

Abstract
Protein aggregation has been implicated in the pathology of several neurodegenerative diseases, and a better understanding of how it proceeds is essential for the development of therapeutic strategies. Recently, the amyloidogenic heptapeptide GNNQQNY has emerged as a molecule of choice for fundamental studies of protein aggregation. A number of experimental and computational studies have examined the structure of the GNNQQNY aggregate. Less work, however, has been aimed at understanding its aggregation pathway. In this study, we present a detailed computational analysis of such a pathway. To that end, transition path sampling Monte Carlo simulations are used to examine the dimerization process. A statistical analysis of the reaction pathways shows that the dimerization reaction proceeds via a zipping mechanism, initiated with the formation of distinct contacts at the third residue (N). Asparagine residues are found to play a key role in the early stages of aggregation. And, contrary to previous belief, it is also shown that the tyrosine terminal group is not required to stabilize the dimer. In fact, an asparagine residue leads to faster aggregation of the peptide.
Introduction
Multiple human diseases including Alzheimer's, Parkinson's, Creutzfeldt-Jacob's, and Huntington's are associated with the aggregation of proteins (1,2). The postmortem brain cells of patients afflicted with such diseases exhibit insoluble amyloid plaques deposited within the neuronal cells. There is mounting evidence from recent investigations that the main toxic species in these diseases are the prefibrillar oligomers of the proteins that are specific to each disease (3–7). It is also believed that a deeper understanding of the structure and formation process of the oligomeric intermediates would provide useful insights for the development of potentially useful therapeutic strategies.
In this work, we study the dimerization of the fiber-forming peptide GNNQQNY. This seven-residue peptide belongs to the N-terminal domain of the amyloid-forming peptide Sup35. Sup35 is a prion-like protein from yeast that exhibits an ability to form amyloid β-fibrils (8,9). Our choice of this particular peptide is partly motivated by the fact that Nelson et al. (10) have successfully determined the crystal structure of the protein aggregate using x-ray microcrystallography. The protein aggregate exhibits a structure in which individual peptides are arranged to form parallel β-sheets, and then pairs of β-sheets come together to form an amyloid fibril. Despite its reduced length, this seven-residue peptide displays all the amyloid formation characteristics of full-length Sup35 protein, including cooperative aggregation kinetics, binding of the dye Congo red, and a cross x-ray diffraction (11).
Largely as a result of its small length, GNNQQNY has been studied extensively using molecular simulations. Molecular dynamics simulations in an implicit solvent do suggest that the parallel β-sheet arrangement is the most stable structure for the dimer of the peptide (12,13). Such simulations have shown that side-chain contacts play an important role in the stability of the dimer. Other literature studies (14–18) have also considered whether the parallel β-sheet structure of the peptide disintegrates over the course of long molecular-dynamics runs in explicit water.
Our study presents a departure from previous work in two important respects. First, we use a novel Monte Carlo method to determine the actual free energy change associated with the dimerization process in an explicit solvent and, second, we present a rigorous study of the actual dynamics of the dimerization process, which might ultimately dictate what kind of aggregate structures arise and why. It is important to note that the self-association of the peptide into a dimer is an inherently slow process. Several days of computational time are necessary to generate a single aggregation trajectory, and hundreds of trajectories are required to extract a statistically meaningful pathway. To overcome this difficulty, we resort to transition path sampling (TPS) techniques that have been specially conceived for studies of infrequent events (19,20). In the particular case of GNNQQNY dimerization, our TPS simulations reveal that two distinct intramolecular contacts are sufficient to describe the transition state during the dimerization event. After the formation of these contacts, the fate of the peptides is sealed and the dimerization can come to fruition. By analyzing hundreds of TPS-generated trajectories, we are able to analyze the role of water and that of side-chain interactions, as well as the secondary structure of the individual peptide molecules throughout the dimerization reaction.
Methods
The protein was modeled using the GROMACS53a6 force field. The aggregated dimer system was prepared from the x-ray crystal structure of the peptide aggregate (Protein Data Bank code 1YJP) (10). Fig. 1 shows the aggregated dimer structure of the peptide. The 312-atom peptide system was solvated with ∼10,000 simple-point charge water molecules in a periodic cubic box having a 3.6-nm side.
Figure 1.
