

Abstract
Protein sequences evolved to fold in cells, including cotranslational folding of nascent polypeptide chains during their synthesis by the ribosome. The vectorial (N- to C-terminal) nature of cotranslational folding constrains the conformations of the nascent polypeptide chain in a manner not experienced by full-length chains diluted out of denaturant. We are still discovering to what extent these constraints affect later, posttranslational folding events. Here we directly address whether conformational constraints imposed by cotranslational folding affect the partitioning between productive folding to the native structure versus aggregation. We isolated polyribosomes from Escherichia coli cells expressing GFP, analyzed the nascent chain length distribution to determine the number of nascent chains that were long enough to fold to the native fluorescent structure, and calculated the folding yield for these nascent chains upon ribosome release versus the folding yield of an equivalent concentration of full-length, chemically denatured GFP polypeptide chains. We find that the yield of native fluorescent GFP is dramatically higher upon ribosome release of nascent chains versus dilution of full-length chains from denaturant. For kinetically trapped native structures such as GFP, folding correctly the first time, immediately after release from the ribosome, can lead to lifelong population of the native structure, as opposed to aggregation.
Abbreviations used: aa, amino acid; ATP, adenosine triphosphate; BCIP, bromo-chloro-indolyl-phosphate; EDTA, ethylenediaminetetraacetic acid; GdHCl, guanidine hydrochloride; GFP, green fluorescent protein; GFPib, unfolded green fluorescent protein isolated from inclusion bodies; GTP, guanosine triphosphate; IBs, inclusion bodies; IPTG, isopropyl β-D-1-thiogalactopyranoside; MW, molecular weight; nt, nucleotide; NBT, nitro blue tetrazolium chloride; RNase, ribonuclease; rRNA, ribosomal RNA; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; TCEP, tris (2-carboxyethyl) phosphine; TF, trigger factor
Introduction
Newly synthesized polypeptide chains face the challenge of folding efficiently to form their native structures in the complex and crowded environment of the cell (1). For cytosolic proteins, protein folding can be initiated cotranslationally while the polypeptide chain is still tethered to the translating ribosome (2–5). In contrast to protein refolding, where full-length polypeptide chains are typically diluted away from a chemical denaturant, long-range interactions between distal portions of the sequence cannot be established during early stages of protein biosynthesis. Yet, many protein structural topologies, particularly those rich in β-sheet structure, contain such long-range interactions. The amino acid residues that make contacts in a β-sheet are typically located farther apart in the primary protein sequence than the amino acids in an α-helical structure, and many β-sheets are formed from β-strands that are not contiguous in the protein primary structure. Hence, during polypeptide chain synthesis by the ribosome, the formation of native-like contacts in the N-terminal portions of a β-sheet protein may be delayed because the C-terminal contacting residues have not yet been synthesized or are sterically inaccessible in the 100 Å-long ribosome exit tunnel.
Although the nonvectorial topology of many protein structures raises questions regarding how much native-like structure can be formed cotranslationally, it is undeniable that this is the environment and selective pressure under which protein sequence evolution has occurred. Hence, a reasonable question to ask is whether protein sequences have evolved to stabilize intermediates formed under these cotranslational conditions, even though these intermediates may not be preferentially populated when the full-length polypeptide chain is diluted all at once out of a chemical denaturant (6).
Of course, some protein native structures are only marginally stable, and these polypeptide chains are likely to spend their lifetime sampling partially folded or even highly denatured conformations in addition to the native structure. However, a growing number of proteins are being characterized as “kinetically stable”, meaning they have an unusually high energy barrier for unfolding (7). For these proteins, getting the process of folding to work correctly the first time, immediately after synthesis, might ensure lifelong population of the native structure. Intriguingly, β-sheets and other complex structural topologies appear to be overrepresented among kinetically trapped proteins (8).
Indeed, past studies have demonstrated that cotranslational folding of nascent chains can occur more quickly than in vitro refolding (9,10). Investigations of the impact of cotranslational folding on folding yield, however, have focused more on the contributions of molecular chaperones (11,12) or translation on ribosomes from different organisms (9,13,14) to folding yield, rather than directly comparing the yields of correctly folded protein produced in vivo versus in vitro.
We investigated the effect of cotranslational folding on the overall folding efficiency of GFP (15), a protein with a complex β-sheet-rich structural topology. In native GFP, the polypeptide chain forms an 11-stranded β-barrel that wraps around the central α-helix, enabling the autocatalytic formation of the central imidazolinone chromophore (Fig. 1, A and B). GFP folding requires the formation of more than 200 sequence-distant contact pairs (≥12 residues apart, ≤6 Å maximum distance) (16,17), and is a prerequisite for the formation of a functional chromophore (15). Although the GFP native structure is quite stable and exhibits some properties of kinetically trapped proteins, overexpression of wild-type GFP in Escherichia coli leads to misfolding and aggregation (15). Intriguingly, the majority of GFP polypeptide chains isolated from IBs do not contain a chromophore, which suggests that the bulk of GFP misfolding occurs before an initial round of correct folding (18). Because GFP is so broadly used as a fluorescent reporter for cell-based assays, its high tendency to aggregate has produced intense interest in the GFP folding mechanism (18–22). Typical in vitro refolding yields for GFP hover around 50–60% (18–20). However, the contribution of cotranslational folding to the overall efficiency of GFP folding has not been addressed. Here we report the de novo folding efficiency of newly synthesized GFP polypeptide chains released from polysomes, versus folding of full-length GFP upon dilution from denaturant, and present evidence that the vectorial synthesis of GFP contributes to GFP folding efficiency.
Figure 1.
