

Abstract
Axons show a poor regenerative capacity following traumatic central nervous system (CNS) injury, partly due to the expression of inhibitors of axonal outgrowth, of which Nogo-A is considered the most important. We evaluated the acute expression of Nogo-A, the Nogo-66 receptor (NgR) and the novel small proline-rich repeat protein 1A (SPRR1A, previously undetected in brain), following experimental lateral fluid percussion (FP) brain injury in rats. Immunofluorescence with antibodies against Nogo-A, NgR and SPRR1A was combined with antibodies against the neuronal markers NeuN and microtubule-associated protein (MAP)-2 and the oligodendrocyte marker RIP, while Western blot analysis was performed for Nogo-A and NgR. Brain injury produced a significant increase in Nogo-A expression in injured cortex, ipsilateral external capsule and reticular thalamus from days 1–7 post-injury (P < 0.05) compared to controls. Increased expression of Nogo-A was observed in both RIP- and NeuN positive (+) cells in the ipsilateral cortex, in NeuN (+) cells in the CA3 region of the hippocampus and reticular thalamus and in RIP (+) cells in white matter tracts. Alterations in NgR expression were not observed following traumatic brain injury (TBI). Brain injury increased the extent of SPRR1A expression in the ipsilateral cortex and the CA3 at all post-injury time-points in NeuN (+) cells. The marked increases in Nogo-A and SPRR1A in several important brain regions suggest that although inhibitors of axonal growth may be upregulated, the injured brain is also capable of expressing proteins promoting axonal outgrowth following TBI.
Keywords: Nogo-A, Nogo-66 receptor, Small proline-rich repeat protein 1A (SPRR1A), Traumatic brain injury, Oligodendrocytes
Introduction
Traumatic brain injury (TBI) is a major health problem in the industrialized world. In the United States alone, 2.5–6.5 million survivors suffer from the physical, psychological and economical consequences of TBI, and a large number of TBI victims have significant long-term deficits in information processing, perceptual function and memory (Consensus conference, 1999; Levin, 1995, 1998). Axonal damage with widespread distribution, termed diffuse axonal injury (DAI), is an important contributor to the morbidity and mortality seen both clinically and in experimental TBI (Adams et al., 1989; Pierce et al., 1996; Maxwell et al., 1997; Christman et al., 1997), and is characterized by axonal swelling and, ultimately, disconnection. The intracellular DNA fragmentation that has been observed in white matter tracts of TBI patients for up to a year post-injury, may also contribute to continuing damage of the white matter tracts and be linked to the observed progressive atrophy with enlargement of the cerebral ventricles (Williams et al., 2001; Shiozaki et al., 2001). These observations of progressive brain atrophy have been replicated in experimental TBI models (Smith et al., 1997; Pierce et al., 1998; Bramlett and Dietrich, 2002; Bramlett et al., 1997).
Pioneering studies observed that in the central nervous system (CNS), the proximal ends of axons initially try to grow following transection or injury, but then abort and fail to regenerate (Cajal, 1928). More recent studies have demonstrated the capacity for enhanced growth of adult CNS axons when a permissive environment, such as a peripheral nerve transplant, was provided (David and Aguayo, 1981; Benfey and Aguayo, 1982). It was subsequently discovered that dorsal root ganglion neurons extended their axons in vitro across peripheral nervous system (PNS) tissue containing Schwann cells, but avoided oligodendrocytes and the myelin sheath (Schwab and Thoenen, 1985; Caroni and Schwab, 1988b; Caroni et al., 1988a). These and other studies revealed that inhibitors of axonal outgrowth, present in myelin, are among the possible factors underlying the abortive regeneration of mature CNS neurons observed following CNS injury (Huang et al., 1999; Grados-Munro and Fournier, 2003). The importance of myelin-associated glycoprotein (MAG), oligodendrocyte myelin glycoprotein (OMgp) and Nogo-A in CNS injury has been addressed in several recent reviews, due to their proposed role of inhibiting axonal outgrowth (Hunt et al., 2002a; David and Lacroix, 2003; Sandvig et al., 2005; Schwab, 2004).
Nogo-A, the longest isoform of Nogo and a member of the reticulon family, has two hydrophobic transmembrane domains separated by a 66 amino acid residue region (termed Nogo-66) that potently causes growth cone collapse and inhibition of neurite outgrowth in vitro (Grandpre et al., 2000). Nogo-66, together with both MAG and OMgp, bind and activate the same receptor, the neuronal, glycosyl-phosphatidyl-inositol (GPI)-anchored Nogo-66 Receptor (NgR; Fournier et al., 2001; Domeniconi et al., 2002; Liu et al., 2002a; Wang et al., 2002a) with additional molecules including p75NTR, Lingo-1 and TAJ/TROY required for signaling (Mi et al., 2004; Shao et al., 2005; Park et al., 2005).
Important for the inhibitory effects of Nogo-A in vivo is the presence of both Nogo-A and NgR at sites of axon-myelin and synaptic contact (Wang et al., 2002b). However, high levels of Nogo-A mRNA and protein are present in both oligodendro-cytes and neurons (Grandpre et al., 2000; Hunt et al., 2002a; Wang et al., 2002b). Marked recovery and enhancement of axonal regeneration have been found in models of traumatic spinal cord injury (SCI) with pharmaceutical compounds targeting Nogo-A or NgR, indicating an important role for these factors in the lack of regeneration following SCI (Lee et al., 2003; Schwab, 2004). We have recently reported that post-injury treatment with the anti-Nogo-A antibodies 11C7 and 7B12 attenuated cognitive deficits following a lateral fluid percussion (FP) brain injury in rats (Lenzlinger et al., 2005; Marklund et al., 2004), suggesting that inhibition of Nogo-A may be of therapeutic value in TBI. However, there are several unresolved issues with regard to the role of Nogo-A and NgR in acute CNS injury. As an example, NgR mRNA and protein are unevenly distributed in the adult CNS with some important regions lacking NgR (see Hunt et al., 2002a,b) and mechanisms other than Nogo/NgR interactions must explain the lack of axonal regeneration in these areas. In addition, using mice deficient in Nogo-A or NgR, some reports have shown axonal regeneration and marked behavioral improvement following SCI (Simonen et al., 2003; Kim et al., 2003, 2004), whereas other studies did not detect any differences when compared to their wild-type littermates (Zheng et al., 2003, 2005). Other recent reports have suggested a more complex role for Nogo other than associated with inhibition of axonal outgrowth (Jokic et al., 2005; He et al., 2004; Karnezis et al., 2004; Acevado et al., 2004).
Nerve lesion studies have demonstrated that peripheral, but not central, axotomy induces an enhanced axonal growth state, perhaps due to induction of neuronal regeneration-associated genes (RAGs; Neumann and Woolf, 1999; Chong et al., 1999). In a previous report, the gene for the small proline-rich repeat protein 1A (SPRR1A), a highly specific marker for the differentiation of keratinocytes and squamous epithelial cells (Kartasova and van de Putte, 1988), was demonstrated to be the most highly induced gene using a microarray approach following peripheral axotomy (Bonilla et al., 2002). SPRR1A is not expressed in naive neurons at any age and has not previously been demonstrated in brain (Bonilla et al., 2002). Depletion or blockade of the SPRR1A protein has been shown to decrease outgrowth of adult-injured peripheral neurons, the growth of which was enhanced by over-expression of SPRR1A, even at the presence of inhibitory substrates. Low levels of SPRR1A has been observed in the spinal cord following SCI (Bonilla et al., 2002), and treatments targeting and inhibiting NgR with the peptide NEP 1–40 resulted in improved recovery, increased regeneration of severed axons, synapse formation and increased SPRR1A expression in the spinal cord following SCI in mice (Li and Strittmatter, 2003). Collectively, these observations suggest that SPRR1A may either be a marker of and/or a contributor to regenerative attempts by injured CNS axons and that injury-induced expression of SPRR1A may be involved in axonal regeneration in the PNS. Thus, the relation between growth-inhibitory (NgR, Nogo-A) and growth-promoting (SPRR1A) molecules may be an important indicator of the regenerative response following TBI.
To begin to evaluate the importance of inhibitors of axonal outgrowth in TBI, knowledge of their post-injury expression pattern is paramount. Since no information exists, to date, with regard to TBI-induced changes of Nogo-A, NgR and SPRR1A, the present study evaluated the temporal and regional expression of these proteins in the acute post-injury period following lateral FP brain injury in rats.
Material and methods
Animals
Thirty-three adult, male Sprague–Dawley rats (weight 382 + 11, range 354–411 g; Harlan, Inc., Indianapolis, IN) were included in the study and housed on a 12-h light/dark cycle, with access to food and water ad libitum. All procedures described herein were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and were performed in accordance with standards published by the National Research Council (1996).