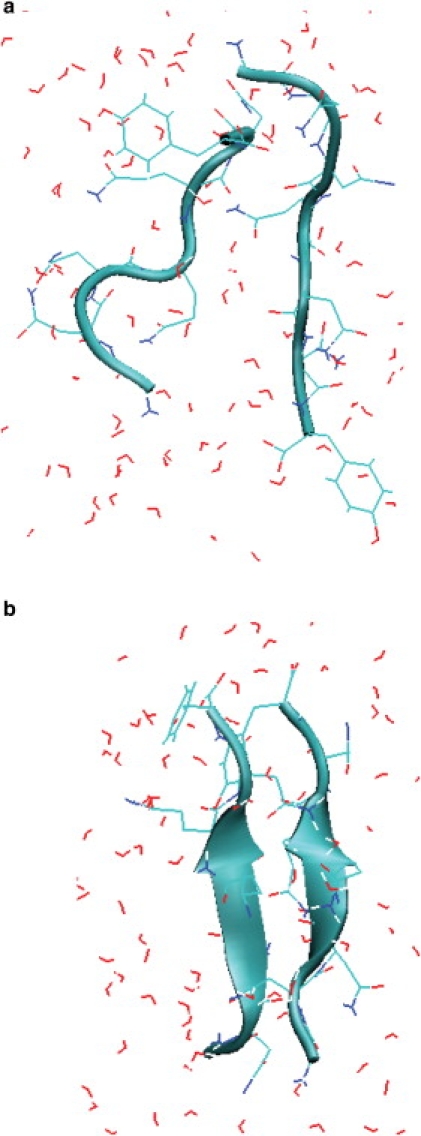
Snapshots of the disaggregated state (a) and the aggregated state (b) of the GNNQQNY peptide.
Molecular dynamics simulations in this work were performed using the GROMACS molecular simulation package (21,22). The software was modified as needed to implement the new free energy simulation techniques described below. Long-range electrostatic interactions were treated with a particle-mesh Ewald sum (23,24). The energy of the aggregated dimer, as constructed from the PDB file 1YJP, was first minimized in a short position-restrained run. Additional water was then added, after which the system was equilibrated for 10 ns at a temperature of 310 K and a pressure of 1 bar using Berendsen coupling (25). The resulting configuration was used as a starting point for subsequent simulations. The disaggregated state of the peptide was generated by raising the temperature of the aggregated state and annealing it back to room temperature.
The relative thermodynamic stability of the aggregated and the disaggregated states of the GNNQQNY peptide was determined from replica exchange umbrella sampling (REUS) simulations (26–28) using a new variant that resorts to a modified interaction potential (29). Briefly, the method involves adding a weak umbrella potential to each of the replicas in a traditional replica exchange molecular dynamics simulation. This umbrella potential is represented as a set of harmonic springs between the native contacts of the two peptides. For our problem, we used 38 replicas between 273 K and 600 K. The additional umbrella potential imposed on the replicas enables better sampling of the aggregated state, whereas the replicas at the higher temperatures efficiently sample the disaggregated state. Labeling the in-register contacts with an index j, the distance between the Cα atoms of the in-register contacts is represented by rj. The corresponding equilibrium distance, r0 was taken to be 0.55 nm. For the seven-residue peptide GNNQQNY, a total of Nc = 7 umbrellas were necessary. Thus, the Hamiltonian for the ith replica is given by
| (1) |
In the above expression, Ki and Ei denote the kinetic and potential energies of replica i, and the sum term represents the umbrella potential, consisting of harmonic springs applied between all the backbone Cα atoms which are in contact. For simplicity, the spring constants kj were all chosen to be equal to k = 100 J/mol/nm2. The transitions between the aggregated state and disaggregated state were monitored using an order parameter ξ defined as
| (2) |
Note that for a given replica, the total umbrella potential is given by
| (3) |
The acceptance criteria for the exchange of configurations between two replicas must be modified from that used in a traditional replica exchange molecular dynamics simulation to account for the additional umbrella potential, and is given by
| (4) |
where Δβ, ΔE, and Δψ represent the difference in the inverse temperature, the difference in total internal energy, and the difference in total umbrella potential of the replicas being considered for an exchange of configurations, respectively. The resulting configurations from the simulation were saved every 4 ps for a total of 10 ns for each window. The potential of mean force or free energy for folding the peptide from a disaggregated state to a parallel β-sheet state was calculated as a function of the order parameter ξ using the weighted histogram analysis method (30). Briefly, we started by calculating the probability histogram Pψ, β(E, ξ) as a function of internal energy E and order parameter ξ using the following equations:
| (5) |
and
| (6) |