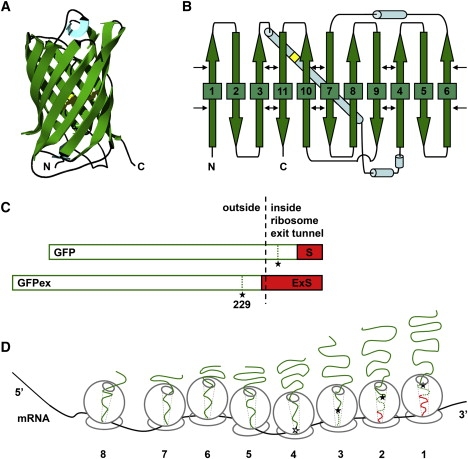
GFP and the GFP-polysome complex. (A) Crystal structure of cycle3 GFP (PDB ID: 1b9c (20)). (B) Native GFP β-strand topology. The β-strands are shown as wide green arrows; α-helices are shown as blue cylinders; chromophore location is shown as a yellow square in the longest α-helix; thin black arrows highlight native structure contacts between noncontiguous β-strands. (C) Schematic of the GFP and GFPex constructs used in this study. The GFP sequence is shown as an open green box. The C-terminal SecM stall sequence (S) and extended stall sequence (ExS) are shown as a red solid box. The black filled stars indicate the position of cycle3 GFP residue 229 in each construct. The black dashed line represents the approximate boundary of the ribosomal surface at the opening of the ribosome exit tunnel. (D) Schematic representation of a GFP-polysome complex. The ribosomes on the mRNA molecule are numbered from 1 to 8, where ribosome 1 is the ribosome closest to the 3′ end of the mRNA and bears a GFP nascent chain synthesized up to the point of translational stalling at the C-terminus of the SecM stall sequence. The ribosome exit tunnel, spanning the entire 50S ribosome subunit, is denoted with light gray dotted lines. The solid green line represents the GFP sequence from residues 1–229, necessary for complete folding and maturation of cycle3 GFP (27). The black filled star represents the relative position of GFP residue 229 in the ribosome tunnel. The synthesis of residue 229 by ribosome 4 is possible but depends on the precise packing of ribosomes 1–3, and hence is denoted with an open star. The dotted line represents the reminder of the cycle3 GFP sequence, which is not required for complete folding and maturation of GFP. The darker solid line represents the SecM stall sequence. This GFP-polysome complex consists of eight ribosomes (the maximum number of nascent chain lengths detected in Fig. 2); however, not all GFP polysomes necessarily consist of eight ribosomes per mRNA.
Materials and Methods
Plasmids
The cycle3 GFP gene (23), plus a sequence encoding a 6 aa N-terminal affinity sequence (not use for the purposes of this work), was cloned into a pET21b-derived plasmid encoding the SecM stall sequence (24). The GFPΔSecM construct was created by site-directed mutagenesis, with the first codon of the SecM stall sequence (TTC) changed to a stop codon (TAA).
Protein expression
Competent E. coli BL21(DE3)pLysS cells (Promega, Madison, WI) transformed with either pET21b/GFP, pET21b/GFPΔSecM, or empty pET21b were grown for 3.5 h (OD600 of 0.4–0.5) at 30°C. Protein expression was induced by the addition of IPTG (Ambion, Foster City, CA) to a final concentration of 0.5 mM. After a 30 min induction, protein expression was halted by adding two 8 mL R buffer (50 mM Tris, pH 7.5; 10 mM MgCl2; 150 mM KCl) ice cubes to the cell suspensions and transferring the flasks to ice (24).
Isolation of ribosomes
Ribosomes and polysomes were isolated as previously described (24), with minor changes. Cells were pelleted, resuspended in 500 μL cold R buffer, and frozen at −80°C for 30 min. Next, the resuspended cells were thawed and treated with lysozyme (1 mg/mL) and subjected to a second freeze/thaw cycle. The thawed lysate was supplemented with 50 mM MgSO4, treated with DNase I (Ambion) for 15 min at 4°C, and spun at 14,000 × g, 4°C until a solid pellet formed. The supernatant was removed and layered onto a 35% sucrose cushion and spun at 437,000 × g for 15 min at 4°C (70.1 Ti rotor; Beckman Coulter, Fullerton, CA). The supernatant and sucrose were removed, and the ribosome/polysome pellet was gently washed and resuspended in freshly prepared R buffer supplemented with 1 mM TCEP (Pierce, Rockford, IL).
Separation of polysomes and 70S ribosomes using size exclusion chromatography
Ribosomes and polysomes were separated using a Sepharose 6B (Sigma-Aldrich, St. Louis, MO) size exclusion column equilibrated with R buffer. Fractions were eluted with R buffer and the ribosome/polysome distribution was analyzed via sucrose density gradient centrifugation (10–50% sucrose density, w/v; 44,300 × g; 18 h; Beckmann SW 28.1 rotor).
SDS-PAGE and Western blotting
Each sample was pretreated with RNase (0.5 mg/mL; Roche, Indianapolis, IN) for 30 min at room temperature to disrupt residual tRNA-nascent chain complexes. Samples were separated by SDS-PAGE, bands were transferred to PVDF membrane (Bio-Rad, Hercules, CA), and the immunoblots were developed with the appropriate antibodies. Rabbit polyclonal antibody against GFP (Novus Biologicals, Littleton, CO) or rabbit anti-TF antibody (a kind gift of B. Bukau) were used as the primary antibody. Goat anti-rabbit AP-conjugated antibody (Novus Biologicals) was used as a secondary antibody. All washes and incubations were performed in phosphate-buffered saline. The GFP bands were visualized after development of an alkaline phosphatase reaction using NBT and BCIP (Promega) as substrates. The intensity of the GFP bands was analyzed with ImageJ software (National Institutes of Health).
Determination of GFP nascent chain length distribution in polysomes
The relative MW of each GFP nascent chain detected on a Western blot was calculated from a standard curve based on the migration distance of the MW markers. The GFP band with the largest MW corresponded to the full-length protein synthesized by ribosome 1, stalled at residue 17 of the SecM stall sequence (corresponding to residue 166 in wild-type SecM, and residue number 260 in the GFP construct used here; Fig. 1, C and D). Nascent chain lengths were determined according the relative MW and the corresponding GFP amino acid sequence.