Surgery and fluid percussion brain injury
The animals were anesthetized (sodium pentobarbital, 60 mg/kg, i.p.) and placed in a stereotaxic frame. The scalp and temporal muscle were reflected, and a 5 mm craniectomy was made on the skull over the left parietal cortex between lambda and bregma, leaving the dura intact. A modified luer-lok cap was cemented (DentSply®, Dentsply International Inc., York, PA, USA) over the craniectomy and filled with saline. A stainless-steel screw was placed anterior to the coronal suture to further anchor the luer-loc and dental cement to the skull bone. Ninety minutes after anesthesia, a subset of the animals (n = 26) were attached to the fluid percussion (FP) device via the luer-lok and subjected to a moderate severity brain injury (2.7 ± 0.3 atm) by the rapid (22 ms) delivery of a pressurized pulse of saline striking the intact dura, deforming the underlying brain tissue as originally described (McIntosh et al., 1989). The luer-lok was then removed and the wound was closed. All animals remained on warming pads to maintain normothermia until they were able to ambulate. Sham-injured animals (n = 7) received anesthesia and all surgical procedures, but did not undergo FP brain injury, served as controls. The same investigator performed all injuries throughout the study. At survival times of 1, 3 or 7 days, 21 surviving brain-injured animals (n = 7 per time-point) and 7 sham-injured animals were reanesthetized with intraperitoneal injection of sodium pentobarbital (200 mg/kg). Animals evaluated for immunohistochemistry (n = 12 brain-injured, 4 sham-injured) were perfused through the heart with heparinized 0.9% saline followed by 4% paraformaldehyde (PFA). The brains were removed, post-fixed at 4°C in PFA for 24 h, transferred to a 30% sucrose solution for 3–4 days and then frozen and kept at −80°C. A separate group of anesthetized animals, used for evaluation of Nogo-A and NgR at 1, 3 and 7 post-injury by immunoblot analysis (n = 9 brain-injured, n = 3 sham-injured), was perfused through the heart with cold saline at +4°C, and decapitated. Each brain was quickly removed from the cranium, a 3 mm coronal section was made and tissue pieces from the ipsilateral hemisphere from the cortex at the maximal site of injury were dissected on a chilled glass plate over dry ice as previously described (Soares et al., 1992). The brain regions were snap-frozen in isopentane (2-methylbutane) at −65°C and stored at −80°C until analyzed.
Antibody overview
The polyclonal, specific, rabbit anti-NgR (raised against a GST-NgR fusion protein, corresponding to residues 27–447 of NgR), anti-Nogo-A (raised against a Nogo-A specific amino acid sequence corresponding to aa 623–640 of rat Nogo-A) and anti-SPRR1A (raised against an SPRR1A-His protein) antibodies were generated and characterized in the laboratory of Dr. Strittmatter and used as previously described in detail in previous publications (Fournier et al., 2001; Wang et al., 2002a,b; Bonilla et al., 2002). A biotin-conjugated goat anti-rabbit secondary antibody (1:2000; Jackson) was used for DAB immunohistochemistry (vide infra). The mouse hybridoma RIP (1:4; developed by Dr. Susan Hockfield, Yale University, New Haven, CT, was obtained from the Developmental Studies Hybridoma Bank and developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA) and rabbit 2,3-cyclic nucleotide 3-phosphodiesterase (CNPase, 1:500; a kind gift from Dr. Sirkowski, Dept. of Neurology, University of Pennsylvania) were used as oligodendrocyte markers. RIP and CNPase were found to colocalize in white matter tracts (data not shown) and RIP was used as an oligodendrocyte marker for all subsequent analyses. Neuronal-specific nuclear protein (NeuN; mouse monoclonal 1:1000; Chemicon, USA) and microtubule-associated protein (MAP-2a,b, 1:1000; mouse monoclonal, Sigma, Saint Louis, MI) were used as neuronal markers. An antibody directed toward glial fibrillary acid protein (GFAP, 1:200; mouse monoclonal, Sigma) was used to label astrocytes. Alexa 488-conjugated anti-mouse (1:200 for RIP, GFAP, NeuN and MAP-2) and 594-conjugated goat anti-rabbit (1:200; for CNPase, SPRR1A, Nogo-A and NgR; both from Molecular Probes, Eugene, OR) were used as secondary fluorescent antibodies. To evaluate nonspecific background staining, negative controls with omission of the primary antibody were included for all analyses.
Immunohistochemistry
Forty micrometer (Am) frozen sections were cut on a sliding microtome (Micron Gmbh, Germany), and transferred to cryoprotectant. Sections at bregma −2.3, −4.3 and −6.3 mm were in 0.1 washed M tris buffer saline (TBS), endogenous peroxidases were quenched and nonspecific binding was blocked with 5% normal horse serum (NHS) and 0.3% Triton-X (Sigma) in TBS. The sections were incubated overnight with the primary antibodies (Nogo-A 1:8000; for NgR 1:3000 and for SPRR1A 1:1500) that were detected using a secondary biotin-conjugated goat anti-rabbit IgG antibody (1:2000; Jackson Immunoresearch, West Grove, PA), applied for 1 h at room temperature. The sections were incubated with the ABC reagent (Vector Laboratories, Burlington, CA, USA) for 1 h at room temperature, washed and developed with 3,3′-diaminobenzidine (DAB; Vector Laboratories). The sections were mounted, dehydrated in graded ethanols, cleared in xylene, coverslipped and analyzed with a light microscope (Nikon Eclipse 8600, Melville, NY). In addition, routine staining using 0.3% cresyl violet was performed at brain sections from identical bregma levels and from all animals included in the study.
For immunofluorescence, similar protocols (vide supra) were used with the following changes; the sections were blocked in a mixture containing 10% NHS, 3% bovine serum albumin (BSA), 0.4% Triton-X and 0.1% glycine. The sections were incubated overnight with the primary antibodies (Nogo-A 1: 3000, NgR 1:1000, SPRR1A 1:500, NeuN 1:1000, MAP-2 1:1000, GFAP 1:200 and RIP 1:4). The secondary Alexa 488- or 594-conjugated antibodies (Molecular Probes, Eugene, OR) were applied for 1 h at a concentration of 1:200 at room temperature. The sections were coverslipped using Fluoromount G (Southern Biotechnology Associates, Inc., Birmingham, AL). The images were analyzed using a microscope (Nikon), and pictures were taken with a Magnafire camera (Dage-MTI, Inc., Michigan City, IN).
For verification of colocalization of Nogo-A/NeuN and SPRR1A/NeuN, and Nogo-A/RIP, additional images were captured with a confocal laser scanning microscope equipped with a Krypton–Argon laser (z-stage 0.5 μm; Radiance 2000MP, Bio-Rad Microscience, Hampstead, UK) connected to a microscope (Nikon eclipse TE300). Resulting image stacks were imported into Image J (National Institute of Mental Health, Bethesda, Maryland, USA) and orthogonal reconstructions were created from 10 μm stacks. Cells were considered double-labeled if the cytoplasmic marker surrounded a nuclear marker when viewed in a x –y cross section, as well as x –z and y –z cross sections produced by orthogonal reconstructions from z-series (z-step, 0.5 μm) taken with ×40 or ×60 objective.
Western blot analysis
For protein extraction, the tissue samples were homogenized in lysis buffer (0.32 M sucrose, 1.0 mM EDTA, 5 mM Tris–HCl pH 7.4, 0.1 mM PMSF, 10 μM leupeptin, 10 μM pepstatin-A and 1 mM β-mercaptoethanol). The homogenate was centrifuged at 10,000×g at 4°C for 10 min and the supernatant was used for this study. Assays to determine the protein concentration were performed by comparison with a known concentration of bovine serum albumin. Sodium dodecyl sulfate-polyacrylamide (SDS) gel electrophoresis (Nogo-A 12%, NgR 8%) was performed and lysate equivalent of 10 μg (Nogo-A) or 25 μg (NgR) of protein from samples from ipsilateral cortex was loaded and run on the gel at 100 V together with a size marker (Kaleidoscope, Amersham Bioscience, Buckinghamshire, England). The protein on the gel was subsequently transferred to a 0.2 μM polyvinylidene fluoride (PVDF) transfer membrane (Bio-Rad, Hercules, CA) in a buffer containing methanol, glycine and Tris base. After the transfer, the membrane was placed in 5% nonfat milk in phosphate buffer saline (PBS) with 1% Tween-20 to block nonspecific binding and was then incubated with primary antibodies overnight at 4°C. The primary antibodies (Nogo-A; 1:10,000, NgR; 1:3000) were the same as those used for immunohistochemistry. After washing, the membrane was incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1, 2000; Jackson) applied for 1 h, and then the signal was detected with a chemiluminescence kit (Amersham). For control of total protein load, a polyclonal rabbit anti alpha-tubulin antibody (Sigma, 1:2000) was added on stripped blots. Images of the blots were captured by a CCD camera (Nikon).