In the above equations, M is used to denote the total number of replicas, m is used as an index to denote a particular replica, and K is used to denote the total number of data points (snapshots) in the mth replica. From the probability distribution Pψ, β(ξ) we calculated the potential of mean force (PMF) according to
| (7) |
TPS simulations require that the stable states between which the reaction of interest occurs be identified in terms of one or more non-overlapping order parameters. For GNNQQNY dimerization, we used the number of in-register contacts (Nc) and the average interstrand distance (dis) as order parameters with which to monitor the extent of the reaction. We performed 10-ns molecular dynamics simulations of the aggregated and disaggregated states of the peptide to determine the boundary conditions for the two states (Fig. 2). Based on those results, the boundary for the aggregated state was set at Nc > 0.7 and dis < 0.65 nm; for the disaggregated state, it was set at Nc < 0.2 and dis > 0.9 nm. For our TPS simulations, we used the constant-path length formulation (31). To find an appropriate length for the reaction path we disaggregated the peptide dimer by performing high temperature simulations at 400 K. It was observed that at 400 K the peptide molecules lose their aggregate structure completely in ∼800 ps. Motivated by the reaction time observed in high temperature simulations, we adopted a somewhat conservative constant path length of 2.5 ns for our TPS simulations at 310 K. Having identified a suitable order-parameter and the length of the trajectories for TPS simulations, we proceeded to generate initial trajectories for subsequent calculations. A starting trajectory was generated by running unfolding NVT simulations at 400 K; subsequent annealing to 310 K was implemented by performing sequential shooting moves in the NVT ensemble and decreasing the shooting temperature by 5 K in every step. Finally, NVE trajectories were shot from the NVT reactive trajectories at 310 K. Ten independent initial trajectories were generated through the procedure just outlined to investigate transitions between aggregated states and disaggregated states. To generate the transition state ensemble (TSE), production TPS runs were performed on each of the trajectories using shooting moves in the NVE ensemble as outlined in Chopra et al. (32). The random momentum perturbations were tuned so as to approach a 30% acceptance rate. A total of 1000 reactive trajectories were generated using 10 independent TPS runs. These 1000 trajectories constitute the transition path ensemble (TPE). Out of a total of 1000 reactive trajectories in the TPE, 100 independent trajectories were selected to construct the TSE. The statistical independence of trajectories was measured using the correlation between the order-parameter time series of any two trajectories; when this correlation decayed below 0.5, two trajectories were deemed to be independent.
Figure 2.
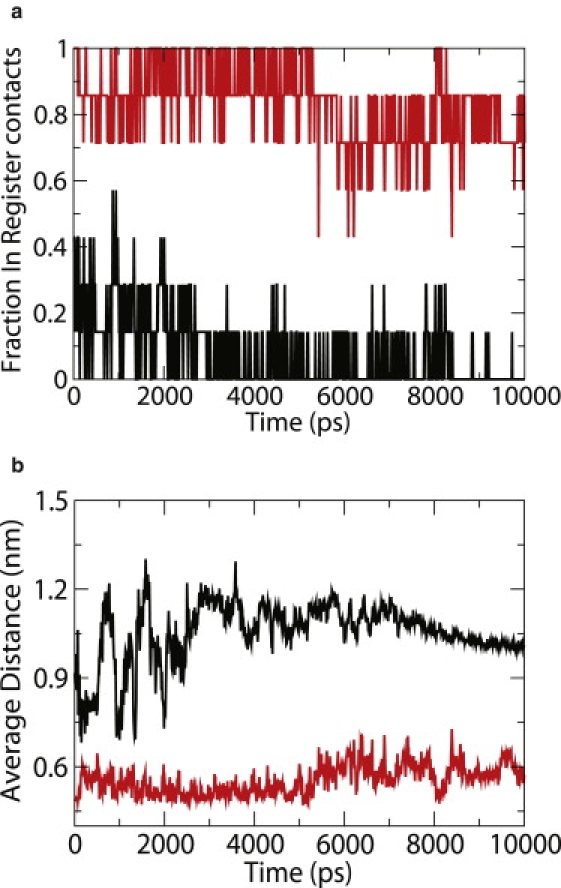
Fraction in-register contacts (a) and average interstrand distance (b) as a function of time for aggregated state (red) and disaggregated state (black).
Frames were saved at intervals of 5 ps for each of the 100 independent reactive trajectories in the TPE. Ten random trajectories of length 1 ns were subsequently shot from each frame of every saved trajectory. The TSE was determined by identifying the frames from which the probability that a random trajectory would end in the aggregated (or disaggregated) state is between 0.35 and 0.65. The TSE determined in this manner was analyzed in terms of a variety of order parameters to identify possible reaction coordinates.