GFP chromophore absorbance spectra
Equivalent amounts (according the absorption at 280 nm) of native purified GFP and unfolded GFP isolated from IBs were used. The absorbance spectra were collected between 250 nm and 500 nm (18) with a Beckman Coulter DU 530 spectrophotometer.
Fluorescence measurements
All fluorescence measurements were performed with a QM-6 fluorescence spectrophotometer (PTI, Birmingham, NJ) equipped with a temperature-equilibrated cuvette holder. Samples were excited at 397 nm and the fluorescence emission signal was recorded between 470 and 550 nm with an integration time of 1 s using 5 nm slit widths. Measurements were performed in a 10 mm cuvette at 20°C.
Purification of native GFP
Native cycle3 GFP was purified according to published procedures (21), with some modifications. The method used is capable of separating native GFP from nonnative, nonfluorescent (but still soluble) GFP. The supernatant collected from the top of the 35% sucrose cushion (see above) was used as a source of soluble GFP. This supernatant contains ribosome-released nascent chains (24,25) and therefore contains GFP chains with the same origin as stalled GFP nascent chains tethered to ribosome. The supernatant was separated on a size exclusion column packed with Sephadex G-75/Superfine resin (Sigma) and equilibrated with GFP purification buffer (GP: 20 mM Tris, pH 7.5; 1 mM EDTA; 1 mM TCEP). The sample was eluted with GP buffer and the fractions were screened for GFP fluorescence. The fractions with the highest specific GFP fluorescence (according to Western blot analysis) were combined, concentrated, loaded onto a DEAE ion exchange column (DEAE Sepharose resin; Sigma-Aldrich), and eluted with a step gradient of 0–0.2 M NaCl in GP buffer (0.02 M NaCl increments). The fractions were collected and analyzed for GFP chromophore fluorescence and protein content. Fractions containing native GFP were pooled and reseparated by size exclusion chromatography. The final GFP concentration was calculated using the GFP molar extinction coefficient at 397 nm (30,000 Lmol−1/cm−1) (26).
Purification of GFP from IBs
Escerichia coli BL21(DE3)pLysS cells transformed with pET21b/GFPΔSecM were grown at 37°C for 3.5 h. Upon induction with IPTG, cultures were incubated at 39°C for 4 h. Protein expression was halted and the cultures were frozen and lysed, as described above. The lysate was spun for 30 min at 14,000 x g, 4°C. The supernatant was removed and the pellet, including cell debris and GFP aggregates, was washed twice with R buffer containing 1% Triton X-100 (Sigma). The final, washed pellet was resuspended in R buffer containing 6 M GdHCl. The GFP concentration was calculated using a standard curve derived from a Western blot prepared with a purified GFP.
Folding experiments with ribosome-released GFP
Folding reactions were initiated by adding 100 mM EDTA (Fisher Scientific, Hanover Park, IL) and 75 μL RNase to a GFP-polysome sample. Each sample originated from an independent ribosome preparation. The average total protein concentration (determined by Bradford assay; see below) and RNA concentration (determined by the absorbance at 260 nm) in each folding reaction was 220 ± 35 μg/mL and 346 ± 63 μg/mL, respectively. The concentration of GFP nascent chains that were long enough to fold and fluoresce (350 ± 43 nM) was calculated from quantification of GFP bands from Western blots using ImageJ software and comparison with a standard curve of purified GFP. The GFP fluorescence spectrum was taken immediately after the addition of EDTA/RNase and monitored until the signal plateaued (∼90 h). Acquisition of GFP fluorescence was orders of magnitude slower than the release of the nascent chains, as measured by the decrease in GFPex anisotropy after EDTA/RNase addition (not shown). The GFP folding yield was calculated as the ratio between the concentration of folded GFP molecules and the total concentration of GFP chains with sufficient length for complete folding and fluorescence (≥229 aa) (27). The concentration of folded, native GFP was calculated by comparing the GFP fluorescence of released chains with the fluorescence of a standard curve of purified native GFP.
Protein assay
The total protein concentration of the folding reactions was determined via Bradford assay (Bio-Rad). The Bradford reaction was developed for 30 min at room temperature. Each sample was prepared in triplicate and the protein concentration was calculated from a standard curve produced with bovine serum albumin (Bio-Rad).
Folding experiments with GFPib
Folding reactions were initiated by 100-fold dilution of GdHCl-denatured GFPib into R buffer containing 100 mM EDTA, 75 μL RNase, and ribosomes purified from cells transformed with empty pET21b vector. The folding reactions were performed in the presence or absence of 1 mM TCEP as a reducing agent. The fluorescence measurements and the GFP folding yield calculations were performed as described for the GFP-polysome complexes.
GFP solubility in vivo
Lysates from cells expressing GFP, GFPΔSecM, or empty vector were prepared as described above for purification of GFP polysomes. The cell lysate was divided in half. One half was untreated and represents the samples denoted as “whole lysate” in Fig. S5 of the Supporting Material. The other half was treated with DNase I (in the presence of 50 mM MgCl2) for 30 min and spun at 14,000 x g for 40 min at 4°C, and the supernatant obtained represents the samples denoted as “supernatant” in Fig. S5. The solubility for each construct was calculated as the ratio between the GFP in the supernatant and the GFP in the lysate.
Results
Purification and characterization of polysomes bearing GFP nascent chains
During active protein synthesis, ∼80% of translating E. coli ribosomes are engaged in poly-ribosomes (polysomes) (28), meaning that the earliest cotranslational folding processes can occur while a nascent polypeptide chain is a component of a polysome complex. To most closely reproduce cellular cotranslational folding conditions, we isolated polysomes from actively translating cells expressing polypeptide chains consisting of full-length cycle3 GFP (23) followed by a C-terminal linker encoding the 17-aa SecM translation stall sequence (24,29,30) (Fig. 1 C). The presence of the SecM stall sequence generates GFP nascent chains with progressively shorter chain lengths on polysomes (Fig. S1), but does not skew the overall cellular distribution of ribosomes and polysomes (Fig. S2) (31).