Cell counts and densitometrical assessments
Images were captured at 20× ipsilaterally and, using the same exposure time, contralaterally for densitometrical evaluation of Nogo-A expression in external capsule (EC) at all three evaluated bregma levels per brain. Using image analysis software (MCID/M4; Imaging Research, St. Catherines, Ontario, Canada), a gray level threshold was set in the contralateral EC for detection of Nogo-A immunoreactivity in the evaluated target area, including only the EC, and the percentage of the target area showing optical density over the selected threshold was analyzed. Using the same setting and scanned target area, values from the ipsilateral side were obtained. Data are presented as % detected in ipsilateral EC-% detected in contralateral EC as previously described (Saatman et al., 2001), and the mean for all evaluated sections per animal was determined (Fig. 3).
Fig. 3.
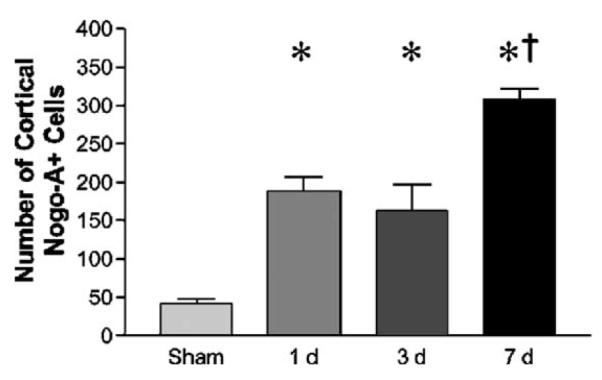
Cell counts of Nogo-A (+) cells in the cortex of sham- and FP brain-injured animals. Brain-injured animals show a significant (*P < 0.05) increase at all evaluated post-injury time-points in the sum of Nogo-A (+) cells from all evaluated bregma levels in cortex ipsilateral to the injury when compared to sham-injured controls. The number of cortical Nogo-A (+) cells was significantly higher at 7 days when compared to 1 and 3 days post-injury and sham-injured animals (†P < 0.05).
To estimate the number of Nogo-A-positive cells in the ipsilateral cortex, reticular thalamus and hilus of the dentate gyrus, each slide was counterstained with 0.3% Cresyl Violet and images were captured at 20× at −2.3, −4.3 and −6.3 mm posterior to bregma (three images from the perilesional cortex and one image from the hilus of the dentate gyrus at each bregma level and two images (rostral and caudal) from the reticular thalamus at bregma level −2.3). For each image, each Nogo-A-positive cell showing Nogo-A staining with a cellular morphology and above background level was manually and exhaustively counted using the MCID image analysis system. The densitometrical evaluation and cell counts were performed by an investigator blinded to the treatment status of each animal.
Statistical analysis
Animal weights, injury-induced apnea, atmospheres and cell counts are presented as mean ± SD. The cell counts and densitometry for the EC are analyzed with a one-way ANOVA and, if significant, followed by Newman–Keuls post hoc test. A P value of <0.05 was considered significant.
Results
Brain injury consistently produced an acute apnea in all animals (28 ± 15 s), and resulted in marked tissue damage observed in the ipsilateral cortex, white matter tracts and hippocampus using cresyl violet-stained sections (data not shown) as has been previously shown in numerous reports using the FP brain injury model (Hicks et al., 1996; Sato et al., 2001). Five animals died in the immediate post-injury period, producing an injury mortality of 18%, consistent with previous reports using the lateral FP brain injury model of moderate severity (Saatman et al., 1997).
Nogo-A expression in cortex
Using Western blot, a single, approximately 200 kDa weak band, corresponding to Nogo-A, was detected in sham-injured animals (Fig. 1) as previously described (Wang et al., 2002b). There were no statistically significant differences in band intensity from 24 h–7 days following FP brain injury when compared with sham-injured controls. In the neocortex of sham-injured animals, numerous Nogo-A-positive cells were observed with weak staining mainly in the perimeter of the cells (Fig. 2A). Strong immunoreactivity for Nogo-A was detected in the habenular complex and the nucleus ruber as previously described (data not shown; Hunt et al., 2002a). Following FP brain injury, numerous Nogo-A immunopositive (Nogo (+)) cells emerged in the immediate vicinity of the lesion (periinjured cortex/penumbra), many of which possessed a clearly neuronal morphology with staining of axons and dentritic processes (Figs. 2B–C). No GFAP/Nogo colabeling was observed (Figs. 2D–F). In the periinjured cortex and central parts of the lesion, adjacent to the external capsule (EC), some Nogo (+) cells were observed to colabel with the oligodendrocytic marker RIP (Figs. 2G–I). Nogo (+) cells frequently colabeled with MAP-2, although occasional Nogo (+)/MAP (−) cells with a neuronal morphology were observed (data not shown). All Nogo (+) cells with neuronal morphology colabeled with NeuN (Figs. 2J–I). Using cell counts, an increased number of Nogo (+) cells with both neuronal and glial morphology were observed from 1 to 7 days post-injury (P < 0.05; Fig. 3). The number of cortical Nogo-A (+) cells was significantly higher at 7 days when compared to 1 and 3 days post-injury and sham-injured animals (P < 0.05).
Fig. 1.

Western blot analysis for Nogo-A and NgR. Protein samples (10 μg for Nogo-A and 25 μg for NgR) were extracted from dissected samples from the ipsilateral cortex of sham- and brain-injured animals from 24 h to 7 days post-injury and resolved in polyacrylamide gels. Single bands emerged at approximately 200 kDa, corresponding to Nogo-A, and 80 kDa, corresponding to NgR, respectively. There were no significant changes between sham- and brain-injured animals. Alpha-tubulin was used as an internal control of protein load.
Fig. 2.

Cortical Nogo-A expression in sham- and brain-injured animals. (A) In the cortex of sham-injured animals, there were frequent Nogo-A (+) cells, with immunoreactivity in the perimeter of the cells (arrowheads, scale bar = 200 μm). Insert shows these cells, displaying a neuronal morphology, at high magnification (scale bar = 50 μm). (B) Fluid percussion (FP) brain injury caused an increased neuronal Nogo-A expression in the vicinity of the lesion (solid arrows). However, there was a reduced staining for Nogo-A in the center of the lesion when compared with the perilesional cortex (asterisk, scale bar = 200 μm), beginning at 24 h post-injury. (B–C) In the cortex of FP brain-injured animals, numerous Nogo-A-positive cells, with neuronal (solid arrows) or glial (open arrows) morphology, emerged beginning at 1 day continuing through 7 days post-injury (Fig. 1C, scale bar = 100 μm). (D–F) In the cortex of FP brain-injured animals, numerous GFAP (+) cells were observed. No Nogo-A/GFAP colabeling was observed (GFAP green (D), Nogo-A red (E), GFAP/Nogo-A colabel in panel F). (G–I) Orthogonal reconstruction of a confocal image showing Nogo-A (+)/RIP (+) cells observed adjacent to the lesion cavity (RIP green (G), Nogo-A red (H), RIP/Nogo colabel yellow (I), open arrows, scale bar = 50 μm). (J–L) In the perimeter of the lesion, Nogo-A (+) cells colabeled with Neu-N. Confocal reconstruction of a NeuN (green, J)/Nogo-A (red, K) colabeled cell in the ipsilateral, injured cortex at 7 days post-injury (L, arrow; scale bar = 50 μm).
Nogo-A expression in white matter tracts
In the white matter tracts of sham-injured animals, weak staining of Nogo-A in oligodendrocytes was observed (Fig. 4A). By 24 h post-injury, markedly increased Nogo-A expression in ipsilateral external capsule was observed (Fig. 4B), in Nogo-A (+) cells colabeling with RIP (Figs. 4D–F). In addition, increased immunostaining for Nogo-A in the ipsilateral fimbriae was observed in 7/12 brain-injured animals from 1–7 days post-injury (Fig. 4C). No changes in Nogo-A expression were observed in the corpus callosum or ipsilateral internal capsule (data not shown). Densitometric analysis revealed a highly significant increase in Nogo-A immunoreactivity in the external capsule from 1 to 7 days in FP brain-injured animals when compared with sham-injured controls (P < 0.05;Fig. 5). The Nogo-A expression was significantly higher in the EC at 7 days versus 3 days post-injury (P < 0.05).
Fig. 4.

Nogo-A expression in ipsilateral white matter tracts in sham- and brain-injured animals. (A) In the external capsule (EC) of sham-injured animals, immunoreactivity for Nogo-A was observed in oligodendrocytes along the white matter fiber tracts (arrows, scale bar = 100 μm). (B) FP brain injury induced a marked increased expression of Nogo-A in the EC ipsilateral to the injury at all time-points evaluated (arrows, scale bar = 100 μm). Insert shows higher magnification of the EC of another brain-injured animal, showing an increased number of Nogo-A (+) cells (arrows, scale bar = 50 μm). (C) Example of increased immunostaining for Nogo-A at 7 days post-injury in the medial, but not lateral part (arrowheads) of the fimbria ipsilateral to the injury, scale bar = 200 μm. (D–F) Confocal reconstruction of the EC of a FP brain-injured animal. An increased number of RIP (+) (D, green)/Nogo-A (+) (E, red)-colabeling cells (F; merged cells shown in yellow, arrows) were observed. Scale bar = 50 μm.