Results and Discussion
The stability of the aggregate (dimer) structure obtained from x-ray crystallography was first examined for the GROMACS53a6 force field using conventional molecular dynamics simulations. Fig. 3 shows the root mean-square deviation of the peptide aggregate (from the x-ray structure) as a function of time. The low root mean-square deviation value indicates that the parallel β-sheet structure represents a relatively stable state for the two-peptide system. The structural stability of the dimer was also assessed by calculating the root mean-square fluctuations of the backbone Cα atoms (Fig. 4). The terminal residues Glycine-1 and Tyrosine-7 exhibit more prominent fluctuations than the residues in the middle. The larger fluctuations at the ends can be attributed to the +1 and −1 charges associated with them. The low fluctuations in the middle indicate that much of the stability of the dimer is provided when the hydrogen bonds formed between the two strands at the central residues. This result is consistent with the simulations of Gsponer et al. (12), Strodel et al. (33), and Vitigliano et al. (34), which showed that the parallel β-sheet structure is a stable conformation for the dimer. It was also observed that the peptide system retains its β-sheet structure throughout the length of the simulations. For this state, the fraction of in-register contacts was found to be always >0.7; the average interstrand distance was found to be <0.65 nm (see Fig. 2). Similarly, we performed molecular dynamics simulation of the disaggregated state, obtained by heating the dimer to 600 K and annealing back to 310 K. The disaggregated state did not return spontaneously into a β-sheet dimer in the 50-ns time range considered in our simulations. This observation serves to emphasize the need for a more elaborate approach such as TPS. The fraction of in-register contacts for the disaggregated state is <0.2, and the average interstrand distance is >0.9 nm throughout the entire duration of the simulation.
Figure 3.
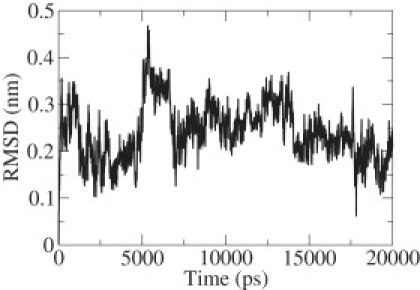
Root mean-square deviation of the parallel β-sheet structure of the peptide as a function of time.
Figure 4.
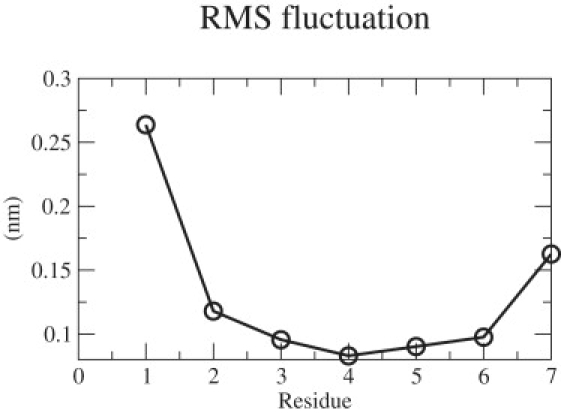
Root mean-square fluctuations of the backbone Cα atoms of GNNQQNY peptide.
The relative stability of the aggregated and disaggregated states of the peptide was determined using REUS simulations. During the course of the simulation, replicas at different temperature were swapped repeatedly. As the system traveled from a low-temperature replica to a high temperature replica, it was found to aggregate and disaggregate multiple times. Fig. 5 shows the free energy changes of the peptide during the aggregated-disaggregated state transformation. At low temperatures, the peptide preferred to be in the aggregated state, whereas at high temperatures the relative free energy was found to favor a disaggregated state. Our results agree with free energy calculations of Gsponer et al. (12) and Vitigliano et al. (34), who used implicit-solvent simulations to conclude that the aggregated state is stable. The actual relative stability (free energy difference) of the aggregated state and disaggregated state at 310 K was measured to be 8.2 kJ/mol, and is slightly higher than that reported in the literature. We believe that the difference in value is due to the use of different force fields (CHARMM19 versus GROMACS53a6) and different solvent conditions (explicit versus implicit) in our studies.
Figure 5.
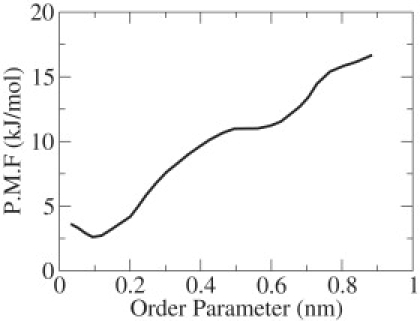
Free energy changes during dimerization of GNNQQNY peptide at 310 K. The order parameter represents deviation of distance between Cα contacts from an ideal value of 0.55 nm.
Although REUS simulations are useful in understanding the relative stability of the aggregated state and the disaggregated state, they do not provide information on the aggregation pathway of the peptides. The complete characterization of the aggregation pathway, i.e., the transition path sampling simulations, was used to generate trajectories connecting our two stable states as outlined above. These so-called reactive trajectories between the stable states constitute the TPE. As mentioned earlier, by focusing only on reactive trajectories, TPS avoids the time spent in a stable free energy minimum waiting for a transition (to another minimum) to occur. As described in Methods, we generated a TPE consisting of 1000 reactive trajectories. Several representative reactive paths from our simulations are shown in Fig. 6.
Figure 6.