To determine the length distribution of GFP nascent chains on E. coli polysomes, polysomes were separated by size exclusion chromatography from 70S ribosomes and released GFP chains, and analyzed by gel electrophoresis. Immunoblotting revealed eight distinct GFP bands with sizes ranging from 19 to 30 kDa, indicating the existence of up to eight ribosomes per GFP mRNA (Figs. 1 D and 2 B, and Table 1). The calculated MW of the largest GFP band (30.8 kDa) corresponds to the MW of the full-length SecM-stalled GFP construct (260 aa, including an N-terminal affinity tag not used in this study). The calculated MW of each GFP band (Table 1) and the amino acid content of the corresponding GFP constructs revealed an average length difference of 12 aa (36 nucleotides) between nascent chains on adjacent ribosomes (Table 1). This calculated difference in GFP nascent chain lengths and the ribosome distribution on the GFP mRNA are remarkably similar to previous findings regarding the distribution of ribosomes on bovine preprolactin and arg-2 mRNAs after translational pausing (32,33). Of interest, the smallest detected GFP band had a calculated MW of ∼19 kDa, corresponding to a difference of 29 aa in length between the nascent chains of ribosomes 7 and 8, equivalent to 87 nt between ribosome centers (Fig. 2 B, band 8, and Table 1). Such ribosome spacing is more characteristic of polysomes formed in vivo in the absence of a translational pause (28), and suggests that ribosome 8, which is most distant from the SecM stall point, is not part of the ribosome cluster formed as a result of translational stalling (Fig. 1 D).
Figure 2.
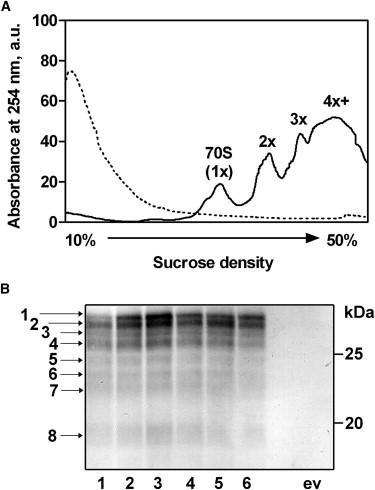
Detection of GFP nascent chains. (A) Sucrose density gradient profile of purified GFP-polysome complexes before (solid line) and after (dashed line) treatment with EDTA and RNase; 1x corresponds to 70S monosomes, 2x corresponds to dimer polysomes, etc., and 4x+ corresponds to polysomes consisting of four or more ribosomes. (B) Anti-GFP Western blot analysis of purified polysome complexes. Lanes 1–6 represent independent GFP-polysome preparations; e.v. represents polysomes prepared from cells transformed with the empty vector. The numbered arrows denote GFP nascent chain bands and correspond to the ribosome numbering in Fig. 1D. The calculated relative MW of each GFP band is shown in Table 1.
Table 1.
GFP nascent chain lengths distribution for GFP polysomes
| Ribosome number | Calculated MW (kDa) | C-terminal truncated residues (aa) | Calculated nascent chain length (aa) | Chain length difference between adjacent ribosomes (aa) |
|---|---|---|---|---|
| 1 | 30.8 | 0 | 260 | - |
| 2 | 29.6 | 12 | 248 | 12 |
| 3 | 28.5 | 22 | 238 | 10 |
| 4 | 27.0 | 36 | 224 | 14 |
| 5 | 25.7 | 47 | 213 | 11 |
| 6 | 24.1 | 62 | 198 | 15 |
| 7 | 22.8 | 73 | 187 | 11 |
| 8 | 19.6 | 102 | 158 | 29 |
GFP nascent chains shown in bold are nearly as long as or longer than 229 aa and therefore capable of forming the imidazolinone chromophore after folding.
Calculation of GFP de novo folding yield requires discrimination between GFP nascent chains that have attained the minimum length necessary for formation of native fluorescent protein (229 residues (27)) from shorter, incomplete GFP nascent chains tethered to the upstream ribosomes (Fig. 1, C and D). The calculated lengths of the GFP nascent chains indicate that ribosomes 1–3, closest to the point of stalling (see numbering scheme in Fig. 1 D), bear GFP nascent chains longer than 229 aa (Fig. 1 D and Table 1). The length of the nascent chain on ribosome 4 (224 aa) is very close to the minimum length required for GFP maturation (Fig. 1 D, open star). Its capacity to form the chromophore after folding will likely depend on the precise packing of ribosomes 1–3, as this will determine the precise stall point for ribosome 4, and hence the length of its nascent chain. Ribosomes 5–8, farthest from the 3′ end of the mRNA (Fig. 1 D), bear nascent chains that are too short to form native, fluorescent GFP. Moreover, in addition to the translational truncation produced by ribosome stacking behind the SecM stall point, the most C-terminal 35–40 aa of every nascent chain will be sequestered inside the ribosome exit tunnel (34,35). This ribosome shielding means that even in the longest GFP nascent chain, borne by ribosome 1, the 17 aa of the SecM stall sequence plus the C-terminal ∼20 residues of the GFP sequence (including residue 229) will be inaccessible for interactions with more N-terminal parts of the nascent chain (Fig. 1, C and D).