Fig. 5.
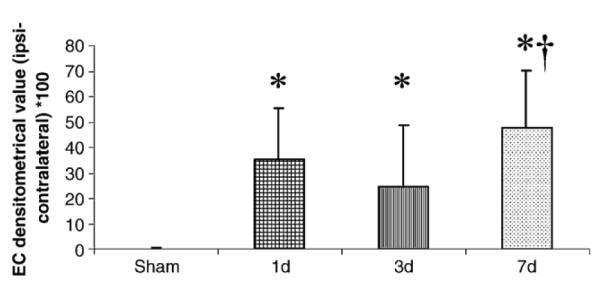
Densitometrical evaluation of Nogo-A staining in the ipsilateral external capsule (EC). At all post-injury time-points and at all bregma levels evaluated, FP brain injury caused a significantly (*P < 0.05) increased Nogo-A-expression when compared with sham-injured controls. The Nogo-A expression was significantly higher in the EC at 7 days versus 3 days post-injury (†P < 0.05).
Nogo-A expression in hippocampus
In sham-injured animals, modest staining for Nogo-A was observed in all hippocampal subfields except for the dentate gyrus where only weak immunoreactivity for Nogo-A was present (Fig. 6A), and Nogo-A (+) cells were observed in the pyramidal cell layer of the hippocampal CA3 region (Fig. 6B, arrows). FP brain injury did not cause marked changes in hippocampal Nogo-A immunoreactivity, except for a few, strongly Nogo-A immunopositive cells in the CA3 region of the hippocampus (Figs. 6C–D). Numerous Nogo-A (+) cells were observed in the hilus of the dentate gyrus of sham-injured animals (Fig. 6E), which colabeled with NeuN (data not shown). By 24 h post-injury, these cells were found to be selectively vulnerable to TBI (Fig. 6F). In the subgranular layer of the dentate gyrus, numerous Nogo (+) processes emerged post-injury (Fig. 6F) that were both MAP-2 (−) and GFAP (−) (6G–I). The decreased number of Nogo-A (+) cells in the hilus of the dentate gyrus ipsilateral to the site of brain injury was confirmed with cell counting (P < 0.05; Fig. 7).
Fig. 6.

Expression of Nogo-A in the hippocampus of sham- and brain-injured animals. (A) In sham-injured animals, all hippocampal subfields showed immunoreactivity for Nogo-A, although only weakly in the dentate gyrus (arrows, scale bar = 0.5 mm). (B) In the CA3 subregion of sham-injured animals, Nogo-A (+) cells was observed in the pyramidal cell layer (arrows, scale bar = 50 μm). (C–D) FP brain injury did not markedly cause an increased expression of hippocampal Nogo-A, although strongly immunoreactive neurons emerged in the CA3 region of the hippocampus (C; arrows; scale bar = 100 μm and insert, scale bar = 50 μm; panel D shows a Nogo-A (+) neuron in the pyramidal cell layer; scale bar = 50 μm). (E–F) In the hilus of the dentate gyrus of sham-injured animals, there were numerous Nogo-A (+) cells (E; arrows, scale bar = 200 μm). In contrast, less Nogo-A (+) cells were observed in the hilus of FP brain-injured animals (F; arrow, scale bar = 200 μm). In addition, numerous Nogo (+) processes emerged post-injury in the subgranular layer of the dentate gyrus (F, arrows). (G–I) These Nogo (+) processes (arrows) were both GFAP- (data not shown) and MAP-2 (–), MAP-2 green (G), Nogo-A red (H) and merge shown in panel I.
Fig. 7.

Cell counts within the hilus of the dentate gyrus. A significantly reduced number of Nogo-A (+) cells were observed in FP brain-injured animals at all evaluated time-points post-injury when compared with sham-injured controls (*P < 0.05).
Nogo-A expression in reticular thalamus
In the reticular thalamus (RT) of sham-injured animals, weak Nogo-A immunoreactivity was observed (Fig. 8A). Beginning at 1 day post-injury, increased numbers of Nogo-A (+) cells were observed in the RT (Figs. 8B–C), that colabeled with both MAP-2 (data not shown) and NeuN (Figs. 8D–F). Cell counting revealed a significant increase in Nogo-A (+) cells in the RT from 1 to 7 days post-injury (P < 0.05;Fig. 9). The number of Nogo-A (+) cells in the RT was significantly higher at 7 days when compared to 1 and 3 days post-injury and sham-injured animals (P < 0.05).
Fig. 8.

Nogo-A expression in the reticular thalamus for sham- and brain-injured animals. (A) Weak immunoreactivity for Nogo-A was observed in the reticular thalamus of sham-injured animals (arrows). (B–C) At all evaluated time-points post-injury, a markedly increased expression of Nogo-A was observed in FP brain-injured animals, showing numerous Nogo-A (+) cells (B; arrows, 3 days post-injury, scale bar = 0.5 mm and C; arrows 7 days post-injury, scale bar = 50 μm). (D–F) Confocal reconstruction of NeuN (+) (green, D)/Nogo-A (+) (red, E) coexpressing (F) cells (arrows). Scale bar = 50 μm.
Fig. 9.
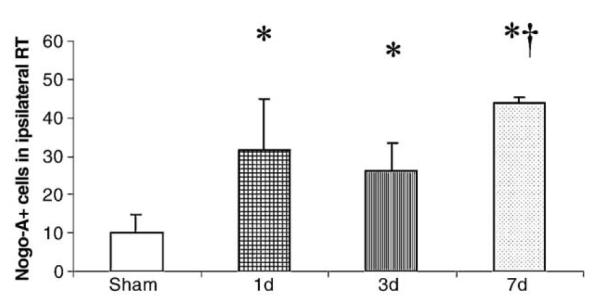
Cell counts in ipsilateral reticular thalamus (RT). At all time-points evaluated, FP brain-injured animals had an increased number of Nogo-A (+) cells (*P < 0.05). The number of Nogo-A (+) cells was significantly higher in the RT at 7 days when compared to 1 and 3 days post-injury and sham-injured animals (†P < 0.05).
NgR expression in sham-injured controls and FP brain-injured animals
Western blot analysis revealed a prominent band of approximately 80 kDa, corresponding to the NgR, in samples from ipsilateral cortex as previously demonstrated (Wang et al., 2002b), although no differences between sham-injured and FP brain-injured animals were observed (Fig. 1).
NgR expression was detected in all layers of the neocortex and the white matter tracts (e.g. external capsule, EC) of brain tissue from sham-injured animals (Figs. 10A–B). No NgR expression of the striatum and hypothalamus was observed (Hunt et al., 2002a,b, data not shown). The staining for NgR was observed predominately in the cytosol of NeuN (+) (data not shown) or MAP-2 (+) cells (Figs. 10D–F) and the negative control sections, with omission of the primary antibody, did not show any unspecific staining. Beginning 1 day following FP brain-injury, there was a decreased immunoreactivity for NgR observed within the lesion (data not shown). No significant changes were observed in the perilesional cortex (Fig. 10C). In 8 of 12 brain-injured animals, an increased expression of NgR was observed in the ipsilateral external capsule (Fig. 10C).
Fig. 10.

NgR immunoreactivity in sham- and FP-brain-injured animals. (A–B) Ipsilateral cortex (Ctx) and external capsule (EC) of a sham-injured animal. Scale bars = 200 μm (A) and 0.5 mm (B). Arrows indicate the weak linear, axonal staining in the external capsule and neuronal staining in the adjacent cortex. (C) NgR immunoreactivity in an FP brain-injured animal. No marked changes were observed post-injury in the cortex, although increased (arrow) NgR immunoreactivity in the EC was frequently observed (scale bar = 0.5 mm). (D–F) Confocal images of MAP-2 (+) (green, D)/NgR (+) (red, E) colabeled cells (F, arrows; scale bar = 50 μm), observed in the cortex of both sham- and FP brain-injured animals.
SPRR1A expression in sham-injured controls and FP brain-injured animals
Although SPRR1A immunoreactivity has not previously been detected in the brain (see Bonilla et al., 2002), occasional and scattered SPRR1A-positive (SP (+)) cells with a neuronal morphology were observed in the neocortex of sham-injured animals (insert, Fig. 11A). Beginning 24 h following lateral FP brain injury, numerous SP (+) cells were observed in the ipsilateral cortex in the vicinity of the lesion (Fig. 11B), and in the CA3 of the ipsilateral hippocampus (Fig. 11C). The majority of SP (+) cells showed a neuronal morphology with staining for SPRR1A in the cytosol, processes and dendrites (Figs. 11D–I). The SP (+) cells were more frequently observed to colabel with NeuN (Figs. 11G–I) than with MAP-2 (Figs. 11D–F). Up to 1–2% of cortical NeuN (+) neurons in sham-injured animals were also found to express SP that increased to, on average, 13% in the perilesional cortex of brain-injured animals.