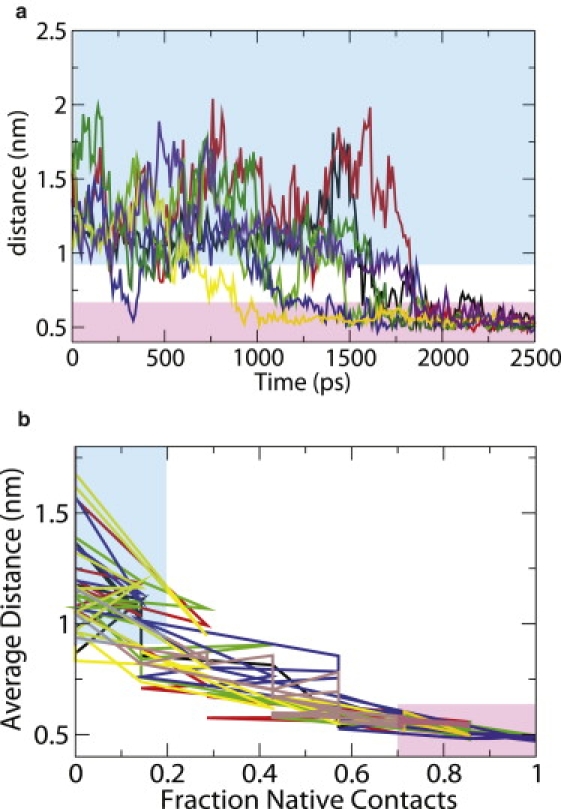
Representative reactive trajectories connecting the aggregated state and the disaggregated state. Blue box indicates the location of disaggregated state, and the pink box indicates the aggregated state of the peptide.
Analysis of the TPE indicates that the aggregation reaction happens through the sequential formation of key contacts between the two strands. More specifically, our results indicate that dimerization begins at residues number 3 and 4, followed by formation of the contacts at the other residues of the molecule (see Fig. 7). The contacts between the terminal residues occur last. The Asparagine-3 and Glutamine-4 residues, where the formation of contacts between the two peptides begins, have large polar side chains and hence assist in the formation and stabilization of these contacts. A TSE was constructed by analyzing all reactive trajectories and by isolating configurations that are as likely to lead to a disaggregated state as they are to lead to the aggregated state (see Methods). Fig. 8 shows a contact map of configurations in the TSE. The contact map provides the probability density that a contact between residues i and j belonging to the two strands arises in the TSE. One can see in the figure that the probability density map is completely dominated by the formation of contacts at residues 3 and 4. This suggests that much of the activation energy needed to undergo a transformation from the disaggregated state to the aggregated state is employed to form these contacts. Once these contacts are formed, it is relatively easy for the system to form more contacts, and end up in an aggregated state.
Figure 7.
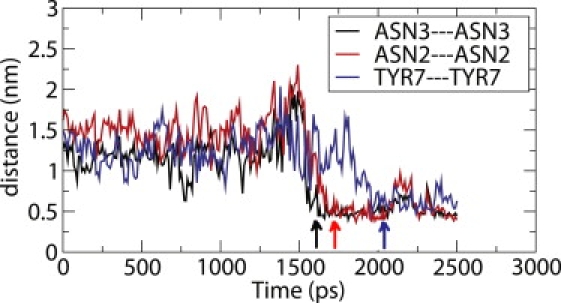
Interstrand distances between corresponding residues in the two chains as a function of time. The time point at which the contacts are formed is marked with an arrow.
Figure 8.
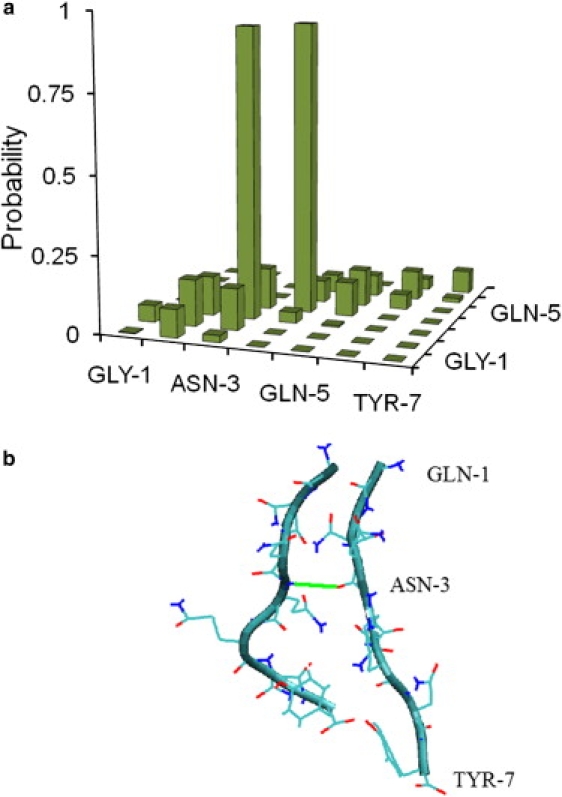
Probability density of the various contacts in transition state ensemble (a); representative snapshot of the transition state (b).