Nascent GFP folds to high yield upon release from the ribosome
We then sought to determine whether these nascent GFP polypeptide chains could achieve their native conformation while tethered to the ribosome. The fluorescence emission spectrum of freshly prepared polysomes was evaluated for GFP chromophore fluorescence as a reporter of native GFP (15). The GFP polysome complexes did not produce the characteristic peak for native GFP (Fig. 3, dashed line). This result demonstrates that even the longest GFP nascent chains, tethered to ribosome 1, cannot fold to a native conformation while their C-terminal residues are conformationally constrained within the ribosomal exit tunnel. Presumably, these residues are sterically unavailable for completion of the hydrogen-bonding network required to form the native GFP β-barrel structure. This conclusion is consistent with previous studies of truncated GFP constructs, which showed that the residues comprising the C-terminal GFP β-strand are necessary for folding and formation of fluorescent protein (27,36). We also constructed a GFP nascent chain with a C-terminal extension to span the ribosome exit tunnel, placing all GFP residues outside the tunnel (GFPex; Fig. 1 C). We purified ribosomes bearing GFPex nascent chains and measured their fluorescence emission properties. In contrast to GFP nascent chains, ribosome-bound GFPex nascent chains exhibited measurable quantities of GFP fluorescence before EDTA/RNase treatment (Fig. 3, dotted line).
Figure 3.
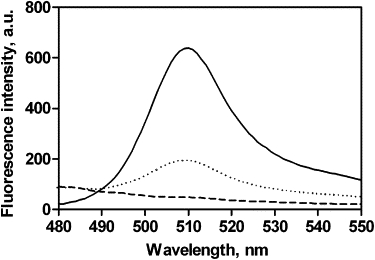
De novo folding of ribosome-released GFP. Fluorescence emission spectra of GFP nascent chains before the addition of EDTA and RNase (dashed line), and after completion of the folding reaction (solid line). Dotted line: Fluorescence emission spectra of ribosome-bound GFPex nascent chains.
The lack of GFP chromophore fluorescence for GFP nascent chains indicates that these ribosome-bound chains cannot reach a completely native conformation, but provides no information on what partial folding might occur, or whether these partially folded conformations are native-like, on-pathway conformations, or misfolded, aggregation-prone conformations, or some combination of the two. We hypothesized that if a GFP nascent chain preferentially adopts a partially folded, on-pathway conformation, it should be able to fold with high efficiency after release from the ribosome, rather than aggregate. To test this idea, we treated GFP polysome complexes with EDTA and RNase to destabilize the ribosomes (Fig. 2 A, dashed line) and release the nascent GFP chains (data not shown). EDTA/RNase treatment of GFP polysome complexes produced a strong GFP fluorescence emission signal (Fig. 3, solid line). To calculate the folding efficiency of these released GFP nascent chains, we compared the amount of native, fluorescent GFP chains with the total amount of GFP nascent chains having the minimum length required for formation of native protein (Fig. 1 D, nascent chains 1–4, and Fig. 2 B). At 20°C, 70% ± 7% of these GFP nascent chains achieved the native fluorescent structure (Fig. 4 A).
Figure 4.
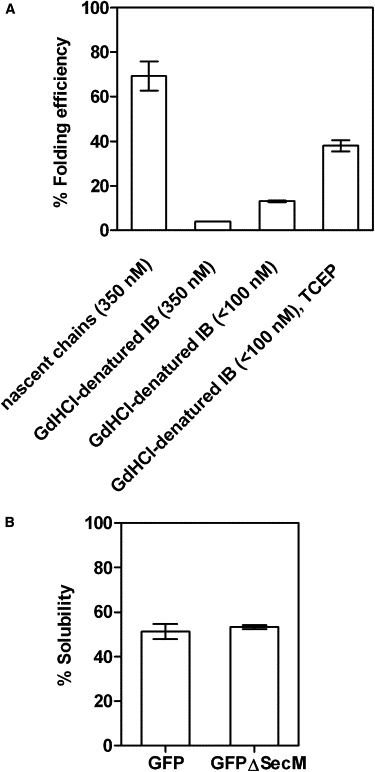
GFP folding efficiency and solubility. (A) Folding efficiency of ribosome-released GFP (nascent chains) or unfolded GFP from IBs in the absence or presence of reducing agent (TCEP). All reaction mixtures contained ribosomes, EDTA, RNase, and TCEP if denoted. The calculated folding efficiency represents the ratio of the amount of native fluorescent GFP to the total amount of GFP chains that are long enough to form a native protein structure (from ribosomes 1–4; see text). Precise quantification of the folding yield of GFPib at concentrations of 350 nM was hindered by the formation of macroscopic aggregates; hence, the reported value is approximate. (B) Effect of SecM stall sequence on GFP solubility. GFPΔSecM lacks the SecM stall sequence at the C-terminus of the GFP construct. The calculated solubility represents the ratio of the amount of soluble protein (in the supernatant) to the amount of total expressed protein (in the cell lysate). In all cases, error bars represent the standard error calculated from a minimum of three independent samples.
Folding of full-length GFP diluted from denaturant is less efficient
Next, we compared the inherent de novo folding efficiency of ribosome-released GFP nascent chains with the folding efficiency of full-length GFP containing the SecM stall sequence (Fig. 1 C) that was forced into IBs by expression at 39°C. In vivo, after ribosome release, these GFP chains are pulled into IBs before chromophore formation occurs (Fig. S4). Because these chains lack a preformed chromophore, their folding can be directly compared with de novo folding of newly synthesized GFP chains, which also lack the chromophore (18,37). Full-length GFPib chains were solubilized in GdHCl, and folding was initiated by dilution out of denaturant into conditions analogous to the folding experiments described above for ribosome-released GFP chains. These folding reactions included polysomes purified from E. coli transformed with the empty vector as a substitute for the ribosomal component of GFP polysome complexes, as previous studies have suggested a direct contribution of ribosome components to in vitro protein refolding yields (38). Yet, in contrast to the folding of ribosome-release nascent GFP chains, folding of GFPib diluted from GdHCl was dramatically less efficient. Folding experiments using GFPib concentrations analogous to the concentrations of nascent GFP in polysome complexes (350 ± 43 nM) resulted in the formation of substantial aggregates and no significant folding yield (Fig. 4 A, the bar without error bars). At lower GFPib concentrations (<100 nM), the calculated folding efficiency for GFPib was 13.0% ± 0.4%, still significantly lower than the folding efficiency of ribosome-released GFP nascent chains (Fig. 4 A).