Fig. 11.

SPRR1A expression in sham- and brain-injured animals. (A) Mainly background staining was observed in the cortex (scale bar = 0.5 mm) of sham-injured animals, although occasional SPRR1A-positive (SP (+)) cells were found (insert, scale bar = 50 μm). (B) SP (+) cells with a neuronal morphology were found in ipsilateral cortex of all animals beginning at 24 h (insert, scale bar 200 μm) continuing through 7 days post-injury (arrows, scale bar = 0.5 mm). (C) Usually, only few SPRR1A-positive cells were detected in the CA3 of the ipsilateral hippocampus, although in one animal, a marked increased number of SP (+) cells (arrows) was found at 7 days post-injury in the CA3 (open arrow) ipsilateral to the injury. Scale bar = 200 μm. (D–F) SP (+) cells (red, E) were frequently MAP-2 (−) post-injury (MAP-2 green (D), merged image in panel F), although a few SP (+)/MAP-2 (+) cells were also observed (arrow; MAP-2 green, SPRR1A red, merge yellow). (G–H) The SP (+) cells were found to colabel with NeuN post-injury, shown here with a confocal reconstruction of NeuN (+) (green, D)/SP (+) (red, E)-coexpressing (F) cell. Scale bar = 50 μm.
Discussion
In the present report, we demonstrate that Nogo-A expression is increased in both neurons and oligodendrocytes in widespread regions throughout the brain following experimental TBI. Furthermore, we demonstrate that the injured brain is capable of increased expression of SPRR1A, a promotor of axonal outgrowth in vitro, in neurons of the cortex and CA3. Increased immunoreactivity for NgR was observed in the ipsilateral (injured) external capsule, while a decreased staining was observed within the lesion cavity of brain-injured animals, likely attributable to post-traumatic cell death.
The marked and widespread increase in Nogo-A immuno-reactivity observed in the present study has implications for the pathophysiology of TBI. Nogo-A is present in all stages of oligodendrocyte differentiation (Huber et al., 2002; Wang et al., 2002b) and is believed to mediate inhibition of neurite outgrowth and axonal regeneration through binding to and activation of neuronal NgR in juxtaposed axons (Wang et al., 2002b; Oertle et al., 2003). In the present report, RIP was used to label oligodendrocytes as previously described (Friedman et al., 1989; Nguyen and Pender, 1999; Wennstrom et al., 2004), and we observed an increased number of Nogo-A (+)/RIP (+)cells in ipsilateral white matter following FP brain injury. Although an increased number of glial cells have been observed in the ipsilateral white matter tracts following FP brain injury (Graham et al., 2000), our data are the first to suggest that the number of oligodendrocytes may increase in selected regions following TBI, as previously demonstrated in models of cerebral ischemia (Dewar et al., 2003; Zaidi et al., 2004), spinal cord injury (McTigue et al., 2001) and cortical resection brain injury (Ludwin, 1984). In a recent report, proliferation of oligodendrocyte precursors, that may differentiate into mature oligodendrocytes following CNS injury (McTigue et al., 2001), was observed in the acute period following experimental TBI (Chen et al., 2003). Since oligodendrocytes and oligodendro-cyte precursor cells have been suggested to exert an inhibitory effect on axonal outgrowth (Chen et al., 2002a,b), an increase in the number of oligodendrocytes may contribute to the lack of axonal regeneration in TBI. However, we cannot exclude the possibility that increased Nogo-A expression in ipsilateral white matter tracts also resulted, in part, from an injury-induced release of Nogo-A into the extracellular matrix after oligoden-drocyte, axon and/or myelin damage (Goldberg and Barres, 2000), as previously described for MAG (Tang et al., 2001).
Although the expression of Nogo-A in neurons under normal conditions has previously been described (Grandpre et al., 2000; Josephson et al., 2001; Huber et al., 2002; Hunt et al., 2002b; Liu et al., 2002b; Wang et al., 2002b; Jin et al., 2003; Mingorance et al., 2004), the function of neuronal Nogo-A remains unclear. However, as a member of the reticulon family of genes (Oertle and Schwab, 2003), Nogo (RTN-4) has been suggested to play a role in cellular physiology (Oertle et al., 2003) and protein trafficking (van de Velde et al., 1994; Grandpre et al., 2000; Teng et al., 2004). We observed increased Nogo-A expression in ipsilateral cortex and reticular thalamus, that may be selectively vulnerable regions following TBI (Ross et al., 1993; Hicks et al., 1996). Part of the increased neuronal immunoreactivity for Nogo-A observed in these regions may be explained by impaired anterograde transport of Nogo-A due to post-traumatic axonal damage, since Nogo-A is detected at the synaptic level under normal physiological conditions (Liu et al., 2003) and has been observed in growing axons of the developing CNS (Tozaki et al., 2002). However, Nogo-66 and Nogo-A mRNA expression may be upregulated in neurons with a preserved capacity for axonal regeneration, implying that Nogo-A may even be a growth-promoting gene when present in neurons (Hunt et al., 2003), such as intrinsic neurons in the reticular thalamus, a region with relatively high capacity for axonal regeneration showing marked upregulation of growth-related genes following axotomy (Anderson et al., 1998; Hunt et al., 2002b, 2003). In the present report, we also observed increased Nogo-A expression in dendritic processes with yet unclear functional significance. The effects of altered neuronal Nogo-A expression in TBI remain to be established.
We observed a decreased number of Nogo-A positive neurons in the hilus of the dentate gyrus ipsilateral to the brain injury. Although a selective downregulation of Nogo-A in hilar neurons cannot be excluded, neuronal cell death in the hilus of the dentate gyrus is a well-known consequence of lateral fluid percussion brain injury (Lowenstein et al., 1992; Hicks et al., 1993; Floyd et al., 2002), likely accounting for the observed changes in Nogo-A expression. In addition, we observed numerous Nogo-A (+) processes in the subgranular layer (SGL) of the dentate gyrus emerging post-injury. The origin of these processes is not clear, although they were found to be both MAP-2 (−) and GFAP (−), suggesting that they are neuroblasts.
Although marked changes in Nogo-A expression were observed with immunohistochemistry, no significant differences were found among the treatment groups using Western blots of cortical samples. We cannot exclude the possibility that the expression of Nogo-A increases due to a shift from a diffuse distribution in processes to a distribution in the cell soma, that may be more apparent by histology. A more likely explanation, however, is that while the levels of Nogo-A are decreased in the necrotic lesion, they are concurrently increased in selected perilesional tissue and the total levels of Nogo-A remain unchanged or show only a modest increase. The changes in Nogo-A observed in the present study are in contrast to the relatively minor changes observed in models of experimental spinal cord injury (SCI; Josephson et al., 2001; Huber et al., 2002; Hunt et al., 2002b, 2003; Wang et al., 2002b). However, in other models of CNS injury such as hippocampal deafferentation, global ischemia and kainic acid lesions, a marked increase of Nogo-A mRNA was observed (Zhou et al., 2003; Meier et al., 2003; Mingorance et al., 2004). These reports indicate that the expression of Nogo-A is not generally injury-induced and imply a role for Nogo-A in some, but not all, CNS injury models. We have recently reported that administration of the neutralizing Nogo-A specific antibodies 11C7 and 7B12 improved cognitive function following FP brain injury (Lenzlinger et al., 2005; Marklund et al., 2004). Since these antibodies bind to the same Nogo-A specific domain as the anti-Nogo-A antibody evaluated in the present report (Wang et al., 2002a,b; Oertle et al., 2003), increased Nogo-A expression may be involved in mediating secondary or delayed damage and contribute to the lack of axonal regeneration following TBI.
NgR has been previously shown to distribute diffusely among neuronal cytoplasmic elements, including intracellular membranes of synaptic vesicles and mitochondria, without labeling either oligodendrocytes or astrocytes (Wang et al., 2002a,b; Oertle et al., 2003; Hunt et al., 2002a), supported by the staining pattern and colocalization of NgR with the neuronal markers NeuN and MAP-2 in both sham- and brain-injured animals in the present study. Although the Nogo-A/NgR interaction implies a localization of NgR to neuronal membranes, we used a protocol in the present report with a relatively high concentration of detergent (Wang et al., 2002a,b), that may contribute to the diffuse cytosolic expression of NgR observed in certain neurons (see Fig. 10). Interestingly, no NgR has been detected in the reticular thalamus (Hunt et al., 2002a,b), where marked increases in Nogo-A occur following TBI. We did not observe marked increases in NgR expression following FP brain injury, consistent with the finding following SCI or cortical lesions (Huber et al., 2002; Josephson et al., 2003). In a subset of animals, a focal increase of NgR expression in the external capsule was observed, that may be caused by impaired axonal transport with accumulation of NgR at the site of axonal injury or an upregulation of NgR in these animals. NgR has been reported to be upregulated early following deafferentiation injury to the hippocampus or global cerebral ischemia (Zhou et al., 2003; Meier et al., 2003; Mingorance et al., 2004). The importance of increased NgR in CNS injury remains to be established, although preserved or increased NgR expression in ipsilateral white matter tracts combined with increased Nogo-A levels may cause further inhibition of axonal regeneration or sprouting following CNS injury (Grandpre et al., 2002; Li and Strittmatter, 2003).