It is of interest to note that the stacking of Asparagine residues identified in the TPE and TSE is a widely observed structural feature that arises in several proteins that exist in a β-helix (35,36). For example, based on parallel tempering molecular dynamics simulations of DFNKF, a different amyloid-forming peptide, Tsai et al. (37) proposed that Asparagine stacking is a bottleneck during the assembly of individual peptide molecules. It should be noted that their results were based on the free energy landscape calculated from parallel tempering molecular dynamics simulations, which does not necessarily correspond to the actual, dynamic, reaction coordinate. Our results, obtained from TPS Monte Carlo, do identify the reactive pathways for dimerization of GNNQQNY, and they also reveal this bottleneck.
For the particular case of GNNQQNY, our results indicate that Glutamine-4 plays an equally important role in the TSE. The stacking of Glutamine residues, though not as widely discussed as that of Asparagine residues, has also been observed in experiments on β-helical structures of Polyglutamine (38). Once the contacts at these two residues are formed, they act as a distance constraint between the two peptides, which reduces the dimensionality of the conformational search required for the formation of additional contacts. A similar mechanism for aggregation (often referred to as dock-and-lock) has been observed during the growth of preformed amyloid fibrils of Amyloid β-peptide (39), and more recently in simulations of the growth of GNNQQNY (40). Although our results agree with the overall mechanism proposed by these authors, there are also important differences. In the work of Reddy et al. (40), all the contacts are formed during the locking stage. In our work, we find that a proper docking event includes the formation of a contact at the Asparagine-3 residue; without the formation of this contact, docking attempts do not lead to aggregation. This difference might be because in a study by Reddy et al. (40), a preformed contact at the Glycine residue was used to reduce computational time, and the presence of this contact could be altering the aggregation pathway. Also note that the conclusions in their work were based on a few molecular-dynamics trajectories, whereas our findings are based on a statistically relevant (large) number of reactive trajectories obtained from TPS simulations performed without constraints.
It has been proposed in the experimental studies by Tjernberg et al. (41) and Azriel and Gazit (42) that the aromatic end residue Tyrosine provides directionality (formation of a parallel versus an antiparallel β-sheet) during self-assembly of the peptides. However, in our simulations, we found that the formation of the contacts at the Tyrosine residues occurs late in the pathway. The formation of the contacts at the other residues was found to determine the progression of aggregation toward the formation of a parallel β-sheet.
The structural characteristics of the TSE were studied by plotting the density of the dihedral angles in the ϕ-ψ space (Fig. 9). We find that conformations belonging to the TSE are populated in the extended state conformation, which resembles more closely the final aggregated state. The extended state conformation is not favored entropically, but in the resulting aggregate, it is stabilized by the main-chain hydrogen bonds. In the transition state, where the main-chain hydrogen bonds are not yet completely formed, the extended state conformation is highly unfavorable and contributes toward the activation energy involved during the aggregation process.
Figure 9.
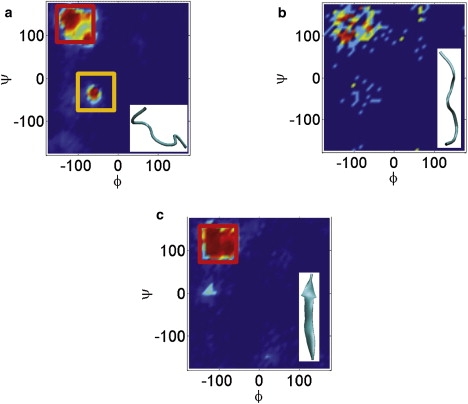
Probability density of the ϕ-ψ dihedral angles in (a) disaggregated state, (b) transition state ensemble, and (c) aggregated state. The location of the red box corresponds to an extended state conformation whereas the yellow box corresponds to a random coil state.
Several computational studies of the GNNQQNY peptide have also suggested that the aromatic Tyrosine residue (Tyr-7)—the only hydrophobic residue in the sequence—is important for aggregation of the peptide and its stability. It has also been suggested that the π-π stacking of the aromatic rings in Tyr is responsible for fibril formation (43,44). An important question to consider is therefore whether Tyr-7 affects the aggregation pathway. We address this question by analyzing the TSE. The structures from the TSE were mutated to have Asparagine instead of Tyrosine as residue number 7. The probability of reaching the disaggregated state and the aggregated state starting from these structures was obtained as explained in Methods. Interestingly, we find that the probability of reaching the aggregated state is in fact enhanced when Tyrosine is mutated into an Asparagine residue (see Fig. 10). Although changing Tyr to Asn makes the peptide more hydrophilic, the interstrand side-chain-to-side-chain interactions promoted by Asparagine residues are more favorable for the formation of a β-sheet. This observation is consistent with remarks by Dobson (1), who has suggested that the main-chain interactions are primarily responsible for peptide aggregation, but the rate of aggregation is known to vary with peptide sequence. Our results indicate that the side-chain interactions are important in the amyloid formation pathway and influence the rate of aggregation of the peptides.
