.Dhani S.U., Bear C.E. Role of intramolecular and intermolecular interactions in ClC channel and transporter function. Pflugers Arch. 2006;451:708–715. doi: 10.1007/s00424-005-1513-4. [DOI] [PubMed] [Google Scholar]
- 16.Zifarelli G., Pusch M. The role of protons in fast and slow gating of the Torpedo chloride channel ClC-0. Eur. Biophys. J. 2009 doi: 10.1007/s00249-008-0393-x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 17.Yusef Y.R., Zúñiga L., Sepúlveda F.V. Removal of gating in voltage-dependent ClC-2 chloride channel by point mutations affecting the pore and C-terminus CBS-2 domain. J. Physiol. 2006;572:173–181. doi: 10.1113/jphysiol.2005.102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 19.Miller C., Nguitragool W. A provisional transport mechanism for a chloride channel-type Cl−/H+ exchanger. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:175–180. doi: 10.1098/rstb.2008.0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Accardi A., Lobet S., Dutzler R. Synergism between halide binding and proton transport in a CLC-type exchanger. J. Mol. Biol. 2006;362:691–699. doi: 10.1016/j.jmb.2006.07.081. [DOI] [PubMed] [Google Scholar]
- 21.Accardi A., Walden M., Miller C. Separate ion pathways in a Cl−/H+ exchanger. J. Gen. Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D., Voth G.A. Proton transport pathway in the ClC Cl−/H+ antiporter. Biophys. J. 2009;97:121–131. doi: 10.1016/j.bpj.2009.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim H.-H., Miller C. Intracellular proton-transfer mutants in a CLC Cl−/H+ exchanger. J. Gen. Physiol. 2009;133:131–138. doi: 10.1085/jgp.200810112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levitt M., Sander C., Stern P.S. Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 1985;181:423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- 25.Rueda M., Chacón P., Orozco M. Thorough validation of protein normal mode analysis: a comparative study with essential dynamics. Structure. 2007;15:565–575. doi: 10.1016/j.str.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 26.Miloshevsky G.V., Jordan P.C. The open state gating mechanism of gramicidin a requires relative opposed monomer rotation and simultaneous lateral displacement. Structure. 2006;14:1241–1249. doi: 10.1016/j.str.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Miloshevsky G.V., Jordan P.C. Open-state conformation of the KcsA K+ channel: Monte Carlo normal mode following simulations. Structure. 2007;15:1654–1662. doi: 10.1016/j.str.2007.09.022. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu H., Iwamoto M., Oiki S. Global twisting motion of single molecular KcsA potassium channel upon gating. Cell. 2008;132:67–78. doi: 10.1016/j.cell.2007.11.040. [DOI] [PubMed] [Google Scholar]
- 29.Thompson A.N., Posson D.J., Nimigean C.M. Molecular mechanism of pH sensing in KcsA potassium channels. Proc. Natl. Acad. Sci. USA. 2008;105:6900–6905. doi: 10.1073/pnas.0800873105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miloshevsky G.V., Hassanein A., Jordan P.C. Slow gating in ClC chloride channels: normal mode analysis. Biophys. J. 2009;96:470a. doi: 10.1016/j.bpj.2009.11.035. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bykova E.A., Zhang X.D., Zheng J. Large movement in the C terminus of ClC-0 chloride channel during slow gating. Nat. Struct. Biol. 2006;13:1115–1119. doi: 10.1038/nsmb1176. [DOI] [PubMed] [Google Scholar]
- 32.Miloshevsky G.V., Jordan P.C. Anion pathway and potential energy profiles along curvilinear bacterial ClC Cl− pores: electrostatic effects of charged residues. Biophys. J. 2004;86:825–835. doi: 10.1016/S0006-3495(04)74158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 34.Leach A.R. Prentice Hall, Harlow; Chelmsford, United Kingdom: 2001. Molecular Modelling: Principles and Applications. [Google Scholar]
- 35.Hager W.W., Zhang H. A new conjugate gradient method with guaranteed descent and an efficient line search. SIAM J. Optim. 2005;16:170–192. [Google Scholar]
- 36.Eckart C. Some studies concerning rotating axes and polyatomic molecules. Phys. Rev. 1935;47:552–558. [Google Scholar]
- 37.Ridder C.J.F. Accurate computation of F′(x) and F′(x) F″(x) Adv. Eng. Software. 1982;4:75–76. [Google Scholar]
- 38.Qiang C., Bahar I. Chapman & Hall/CRC; Boca Raton, FL: 2006. Normal Mode Analysis. In Theory and Applications to Biological and Chemical Systems. [Google Scholar]
- 39.Ma J. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure. 2005;13:373–380. doi: 10.1016/j.str.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Lu M., Ma J. The role of shape in determining molecular motions. Biophys. J. 2005;89:2395–2401. doi: 10.1529/biophysj.105.065904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elvington S.M., Liu C.W., Maduke M.C. Substrate-driven conformational changes in ClC-ec1 observed by fluorine NMR. EMBO J. 2009;28:3090–3102. doi: 10.1038/emboj.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abramson J., Smirnova I., Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 43.Engh A.M., Maduke M. Cysteine accessibility in ClC-0 supports conservation of the ClC intracellular vestibule. J. Gen. Physiol. 2005;125:601–617. doi: 10.1085/jgp.200509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walden M., Accardi A., Miller C. Uncoupling and turnover in a Cl−/H+ exchange transporter. J. Gen. Physiol. 2007;129:317–329. doi: 10.1085/jgp.200709756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jayaram H., Accardi A., Miller C. Ion permeation through a Cl−-selective channel designed from a CLC Cl−/H+ exchanger. Proc. Natl. Acad. Sci. USA. 2008;105:11194–11199. doi: 10.1073/pnas.0804503105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weinreich F., Jentsch T.J. Pores formed by single subunits in mixed dimers of different CLC chloride channels. J. Biol. Chem. 2001;276:2347–2353. doi: 10.1074/jbc.M005733200. [DOI] [PubMed] [Google Scholar]
- 47.Zdebik A.A., Zifarelli G., Pusch M. Determinants of anion-proton coupling in mammalian endosomal CLC proteins. J. Biol. Chem. 2008;283:4219–4227. doi: 10.1074/jbc.M708368200. [DOI] [PubMed] [Google Scholar]
- 48.Accardi A., Kolmakova-Partensky L., Miller C. Ionic currents mediated by a prokaryotic homologue of CLC Cl− channels. J. Gen. Physiol. 2004;123:109–119. doi: 10.1085/jgp.200308935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguitragool W., Miller C. Inaugural Article: CLC Cl−/H+ transporters constrained by covalent cross-linking. Proc. Natl. Acad. Sci. USA. 2007;104:20659–20665. doi: 10.1073/pnas.0708639104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suhre K., Sanejouand Y.H. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tama F., Gadea F.X., Sanejouand Y.H. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 52.Duffield M., Rychkov G., Roberts M. Involvement of helices at the dimer interface in ClC-1 common gating. J. Gen. Physiol. 2003;121:1–14. doi: 10.1085/jgp.20028741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguitragool W., Miller C. Uncoupling of a CLC Cl−/H+ exchange transporter by polyatomic anions. J. Mol. Biol. 2006;362:682–690. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.