The success of the regenerative attempts by the injured CNS also appears dependent on the capacity of injured neurons to express intrinsic molecular machinery required for neurite elongation, generally thought to be inhibited in mature intact CNS neurons (Smith and Skene, 1997; Bouslama-Oueghlani et al., 2003). Adult neuronal populations upregulate regeneration-associated genes (RAGs) after axotomy (Tetzlaff et al., 1991; Schaden et al., 1994; Bonilla et al., 2002; Tanabe et al., 2003; Fischer et al., 2004), and axonal sprouting following CNS injury, including TBI, may be accompanied by reinduction of genes ordinarily associated with developmental axonal growth (Emery et al., 2000; Abankwa et al., 2002; Emery et al., 2003; Mason et al., 2003). Over-expression of SPRRR1A can promote axonal outgrowth in vitro (Bonilla et al., 2002), and following FP brain injury, we observed an increased expression of SPRR1A(+)/NeuN (+) neurons in injured cortex and, to lesser extent, CA3 of the hippocampus although frequent SPRR1A (+)/MAP-2 (−) cells were also evident. MAP-2 expression has been shown to decrease in damaged brain areas early following FP brain injury (Hicks et al., 1995, 1997; Saatman et al., 1998), although recent reports suggest that the loss of MAP2 is transient and may not occur only in dying cells following mild TBI (Folkerts et al., 1998; Huh et al., 2003). In our study, SPRR1A-positive neurons showed preserved morphology with intact processes, suggesting that SPRR1A is not increased solely in dying neurons. Pharmacological inhibition of NgR following SCI induced numerous SPRR1A-positive axons, suggesting that NgR signaling may limit axonal outgrowth both by inhibitory effects on axon growth and also by suppression of genes, including SPRR1A, possibly linked to successful axonal regeneration (Li and Strittmatter, 2003). In the present report, we observed only a limited number of cortical SPRR1A-positive cells following FP brain injury, suggesting that brain injury induced increased but limited expression of important RAGs, likely insufficient to overcome potent increases of the expression of Nogo-A and the presence of a glial scar (Hill et al., 1996; Silver and Miller, 2004). To our knowledge, this study is the first to detect SPRR1A in both the normal and injured adult brain.
The present report documents that the expression of Nogo-A, but not NgR, is selectively increased following FP brain injury in the rat. In addition, SPRR1A, a novel promotor of axonal outgrowth, was increased in the ipsilateral cortex and CA3 of the hippocampus of FP brain-injured animals, suggesting that factors capable of both inhibiting and promoting axonal outgrowth are upregulated in TBI. These events may have important implications for the role of Nogo-A and SPRR1A as treatment targets in TBI.
Acknowledgments
Supported by NIH grants NS RO1-40978 (TKM), NS P50-08803 (TKM) and MH T32-17168 (CTF), and a Merit Review Grant from the Veterans Administration (TKM). Niklas Marklund is partly supported by a grant from the Swedish Brain Foundation. We thank Diego Morales, Rachel Hoover, Kristie Soltesz, Carrie Keck, Hilaire Thompson and David LeBold for excellent technical support, Dr. Kathryn Saatman for constructive criticism to the manuscript and Mrs. Jeanne Marks for editing of the manuscript.
References
- Abankwa D, Kury P, Muller HW. Dynamic changes in gene expression profiles following axotomy of projection fibres in the mammalian CNS. Mol. Cell. Neurosci. 2002;21:421–435. doi: 10.1006/mcne.2002.1183. [DOI] [PubMed] [Google Scholar]
- Acevado L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC. A new role for Nogo as a regulator of vascular remodeling. Nat. Med. 2004;10(4):382–388. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McClellan DR. Diffuse axonal injury in head injury, definition, diagnosis, and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Anderson PN, Campbell G, Zhang Y, Lieberman AR. Cellular and molecular correlates of the regeneration of adult mammalian CNS axons into peripheral nerve grafts. Prog. Brain Res. 1998;117:211–232. doi: 10.1016/s0079-6123(08)64018-2. [DOI] [PubMed] [Google Scholar]
- Benfey M, Aguayo AJ. Extensive elongation of axons from rat brain into peripheral nerve grafts. Nature. 1982;296:150–152. doi: 10.1038/296150a0. [DOI] [PubMed] [Google Scholar]
- Bonilla IE, Tanabe K, Strittmatter SM. Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J. Neurosci. 2002;22:1303–1315. doi: 10.1523/JNEUROSCI.22-04-01303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouslama-Oueghlani L, Wehrle R, Sotelo C, Dusart I. The developmental loss of the ability of Purkinje cells to regenerate their axons occurs in the absence of myelin, an in vitro model to prevent myelination. J. Neurosci. 2003;23:8318–8329. doi: 10.1523/JNEUROSCI.23-23-08318.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Kraydieh S, Green EJ, Dietrich WD. Temporal and regional patterns of axonal damage following traumatic brain injury, a beta-amyloid precursor protein immunocytochemical study in rats. J. Neuro pathol. Exp. Neurol. 1997;56:1132–1141. doi: 10.1097/00005072-199710000-00007. [DOI] [PubMed] [Google Scholar]
- Cajal SR. In: Degeneration and Regeneration of the Nervous System. May RM, editor. Oxford Univ. Press; London: 1928. [Google Scholar]
- Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85–96. doi: 10.1016/0896-6273(88)90212-7. [DOI] [PubMed] [Google Scholar]
- Caroni P, Savio T, Schwab ME. Central nervous system regeneration, oligodendrocytes and myelin as non-permissive substrates for neurite growth. Prog. Brain Res. 1988;78:363–370. doi: 10.1016/s0079-6123(08)60305-2. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Negra M, Levine A, Ughrin Y, Levine JM. Oligodendrocyte precursor cells, reactive cells that inhibit axon growth and regeneration. J. Neurocytol. 2002a;31:481–495. doi: 10.1023/a:1025791614468. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Ughrin Y, Levine JM. Inhibition of axon growth by oligodendrocyte precursor cells. Mol. Cell. Neurosci. 2002b;20:125–139. doi: 10.1006/mcne.2002.1102. [DOI] [PubMed] [Google Scholar]
- Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp. Neurol. 2003;182:87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- Chong MS, Woolf CJ, Haque NS, Anderson PN. Axonal regeneration from injured dorsal roots into the spinal cord of adult rats. J. Comp. Neurol. 1999;410:42–54. [PubMed] [Google Scholar]
- Christman CW, Salvant JB, Walker SA, Povlishock JT. Characterization of a prolonged regenerative attempt by diffusely injured axons following traumatic brain injury in the adult cat, a light and electron microscopic immunocytochemical study. Acta Neuropathol. (Berl.) 1997;94:329–337. doi: 10.1007/s004010050715. [DOI] [PubMed] [Google Scholar]
- Consensus conference Rehabilitation of persons with traumatic brain injury. NIH Consensus Development Panel on Rehabilitation of Persons With Traumatic Brain Injury. JAMA. 1999;282:974–983. [PubMed] [Google Scholar]
- David S, Aguayo AJ. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- David S, Lacroix S. Molecular approaches to spinal cord repair. Annu. Rev. Neurosci. 2003;26:411–440. doi: 10.1146/annurev.neuro.26.043002.094946. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP. Oligodendrocytes and ischemic brain injury. J. Cereb. Blood Flow Metab. 2003;23:263–274. doi: 10.1097/01.WCB.0000053472.41007.F9. [DOI] [PubMed] [Google Scholar]
- Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin M. Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron. 2002;35:283–290. doi: 10.1016/s0896-6273(02)00770-5. [DOI] [PubMed] [Google Scholar]
- Emery DL, Raghupathi R, Saatman KE, Fischer I, Grady MS, McIntosh TK. Bilateral growth-related protein expression suggests a transient increase in regenerative potential following brain trauma. J. Comp. Neurol. 2000;424:521–531. [PubMed] [Google Scholar]
- Emery DL, Royo NC, Fischer I, Saatman KE, McIntosh TK. Plasticity following injury to the adult central nervous system, is recapulation of a developmental state worth promoting? J. Neurotrauma. 2003;20:1271–1292. doi: 10.1089/089771503322686085. [DOI] [PubMed] [Google Scholar]
- Fischer D, He Z, Benowitz LI. Counteracting the Nogo receptor enhances optic nerve regeneration if retinal ganglion cells are in an active growth state. J. Neurosci. 2004;24:1646–1651. doi: 10.1523/JNEUROSCI.5119-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd CL, Golden KM, Black RT, Hamm RJ, Lyeth BG. Craniectomy position affects Morris water maze performance and hippocampal cell loss after parasagittal fluid percussion. J. Neurotrauma. 2002;19:303–316. doi: 10.1089/089771502753594873. [DOI] [PubMed] [Google Scholar]