GFP has two cysteine residues, both of which are reduced in the native structure (20). To estimate the maximum possible folding efficiency for GFPib in the presence of destabilized polysomes, we supplemented the GFPib folding buffer with 1 mM TCEP as a reducing agent. The folding efficiency of these free, denatured chains increased to 38% ± 3%, still significantly lower than the folding efficiency of ribosome-released GFP nascent chains (Fig. 4 A).
Nascent GFP folding and molecular chaperones
The high folding efficiency of ribosome-released GFP nascent chains relative to chemically denatured GFP chains isolated from IBs indicates that the nascent GFP polypeptide chains populate a distribution of partially folded conformations that is different from the distribution of conformations populated by free GFP polypeptide chains upon dilution from denaturant. Cotranslational formation of foldable, on-pathway conformations has been reported for nascent chains of other proteins, including firefly luciferase, ricin, rhodanese, and P22 tailspike protein (11,25,39–41). For efficient folding, however, the ribosome-released polypeptide chains of many proteins required the presence of ATP and/or GTP as an energy source for ATP-dependent molecular chaperones. In contrast, the nascent GFP folding reactions described here were performed in the absence of ATP, and the GFP folding yield was not affected by the addition of ATP (Fig. 5 A), which suggests that the increased efficiency of the GFP folding process is independent of chaperones such as DnaK and/or GroEL/S.
Figure 5.
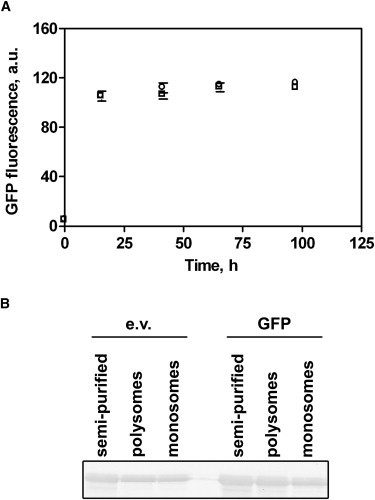
Nascent GFP folding and molecular chaperones. (A) De novo folding of ribosome-released GFP is independent of the presence (open circles) or absence (open squares) of 1 mM ATP. (B) GFP-polysome complexes do not preferentially recruit TF. Similar concentrations of TF were detected on ribosomes isolated from cells transformed with either empty pET21b vector (e.v.) or GFP-pET21b vector (GFP). Each lane contains an equal amount of ribosomes, determined by absorbance at 260 nm.
TF is an ATP-independent, ribosome-associated bacterial chaperone (42–47). When associated to the ribosome, TF is the first chaperone to interact with newly synthesized nascent chains (42–47). Studies have shown increased recruitment of TF to bacterial ribosomes bearing unstructured, hydrophobic nascent chains (25,43,48). To address whether TF is involved in the folding of ribosome-released GFP polypeptide chains, we compared the amount of ribosome-associated TF on polysome complexes isolated from cells expressing GFP with the amount of TF on polysomes isolated from E. coli transformed with the empty vector. Polysomes isolated from cells expressing GFP were not associated with significantly more TF than polysomes from control cells (Fig. 5 B). This result suggests that TF is not selectively recruited to GFP nascent chains. This result is in agreement with other studies that showed successful folding of GFP in an assembled translation system that lacked molecular chaperones (25,49,50), and reduced association of TF with ribosomes expressing GFP (48).
GFP folding yield is not affected by the SecM stall sequence in vivo
Finally, given that some C-terminal protein extensions affect GFP solubility and folding in the cell (51–54), we evaluated whether the SecM stall sequence itself influences the folding abilities of nascent GFP chains. Due to the transient nature of SecM-induced ribosome stalling (29,30), polypeptide chains synthesized with a stall sequence are ultimately released from the ribosome and accumulate in the bacterial cytosol (24,25,55). To test whether SecM-induced translation stalling can increase GFP folding yield, we compared the in vivo folding yield of SecM-stalled GFP with that of GFP expressed without the stall sequence (referred to here as GFPΔSecM), as determined by the solubility of GFP and GFPΔSecM under the conditions used for purification of GFP polysomes. Although deletion of the SecM stall sequence allows higher turnover of the translational components of the cell, resulting in increased GFP synthesis (Fig. S5), the fraction of soluble protein was not significantly different for the two constructs (51% ± 3% for GFP versus 53% ± 1% for GFPΔSecM; Fig. 4 B). The calculated GFP solubility corresponds to published data (21,52,54) and suggests that the C-terminal SecM stall sequence does not affect the folding abilities of GFP.
Conclusions
Our results demonstrate that ribosome-released GFP folds to a significantly higher yield than chemically denatured GFP isolated from IBs. Although we do not know what specific conformations are adopted by the GFP nascent chains, it is clear that vectorial synthesis of the polypeptide chain by the ribosome facilitates the acquisition of conformations that are significantly more likely to produce native GFP than the conformations populated among full-length GFP polypeptide chains that fold after dilution from chemical denaturant. Of interest, this high GFP folding yield was obtained in a complex environment containing ribosomal proteins, rRNA fragments, and truncated, folding-incompetent GFP polypeptide chains. Moreover, although GFP is a eukaryotic protein, it was translated here on prokaryotic ribosomes. However, despite the complex environment and heterologous expression system, the starting point for folding of these GFP nascent chains more closely resembles the in vivo conditions under which the GFP sequence evolved to fold, rather than refolding in vitro (2,4–6).
Acknowledgments
We thank Kay Finn for technical assistance, and Ian Sander and the other members of the Clark group for helpful discussions. The anti-TF antibody was a generous gift from Bernd Bukau.