Figure 10.
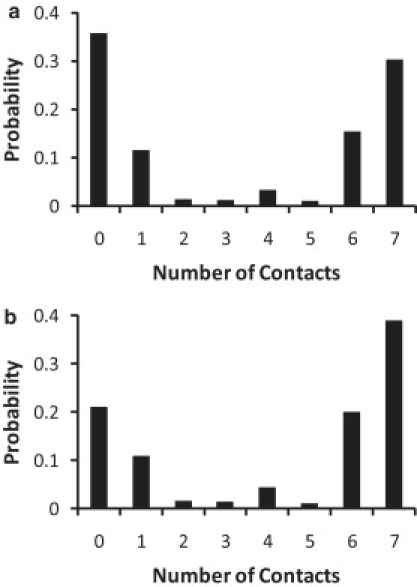
Probability density of reaching a aggregated/disaggregated state starting from a conformation in the transition state ensemble in (a) wild-type, GNNQQNY, and (b) mutated peptide, GNNQQNN.
Conclusions
We have studied the dimerization of GNNQQNY peptide using atomistic simulations in explicit water. We used a new variant of parallel tempering simulations to determine the free energy for dimerization of the peptide. It was found that the dimer is more stable, with a dimerization free energy of ∼3.3 kT. TPS simulations were used to gain insight into the aggregation pathway of the GNNQQNY peptide. Analysis of one-hundred uncorrelated reactive trajectories revealed that the transition state along the transition path involves the formation of key contacts at Asparagine-3 and Glutamine-4. Once these contacts are formed, the binding of other residues occurs in a sequential manner with a relatively large probability. This indicates to us that the formation of contacts at these residues represents the relevant bottleneck during GNNQQNY aggregation. The biological relevance of this finding could be significant. Indeed, a majority of the proteins involved in protein aggregation diseases are rich in Asparagine residues. Our results suggest that propensity for aggregation might largely depend on how favorable ASN-ASN interactions are in these proteins. Our reactive trajectories also indicate that glutamine residues play an important role in the aggregation pathway, consistent with earlier experimental observations on polyglutamine peptides.
Our simulations show that, from a kinetic perspective, Tyrosine may be less important than previously thought, at least in the early stages (dimer formation) of aggregation. An analysis of reactive trajectories sows that Tyrosine stacking occurs much later in the pathway of GNNQQNY aggregation, when the peptide is already committed to form a dimer. Thus, when aiming to improve stability of proteins, one should consider designing molecules that disrupt interactions at the Asparagine residue, and not only at the end Tyrosine.
Acknowledgments
This work was supported through the University of Wisconsin-Madison Materials Research Science and Engineering Center on Nanostructured Interfaces and grant No. CBET-0755730.
References
- 1.Dobson C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Walsh D.M., Selkoe D.J. A β oligomers—a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 4.Klein W.L., Stine W.B., Jr., Teplow D.B. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol. Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 6.Silveira J.R., Raymond G.J., Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kayed R., Head E., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 8.Wickner R.B. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 9.Patino M.M., Liu J.J., Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 10.Nelson R., Sawaya M.R., Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balbirnie M., Grothe R., Eisenberg D.S. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated β-sheet structure for amyloid. Proc. Natl. Acad. Sci. USA. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gsponer J., Haberthür U., Caflisch A. The role of side-chain interactions in the early steps of aggregation: molecular dynamics simulations of an amyloid-forming peptide from the yeast prion Sup35. Proc. Natl. Acad. Sci. USA. 2003;100:5154–5159. doi: 10.1073/pnas.0835307100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cecchini M., Rao F., Caflisch A. Replica exchange molecular dynamics simulations of amyloid peptide aggregation. J. Chem. Phys. 2004;121:10748–10756. doi: 10.1063/1.1809588. [DOI] [PubMed] [Google Scholar]
- 14.Zheng J., Ma B., Nussinov R. Structural stability and dynamics of an amyloid-forming peptide GNNQQNY from the yeast prion sup-35. Biophys. J. 2006;91:824–833. doi: 10.1529/biophysj.106.083246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z., Chen H., Lai L. Molecular dynamics simulations on the oligomer-formation process of the GNNQQNY peptide from yeast prion protein Sup35. Biophys. J. 2007;93:1484–1492. doi: 10.1529/biophysj.106.100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipfert J., Franklin J., Doniach S. Protein misfolding and amyloid formation for the peptide GNNQQNY from yeast prion protein Sup35: simulation by reaction path annealing. J. Mol. Biol. 2005;349:648–658. doi: 10.1016/j.jmb.2005.03.083. [DOI] [PubMed] [Google Scholar]