- Folkerts MM, Berman RF, Muizelaar JP, Rafols JA. Disruption of MAP-2 immunostaining in rat hippocampus after traumatic brain injury. J. Neurotrauma. 1998;15:349–363. doi: 10.1089/neu.1998.15.349. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Grandpre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Friedman B, Hockfield S, Black JA, Woodruff KA, Waxman SG. In situ demonstration of mature oligodendrocytes and their processes, an immunocytochemical study with a new monoclonal antibody, Rip. Glia. 1989;2:380–390. doi: 10.1002/glia.440020510. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Barres BA. Nogo in nerve regeneration. Nature. 2000;403:369–370. doi: 10.1038/35000309. [DOI] [PubMed] [Google Scholar]
- Grados-Munro EM, Fournier AE. Myelin-associated inhibitors of axon regeneration. J. Neurosci. Res. 2003;74:479–485. doi: 10.1002/jnr.10803. [DOI] [PubMed] [Google Scholar]
- Graham DI, Raghupathi R, Saatman KE, Meaney DF, McIntosh TK. Tissue tears in the white matter after lateral fluid percussion brain injury in the rat, relevance to human brain injury. Acta Neuropathol. 2000;99:117–124. doi: 10.1007/pl00007414. [DOI] [PubMed] [Google Scholar]
- Grandpre T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–444. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- Grandpre T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat. Med. 2004;10(9):959–965. doi: 10.1038/nm1088. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Smith DH, Lowenstein DH, Saint Marie RL, McIntosh TK. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J. Neurotrauma. 1993;10:405–414. doi: 10.1089/neu.1993.10.405. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Smith DH, McIntosh TK. Temporal response and effects of excitatory amino acid antagonism on microtubule-associated protein 2 immunoreactivity following experimental brain injury in rats. Brain Res. 1995;678:151–160. doi: 10.1016/0006-8993(95)00179-t. [DOI] [PubMed] [Google Scholar]
- Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. (Berl.) 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Baldwin SA, Scheff SW. Serum extravasation and cytoskeletal alterations following traumatic brain injury in rats. Mol. Chem. Neuropathol. 1997;32:1–16. doi: 10.1007/BF02815164. [DOI] [PubMed] [Google Scholar]
- Hill SJ, Barbarese E, McIntosh TK. Regional heterogeneity in the response of astrocytes following traumatic brain injury in the adult rat. J. Neuropathol. Exp. Neurol. 1996;55:1221–1229. doi: 10.1097/00005072-199612000-00005. [DOI] [PubMed] [Google Scholar]
- Huang DW, McKerracher L, Braun PE, David S. A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron. 1999;24:639–647. doi: 10.1016/s0896-6273(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brosamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J. Neurosci. 2002;22:3553–3567. doi: 10.1523/JNEUROSCI.22-09-03553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JW, Raghupathi R, Laurer HL, Helfaer MA, Saatman KE. Transient loss of microtubule-associated protein 2 immunore-activity after moderate brain injury in mice. J. Neurotrauma. 2003;20:975–984. doi: 10.1089/089771503770195821. [DOI] [PubMed] [Google Scholar]
- Hunt D, Coffin RS, Anderson PN. The Nogo receptor, its ligands and axonal regeneration in the spinal cord; a review. J. Neurocytol. 2002a;31:93–120. doi: 10.1023/a:1023941421781. [DOI] [PubMed] [Google Scholar]
- Hunt D, Mason MR, Campbell G, Coffin R, Anderson PN. Nogo receptor mRNA expression in intact and regenerating CNS neurons. Mol. Cell. Neurosci. 2002b;20:537–552. doi: 10.1006/mcne.2002.1153. [DOI] [PubMed] [Google Scholar]
- Hunt D, Coffin RS, Prinjha RK, Campbell G, Anderson PN. Nogo-A expression in the intact and injured nervous system. Mol. Cell. Neurosci. 2003;24:1083–1102. doi: 10.1016/j.mcn.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Jin WL, Liu YY, Liu HL, Yang H, Wang Y, Jiao XY, Ju G. Intraneuronal localization of Nogo-A in the rat. J. Comp. Neurol. 2003;458:1–10. doi: 10.1002/cne.10547. [DOI] [PubMed] [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Pradat PF, Dupuis L, Echaniz-Laguna A, Muller A, Dubourg O, Seilhean D, Hauw JJ, Loeffler JP, Meininger V. Nogo expression in muscle correlates with amyotrophic lateral sclerosis severity. Ann. Neurol. 2005;57(4):553–556. doi: 10.1002/ana.20420. [DOI] [PubMed] [Google Scholar]
- Josephson A, Widenfalk J, Widmer HW, Olson L, Spenger C. NOGO mRNA expression in adult and fetal human and rat nervous tissue and in weight drop injury. Exp. Neurol. 2001;169:319–328. doi: 10.1006/exnr.2001.7659. [DOI] [PubMed] [Google Scholar]
- Josephson A, Trifunovski A, Scheele C, Widenfalk J, Wahlestedt C, Brene S, Olson L, Spenger C. Activity-induced and developmental downregulation of the Nogo receptor. Cell Tissue Res. 2003;311:333–342. doi: 10.1007/s00441-002-0695-8. [DOI] [PubMed] [Google Scholar]
- Karnezis T, Mandemakers W, McQualter JL, Zheng B, Ho PP, Jordan KA, Murray BM, Barres B, Tessier-Lavigne M, Bernard CC. The neurite outgrowth inhibitor Nogo A is involved in autoimmune-mediated demyelination. Nat. Neurosci. 2004;7(7):736–744. doi: 10.1038/nn1261. [DOI] [PubMed] [Google Scholar]
- Kartasova T, van de Putte P. Isolation, characterization, and UV-stimulated expression of two families of genes encoding polypeptides of related structure in human epidermal keratinocytes. Mol. Cell. Biol. 1988;8:2195–2203. doi: 10.1128/mcb.8.5.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPre T, Qiu D, Strittmatter SM. Axon regeneration in young adult mice lacking Nogo-A/B. Neuron. 2003;38(2):187–199. doi: 10.1016/s0896-6273(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44(3):439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Lee DH, Strittmatter SM, Sah DW. Targeting the Nogo receptor to treat central nervous system injuries. Nat. Rev., Drug Discov. 2003;2:872–878. doi: 10.1038/nrd1228. [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Shimizu S, Marklund N, Thompson HJ, Schwab ME, Saatman KE, Hoover RC, Bareyre FM, Motta M, Luginbuhl A, Pape R, Clouse AK, Morganti-Kossmann C, McIntosh TK. Delayed inhibition of Nogo-A does not alter injury-induced axonal sprouting but enhances recovery of cognitive function following experimental traumatic brain injury in rats. Neuroscience. 2005;134:1047–1056. doi: 10.1016/j.neuroscience.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Levin HS. Neurobehavioral outcome of closed head injury, implications for clinical trials. J. Neurotrauma. 1995;12:601–610. doi: 10.1089/neu.1995.12.601. [DOI] [PubMed] [Google Scholar]
- Levin HS. Cognitive function outcomes after traumatic brain injury. Curr. Opin. Neurol. 1998;11(6):643–646. doi: 10.1097/00019052-199812000-00006. Review. [DOI] [PubMed] [Google Scholar]
- Li S, Strittmatter SM. Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J. Neurosci. 2003;23:4219–4227. doi: 10.1523/JNEUROSCI.23-10-04219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BP, Fournier A, Grandpre T, Strittmatter SM. Myelinassociated glycoprotein as a functional ligand for the Nogo-66 receptor. Science. 2002a;297:1190–1193. doi: 10.1126/science.1073031. [DOI] [PubMed] [Google Scholar]
- Liu H, Ng CE, Tang BL. Nogo-A expression in mouse central nervous system neurons. Neurosci. Lett. 2002b;328:257–260. doi: 10.1016/s0304-3940(02)00528-1. [DOI] [PubMed] [Google Scholar]
- Liu YY, Jin WL, Liu HL, Ju G. Electron microscopic localization of Nogo-A at the postsynaptic active zone of the rat. Neurosci. Lett. 2003;346:153–156. doi: 10.1016/s0304-3940(03)00508-1. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury, a potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwin SK. Proliferation of mature oligodendrocytes after trauma to the central nervous system. Nature. 1984;308:274–275. doi: 10.1038/308274a0. [DOI] [PubMed] [Google Scholar]
- Marklund N, Fulp CT, Bareyre FM, Royo NC, Schwab ME, Puri R, Hoover RC, Soltesz K, McMillan A, Mir AK, Spangler Z, Millard M, Keck C, LeBold D, McIntosh TK. The Nogo-A neutralizing antibody 7B12 improves motor and cognitive functional outcome following traumatic brain injury in rats. Society for Neuroscience; San Diego, CA, USA: 2004. Abstract No 230.17. [Google Scholar]