This work was supported by an award from the National Institutes of Health (GM 74807).
Supporting Material
References
- 1.Ellis R.J. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 2001;26:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 2.Fedorov A.N., Baldwin T.O. Cotranslational protein folding. J. Biol. Chem. 1997;272:32715–32718. doi: 10.1074/jbc.272.52.32715. [DOI] [PubMed] [Google Scholar]
- 3.Komar A.A. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2009;34:16–24. doi: 10.1016/j.tibs.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Kramer G., Ramachandiran V., Hardesty B. Cotranslational folding—omnia mea mecum porto? Int. J. Biochem. Cell Biol. 2001;33:541–553. doi: 10.1016/s1357-2725(01)00044-9. [DOI] [PubMed] [Google Scholar]
- 5.Evans M.S., Clarke T.F., 4th, Clark P.L. Conformations of co-translational folding intermediates. Protein Pept. Lett. 2005;12:189–195. doi: 10.2174/0929866053005908. [DOI] [PubMed] [Google Scholar]
- 6.Clark P.L. Protein folding in the cell: reshaping the folding funnel. Trends Biochem. Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Xia K., Manning M., Colón W. Identifying the subproteome of kinetically stable proteins via diagonal 2D SDS/PAGE. Proc. Natl. Acad. Sci. USA. 2007;104:17329–17334. doi: 10.1073/pnas.0705417104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manning M., Colón W. Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. Biochemistry. 2004;43:11248–11254. doi: 10.1021/bi0491898. [DOI] [PubMed] [Google Scholar]
- 9.Kolb V.A., Makeyev E.V., Spirin A.S. Co-translational folding of an eukaryotic multidomain protein in a prokaryotic translation system. J. Biol. Chem. 2000;275:16597–16601. doi: 10.1074/jbc.M002030200. [DOI] [PubMed] [Google Scholar]
- 10.Fedorov A.N., Baldwin T.O. Process of biosynthetic protein folding determines the rapid formation of native structure. J. Mol. Biol. 1999;294:579–586. doi: 10.1006/jmbi.1999.3281. [DOI] [PubMed] [Google Scholar]
- 11.Frydman J., Nimmesgern E., Hartl F.U. Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature. 1994;370:111–117. doi: 10.1038/370111a0. [DOI] [PubMed] [Google Scholar]
- 12.Svetlov M.S., Kommer A., Spirin A.S. Effective cotranslational folding of firefly luciferase without chaperones of the Hsp70 family. Protein Sci. 2006;15:242–247. doi: 10.1110/ps.051752506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicola A.V., Chen W., Helenius A. Co-translational folding of an alphavirus capsid protein in the cytosol of living cells. Nat. Cell Biol. 1999;1:341–345. doi: 10.1038/14032. [DOI] [PubMed] [Google Scholar]
- 14.Netzer W.J., Hartl F.U. Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature. 1997;388:343–349. doi: 10.1038/41024. [DOI] [PubMed] [Google Scholar]
- 15.Tsien R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 16.Kamagata K., Arai M., Kuwajima K. Unification of the folding mechanisms of non-two-state and two-state proteins. J. Mol. Biol. 2004;339:951–965. doi: 10.1016/j.jmb.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Andrews B.T., Schoenfish A.R., Jennings P.A. The rough energy landscape of superfolder GFP is linked to the chromophore. J. Mol. Biol. 2007;373:476–490. doi: 10.1016/j.jmb.2007.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reid B.G., Flynn G.C. Chromophore formation in green fluorescent protein. Biochemistry. 1997;36:6786–6791. doi: 10.1021/bi970281w. [DOI] [PubMed] [Google Scholar]
- 19.Ward W.W., Bokman S.H. Reversible denaturation of Aequorea green-fluorescent protein: physical separation and characterization of the renatured protein. Biochemistry. 1982;21:4535–4540. doi: 10.1021/bi00262a003. [DOI] [PubMed] [Google Scholar]
- 20.Battistutta R., Negro A., Zanotti G. Crystal structure and refolding properties of the mutant F99S/M153T/V163A of the green fluorescent protein. Proteins. 2000;41:429–437. doi: 10.1002/1097-0134(20001201)41:4<429::aid-prot10>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 21.Fukuda H., Arai M., Kuwajima K. Folding of green fluorescent protein and the cycle3 mutant. Biochemistry. 2000;39:12025–12032. doi: 10.1021/bi000543l. [DOI] [PubMed] [Google Scholar]
- 22.Jackson S.E., Craggs T.D., Huang J.R. Understanding the folding of GFP using biophysical techniques. Expert Rev. Proteomics. 2006;3:545–559. doi: 10.1586/14789450.3.5.545. [DOI] [PubMed] [Google Scholar]
- 23.Crameri A., Whitehorn E.A., Stemmer W.P. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat. Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 24.Evans M.S., Ugrinov K.G., Clark P.L. Homogeneous stalled ribosome nascent chain complexes produced in vivo or in vitro. Nat. Methods. 2005;2:757–762. doi: 10.1038/nmeth790. [DOI] [PubMed] [Google Scholar]
- 25.Evans M.S., Sander I.M., Clark P.L. Cotranslational folding promotes β-helix formation and avoids aggregation in vivo. J. Mol. Biol. 2008;383:683–692. doi: 10.1016/j.jmb.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chalfie M., Kain S. Wiley-Liss; New York: 1998. Green Fluorescent Protein: Properties, Applications, and Protocols. [Google Scholar]
- 27.Li X., Zhang G., Huang C.C. Deletions of the Aequorea victoria green fluorescent protein define the minimal domain required for fluorescence. J. Biol. Chem. 1997;272:28545–28549. doi: 10.1074/jbc.272.45.28545. [DOI] [PubMed] [Google Scholar]
- 28.Neidhardt F.C., Curtiss R. ASM Press; Washington, DC: 1996. Escherichia coli and Salmonella: Cellular and Molecular Biology. [Google Scholar]