- 17.Esposito L., Pedone C., Vitagliano L. Molecular dynamics analyses of cross-β-spine steric zipper models: β-sheet twisting and aggregation. Proc. Natl. Acad. Sci. USA. 2006;103:11533–11538. doi: 10.1073/pnas.0602345103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berryman J.T., Radford S.E., Harris S.A. Thermodynamic description of polymorphism in Q- and N-rich peptide aggregates revealed by atomistic simulation. Biophys. J. 2009;97:1–11. doi: 10.1016/j.bpj.2009.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dellago C., Bolhuis P., Geissler P. Transition path sampling. Adv. Chem. Phys. 2002;123:1–78. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 20.Bolhuis P.G., Chandler D., Geissler P.L. Transition path sampling: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 21.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 22.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 23.Darden T., York D., Pedersen L. Particle mesh Ewald: an N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089. [Google Scholar]
- 24.Essmann U., Perera L., Pedersen L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 25.Berendsen H., Postma J., Haak J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 26.Sugita Y., Kitao A., Okamoto Y. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 2000;113:6042–6052. [Google Scholar]
- 27.Yan Q., de Pablo J. Hyperparallel tempering Monte Carlo simulation of polymeric systems. J. Chem. Phys. 2000;113:1276–1282. [Google Scholar]
- 28.Faller R., Yan Q., de Pablo J. Multicanonical parallel tempering. J. Chem. Phys. 2002;116:5419–5423. [Google Scholar]
- 29.Reddy A.S., Izmitli A., de Pablo J.J. Effect of trehalose on amyloid β (29-40)-membrane interaction. J. Chem. Phys. 2009;131:085101. doi: 10.1063/1.3193726. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S., Rosenberg J., Kollman P. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 31.Bolhuis P., Chandler D. Transition path sampling of cavitation between molecular scale solvophobic surfaces. J. Chem. Phys. 2000;113:8154–8160. [Google Scholar]
- 32.Chopra M., Reddy A.S., de Pablo J.J. Folding of polyglutamine chains. J. Chem. Phys. 2008;129:135102. doi: 10.1063/1.2980043. [DOI] [PubMed] [Google Scholar]
- 33.Strodel B., Whittleston C.S., Wales D.J. Thermodynamics and kinetics of aggregation for the GNNQQNY peptide. J. Am. Chem. Soc. 2007;129:16005–16014. doi: 10.1021/ja075346p. [DOI] [PubMed] [Google Scholar]
- 34.Vitagliano L., Esposito L., De Simone A. Stability of single sheet GNNQQNY aggregates analyzed by replica exchange molecular dynamics: antiparallel versus parallel association. Biochem. Biophys. Res. Commun. 2008;377:1036–1041. doi: 10.1016/j.bbrc.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins J., Pickersgill R. The architecture of parallel β-helices and related folds. Prog. Biophys. Mol. Biol. 2001;77:111–175. doi: 10.1016/s0079-6107(01)00013-x. [DOI] [PubMed] [Google Scholar]
- 36.Tsai H.H., Gunasekaran K., Nussinov R. Sequence and structure analysis of parallel β helices: implication for constructing amyloid structural models. Structure. 2006;14:1059–1072. doi: 10.1016/j.str.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Tsai H.H., Reches M., Nussinov R. Energy landscape of amyloidogenic peptide oligomerization by parallel-tempering molecular dynamics simulation: significant role of Asn ladder. Proc. Natl. Acad. Sci. USA. 2005;102:8174–8179. doi: 10.1073/pnas.0408653102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perutz M.F., Finch J.T., Lesk A. Amyloid fibers are water-filled nanotubes. Proc. Natl. Acad. Sci. USA. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen P.H., Li M.S., Thirumalai D. Monomer adds to preformed structured oligomers of Aβ-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:111–116. doi: 10.1073/pnas.0607440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy G., Straub J.E., Thirumalai D. Dynamics of locking of peptides onto growing amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2009;106:11948–11953. doi: 10.1073/pnas.0902473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tjernberg L.O., Näslund J., Nordstedt C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 42.Azriel R., Gazit E. Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide. An experimental support for the key role of the phenylalanine residue in amyloid formation. J. Biol. Chem. 2001;276:34156–34161. doi: 10.1074/jbc.M102883200. [DOI] [PubMed] [Google Scholar]
- 43.Gazit E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16:77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- 44.Makin O.S., Atkins E., Serpell L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]