- Mason MR, Lieberman AR, Anderson PN. Corticospinal neurons up-regulate a range of growth-associated genes following intracortical, but not spinal, axotomy. Eur. J. Neurosci. 2003;18:789–802. doi: 10.1046/j.1460-9568.2003.02809.x. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Povlishock JT, Graham DI. A mechanistic analysis of nondisruptive axonal injury, a review. J. Neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Faden AI. Traumatic brain injury in the rat, characterization of a lateral fluid percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Wei P, Stokes BT. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J. Neurosci. 2001;21:3392–3400. doi: 10.1523/JNEUROSCI.21-10-03392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Brauer AU, Heimrich B, Schwab ME, Nitsch R, Savaskan NE. Molecular analysis of Nogo expression in the hippocampus during development and following lesion and seizure. FASEB J. 2003;17:1153–1155. doi: 10.1096/fj.02-0453fje. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB. LINGO-1 is a component of the Nogo-66/p75 signaling complex. Nat. Neurosci. 2004;17(3):221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Mingorance A, Fontana X, Sole M, Burgaya F, Urena JM, Teng FY, Tang BL, Hunt D, Anderson PN, Bethea JR, Schwab ME, Soriano E, Del Rio JA. Regulation of Nogo and Nogo receptor during the development of the entorhino-hippocampal pathway and after adult hippocampal lesions. Mol. Cell. Neurosci. 2004;26:34–49. doi: 10.1016/j.mcn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- National Research Council . Guide for the Care and Use of Laboratory Animals. National Academy Press; Washington, DC: 1996. pp. 1–118. [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Pender MP. Survival and mitosis of myelinating oligodendrocytes in experimental autoimmune encephalomyelitis, an immunocytochemical study with Rip antibody. Acta Neuropathol. (Berl.) 1999;98:39–47. doi: 10.1007/s004010051049. [DOI] [PubMed] [Google Scholar]
- Oertle T, Schwab ME. Nogo and its paRTNers. Trends Cell Biol. 2003;13:187–194. doi: 10.1016/s0962-8924(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L, Brosamle C, Kaupmann K, Vallon R. Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J. Neurosci. 2003;23:5393–5406. doi: 10.1523/JNEUROSCI.23-13-05393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, Garcia KC, He Z. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45(5):345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Pierce JES, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and amyloid b peptide following experimental brain injury in the rat. J. Neurosci. 1996;16(3):1083–1090. doi: 10.1523/JNEUROSCI.16-03-01083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JES, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral, and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Ross DT, Graham DI, Adams JH. Selective loss of neurons from the thalamic reticular nucleus following severe human head injury. J. Neurotrauma. 1993;10:151–165. doi: 10.1089/neu.1993.10.151. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, Sanderson KL, Voddi M, McIntosh TK. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp. Neurol. 1997;147:418–427. doi: 10.1006/exnr.1997.6629. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Graham DI, McIntosh TK. The neuronal cytoskeleton is at risk after mild and moderate brain injury. J. Neurotrauma. 1998;15:1047–1058. doi: 10.1089/neu.1998.15.1047. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bareyre FM, Grady MS, McIntosh TK. Acute cytoskeletal alterations and cell death induced by experimental brain injury are attenuated by magnesium treatment and exacerbated by magnesium deficiency. J. Neuropathol. Exp. Neurol. 2001;60(2):183–194. doi: 10.1093/jnen/60.2.183. [DOI] [PubMed] [Google Scholar]
- Sandvig A, Berry M, Barrett LB, Butt A, Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. 2005;46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- Sato M, Chang E, Igarashi T, Noble LJ. Neuronal injury and loss after traumatic brain injury, time course and regional variability. Brain Res. 2001;917:45–54. doi: 10.1016/s0006-8993(01)02905-5. [DOI] [PubMed] [Google Scholar]
- Schaden H, Stuermer CA, Bahr M. GAP-43 immunoreactivity and axon regeneration in retinal ganglion cells of the rat. J. Neurobiol. 1994;25:1570–1578. doi: 10.1002/neu.480251209. [DOI] [PubMed] [Google Scholar]
- Schwab ME. Nogo and axon regeneration. Curr. Opin. Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Thoenen H. Dissociated neurons regenerate into sciatic but not optic nerve explants in culture irrespective of neurotrophic factors. J. Neurosci. 1985;5:2415–2423. doi: 10.1523/JNEUROSCI.05-09-02415.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, Murray B, Jung V, Pepinsky RB, Mi S. TAY/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45(3):353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Shiozaki T, Akai H, Taneda M, Hayakata T, Aoki M, Oda J, Tanaka H, Hiraide A, Shimazu T, Sugimoto H. Delayed hemispheric neuronal loss in severely head-injured patients. J. Neurotrauma. 2001;18:665–674. doi: 10.1089/089771501750357618. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat. Rev., Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME. Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron. 2003;38(2):201–211. doi: 10.1016/s0896-6273(03)00226-5. [DOI] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Chen X-H, Pierce JES, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK. Progressive atrophy and neuronal death for one year following brain trauma in the rat. J. Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- Soares HD, Thomas M, Cloherty K, McIntosh TK. Development of prolonged focal cerebral edema and regional cation change following experimental brain injury in the rat. J. Neurochem. 1992;58:1845–1852. doi: 10.1111/j.1471-4159.1992.tb10061.x. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Bonilla I, Winkles JA, Strittmatter SM. Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J. Neurosci. 2003;23:9675–9686. doi: 10.1523/JNEUROSCI.23-29-09675.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Qiu J, Nikulina E, Filbin MT. Soluble myelin-associated glycoprotein released from damaged white matter inhibits axonal regeneration. Mol. Cell. Neurosci. 2001;18:259–269. doi: 10.1006/mcne.2001.1020. [DOI] [PubMed] [Google Scholar]
- Teng FY, Ling BM, Tang BL. Inter- and intracellular interactions of Nogo: new findings and hypothesis. J. Neurochem. 2004;89(4):801–806. doi: 10.1111/j.1471-4159.2004.02366.x. [DOI] [PubMed] [Google Scholar]
- Tetzlaff W, Alexander SW, Miller FD, Bisby MA. Response of facial and rubrospinal neurons to axotomy, changes in mRNA expression for cytoskeletal proteins and GAP-43. J. Neurosci. 1991;11:2528–2544. doi: 10.1523/JNEUROSCI.11-08-02528.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozaki H, Kawasaki T, Takagi Y, Hirata T. Expression of Nogo protein by growing axons in the developing nervous system. Brain Res. Mol. Brain Res. 2002;104:111–119. doi: 10.1016/s0169-328x(02)00172-9. [DOI] [PubMed] [Google Scholar]
- van de Velde HJ, Roebroek AJ, Senden NH, Ramaekers FC, Van de Ven WJ. NSP-encoded reticulons, neuroendocrine proteins of a novel gene family associated with membranes of the endoplasmic reticulum. J. Cell Sci. 1994;107(Pt 9):2403–2416. doi: 10.1242/jcs.107.9.2403. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002a;417:941–944. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J. Neurosci. 2002b;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennstrom M, Hellsten J, Tingstrom A. Electroconvulsive seizures induce proliferation of NG2-expressing glial cells in adult rat amygdala. Biol. Psychiatry. 2004;55:464–471. doi: 10.1016/j.biopsych.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Williams S, Raghupathi R, MacKinnon MA, McIntosh TK, Saatman KE, Graham DI. In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathol. 2001;102:581–590. doi: 10.1007/s004010100410. [DOI] [PubMed] [Google Scholar]
- Zaidi AU, Bessert DA, Ong JE, Xu H, Barks JD, Silverstein FS, Skoff RP. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–390. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M. Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron. 2003;38(2):213–224. doi: 10.1016/s0896-6273(03)00225-3. [DOI] [PubMed] [Google Scholar]
- Zheng B, Atwal J, Ho C, Case L, He XL, Garcia KC, Steward O, Tessier-Lavigne M. Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc. Natl. Acad. Sci. U. S. A. 2005;102(4):1205–1210. doi: 10.1073/pnas.0409026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Li Y, Nanda A, Zhang JH. HBO suppresses Nogo-A, Ng-R, or RhoA expression in the cerebral cortex after global ischemia. Biochem. Biophys. Res. Commun. 2003;309:368–376. doi: 10.1016/j.bbrc.2003.08.006. [DOI] [PubMed] [Google Scholar]