- 29.Nakatogawa H., Ito K. The ribosomal exit tunnel functions as a discriminating gate. Cell. 2002;108:629–636. doi: 10.1016/s0092-8674(02)00649-9. [DOI] [PubMed] [Google Scholar]
- 30.Nakatogawa H., Ito K. Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol. Cell. 2001;7:185–192. doi: 10.1016/s1097-2765(01)00166-6. [DOI] [PubMed] [Google Scholar]
- 31.Brandt F., Etchells S.A., Baumeister W. The native 3D organization of bacterial polysomes. Cell. 2009;136:261–271. doi: 10.1016/j.cell.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 32.Wolin S.L., Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 1988;7:3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sachs M.S., Wang Z., Jacobson A. Toeprint analysis of the positioning of translation apparatus components at initiation and termination codons of fungal mRNAs. Methods. 2002;26:105–114. doi: 10.1016/S1046-2023(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 34.Nissen P., Hansen J., Steitz T.A. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 35.Lu J., Deutsch C. Folding zones inside the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 2005;12:1123–1129. doi: 10.1038/nsmb1021. [DOI] [PubMed] [Google Scholar]
- 36.Cabantous S., Terwilliger T.C., Waldo G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 37.Siemering K.R., Golbik R., Haseloff J. Mutations that suppress the thermosensitivity of green fluorescent protein. Curr. Biol. 1996;6:1653–1663. doi: 10.1016/s0960-9822(02)70789-6. [DOI] [PubMed] [Google Scholar]
- 38.Das B., Chattopadhyay S., Dasgupta C. In vitro protein folding by ribosomes from Escherichia coli, wheat germ and rat liver: the role of the 50S particle and its 23S rRNA. Eur. J. Biochem. 1996;235:613–621. doi: 10.1111/j.1432-1033.1996.00613.x. [DOI] [PubMed] [Google Scholar]
- 39.Kudlicki W., Kitaoka Y., Hardesty B. Elongation and folding of nascent ricin chains as peptidyl-tRNA on ribosomes: the effect of amino acid deletions on these processes. J. Mol. Biol. 1995;252:203–212. doi: 10.1006/jmbi.1995.0488. [DOI] [PubMed] [Google Scholar]
- 40.Clark P.L., King J. A newly synthesized, ribosome-bound polypeptide chain adopts conformations dissimilar from early in vitro refolding intermediates. J. Biol. Chem. 2001;276:25411–25420. doi: 10.1074/jbc.M008490200. [DOI] [PubMed] [Google Scholar]
- 41.Kudlicki W., Odom O.W., Hardesty B. Activation and release of enzymatically inactive, full-length rhodanese that is bound to ribosomes as peptidyl-tRNA. J. Biol. Chem. 1994;269:16549–16553. [PubMed] [Google Scholar]
- 42.Buskiewicz I., Deuerling E., Wintermeyer W. Trigger factor binds to ribosome-signal-recognition particle (SRP) complexes and is excluded by binding of the SRP receptor. Proc. Natl. Acad. Sci. USA. 2004;101:7902–7906. doi: 10.1073/pnas.0402231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaiser C.M., Chang H.C., Barral J.M. Real-time observation of trigger factor function on translating ribosomes. Nature. 2006;444:455–460. doi: 10.1038/nature05225. [DOI] [PubMed] [Google Scholar]
- 44.Merz F., Boehringer D., Deuerling E. Molecular mechanism and structure of trigger factor bound to the translating ribosome. EMBO J. 2008;27:1622–1632. doi: 10.1038/emboj.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rutkowska A., Mayer M.P., Bukau B. Dynamics of trigger factor interaction with translating ribosomes. J. Biol. Chem. 2008;283:4124–4132. doi: 10.1074/jbc.M708294200. [DOI] [PubMed] [Google Scholar]
- 46.Ferbitz L., Maier T., Ban N. Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature. 2004;431:590–596. doi: 10.1038/nature02899. [DOI] [PubMed] [Google Scholar]
- 47.Tomic S., Johnson A.E., Etchells S.A. Exploring the capacity of trigger factor to function as a shield for ribosome bound polypeptide chains. FEBS Lett. 2006;580:72–76. doi: 10.1016/j.febslet.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 48.Agashe V.R., Guha S., Barral J.M. Function of trigger factor and DnaK in multidomain protein folding: increase in yield at the expense of folding speed. Cell. 2004;117:199–209. doi: 10.1016/s0092-8674(04)00299-5. [DOI] [PubMed] [Google Scholar]
- 49.Uemura S., Iizuka R., Funatsu T. Single-molecule imaging of full protein synthesis by immobilized ribosomes. Nucleic Acids Res. 2008;36:e70. doi: 10.1093/nar/gkn338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimizu Y., Inoue A., Ueda T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 51.Waldo G.S., Standish B.M., Terwilliger T.C. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 52.Sacchetti A., Cappetti V., Alberti S. Green fluorescent protein variants fold differentially in prokaryotic and eukaryotic cells. J. Cell. Biochem. 2001;81:117–128. doi: 10.1002/jcb.1091. [DOI] [PubMed] [Google Scholar]
- 53.Sacchetti A., Alberti S. Protein tags enhance GFP folding in eukaryotic cells. Nat. Biotechnol. 1999;17:1046. doi: 10.1038/14990. [DOI] [PubMed] [Google Scholar]
- 54.Chang H.C., Kaiser C.M., Barral J.M. De novo folding of GFP fusion proteins: high efficiency in eukaryotes but not in bacteria. J. Mol. Biol. 2005;353:397–409. doi: 10.1016/j.jmb.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 55.Muto H., Nakatogawa H., Ito K. Genetically encoded but nonpolypeptide prolyl-tRNA functions in the A site for SecM-mediated ribosomal stall. Mol. Cell. 2006;22:545–552. doi: 10.1016/j.molcel.2006.03.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.