

Abstract
A high-throughput 96-well plate-based method for the rapid induction of endogenous prophages from individual bacterial strains was developed. The detection of endogenous prophages was achieved by the filtration of the culture liquor following norfloxacin induction and subsequent PCRs targeting bacteriophage-carried gene markers. The induction method was tested on 188 putative Shiga toxin (Stx)-producing Escherichia coli (STEC) strains and demonstrated the ability to detect both lambdoid and stx-carrying bacteriophages in strains for which plaques were not observed via plaque assay. Lambdoid bacteriophages were detected in 37% of the induced phage preparations via amplification of the Q gene, and Stx1- and Stx2-encoding phages were detected in 2 and 14% of the strains, respectively. The method therefore provided greater sensitivity for the detection of Stx and other lambdoid bacteriophage populations carried by STEC strains than that for the established method of plaque assay using bacterial indicator strains, enabling, for the first time, large-scale bacteriophage population and diversity studies.
The year 1982 marked the first incident in which an unusual serogroup of Escherichia coli, O157:H7, was identified as the causative agent of an outbreak of a form of food poisoning that resulted in fatalities due to Shiga toxin-linked complications (7, 9, 11). The gene encoding Shiga toxin (stx) had been acquired by Escherichia coli O157:H7 following infection with a bacteriophage carrying the stx genes (Stx phage) (8). In less than 20 years, Shiga toxigenic potential has spread across more than 500 serogroups of E. coli as well as other members of the Enterobacteriaceae, and even to a strain of Acinetobacter haemolyticus (1). Additionally, it has been shown that certain pathogenic members of the Enterobacteriaceae can support the adsorption of Stx phages to their cell surface (e.g., Salmonella enteritidis serovars Cholerasuis, Typhi, and Typhimurium) (17). Stx phages are a heterogeneous group of lambdoid bacteriophage mosaics that need only share the characteristic of carrying the stx operon (1). Attempts to define the epidemiological role of Stx phages in outbreaks of Stx-mediated disease has been hampered by the difficulties associated with the isolation and purification of these phages, and the fact that they must be routinely handled under biological safety level 3 conditions in the United Kingdom and a number of other countries.
Traditional phage propagation methods rely upon the infection of a bacterial indicator strain with a suspension of bacteriophages, and plaque assays have been used previously to characterize Stx phages induced from environmental STEC strains (18). However, plaque assay-based methods fail to detect all phages in a given environment (5, 14), primarily because bacterial indicator strains cannot support infection by all bacteriophage types (14). In addition, phage infection may result in lysogenic conversion without phage replication, and therefore no plaques are observed. The latter has been described in two studies of Stx phages in which all phage infections resulted in the production of lysogens (3, 16), and indeed lambdoid Stx phages have been described as exhibiting a preference for such lysogenic infection (3). Phage propagation techniques are time-consuming and laborious, making the analysis of hundreds/thousands of bacterial strains unfeasible, particularly in countries where Stx phages must be handled under biological safety level 3 conditions. As bacteriophages are known to control the population structure of bacteria, affect the fitness/virulence of their host, and effect horizontal gene transfer (1), the lack of the capability to study large phage populations hinders our understanding of the impact these important biological entities have on the biosphere. The application of molecular biological techniques, such as PCR and quantitative PCR (qPCR), to phage-carried genes represents an alternative to traditional phage propagation techniques and has demonstrated the ability to increase the sensitivity of the detection of lambdoid phages and the subset known as Stx phages (14). Here, we describe the development of a new and rapid bacteriophage assay method that combines the norfloxacin induction of phages from their bacterial lysogens (2, 3, 6) with the PCR detection of target phage groups (Stx and lambdoid). Inductions, which can be tailored to any target groups of phages, are performed in 96-well microtiter plates, and induced phage preparations were stored in 96-well PCR plates for further characterization via PCR screening or phage propagation.
To validate this method, presumptive E. coli strains, which were used as a source for bacteriophages, were isolated from water samples collected between July and August 2008 from several water courses on a beef cattle farm (Cheshire, United Kingdom) known to routinely harbor STEC strains (12-14, 17, 18). These water samples were obtained from three different freshwater ponds located in pasture land and one level-controlled trough fed with potable water, all of which were used by cattle for drinking. Water samples (250 ml) were centrifuged at 6,000 × g for 5 min to concentrate cells and particulate matter, and the subsequent pellet was used to inoculate 10-ml aliquots of sterile MacConkey broth. Following overnight incubation, a serial dilution of each culture was streaked onto eosin methylene blue (EMB) agar and incubated overnight. Colonies with dark coloration and possessing a green metallic sheen on EMB agar (presumptive E. coli) were picked and subsequently patched onto fresh EMB plates.
Colony PCR subsequently was performed on environmental isolates using a degenerate stx gene-targeted primer set (StxA and StxB) (Table 1) and Phusion High-Fidelity DNA polymerase (Finnzymes), as previously described (18), to detect isolates carrying an stx operon. Stx-positive (stx+) bacterial isolates identified by PCR subsequently were cultured overnight in phage buffer (PB; sterile Luria broth supplemented with 10 mM CaCl2) and stored at −80°C in 25% (vol/vol) glycerol.
TABLE 1.
PCR primer sets used in this study
| Primer | Sequencea (5′ to 3′) | Gene target | Annealing temp (°C) | Amplicon size (bp) | Reference or source |
|---|---|---|---|---|---|
| Degen StxA | TTTGTYACYGTSAYAGCWGAAG | stx | 47.5 | ∼675 | 18 |
| Degen StxB | TYMTCATTATAYTTDGWRWACT | ||||
| Slt1-F | CGCTGTTGTACCTGGAAAGG | stx1 | 50.0 | ∼254 | 10 |
| Slt1-R | CGCTCTGCTGTGACAGTGAC | ||||
| Slt2-F | GCTTCTGCTGTGACAGTGAC | stx2 | 50.0 | ∼184 | 4 |
| Slt2-R | TCCATGACAACGGACAGCAG | ||||
| Q ATG 5′ | ATGTTCTTATGGTTCACCG | Lambdoid bacteriophage Q gene | 55.5 | ∼435 | 18 |
| Q 3′ | TTACGATCGTAAACTATTTTT | ||||
| GapA 3′ | CGCTCACATCTCCAAATAA | Bacterial gapA gene | 55.0 | ∼1,000 | This study |
| GapA 5′ | ATGACTATCAAAGTAGGTATC |
Ambiguities: K = G or T; S = G or C; W = A or T; Y = C or T; H = A, C, or T; R = A or G.
Each putative STEC strain was inoculated into a single well of a 96-well plate containing 200 μl PB and incubated overnight at 37°C (only 94 of the 96 wells were used in this manner; the last two wells were left empty to serve as space holders for PCR controls in subsequent steps). A schematic diagram of the methodology is shown in Fig. 1. For bacteriophage induction, 10 μl of an overnight culture of each strain was transferred to the same position in a fresh 96-well plate containing 190 μl PB per well (5% inoculum) using a multichannel pipette. Each plate was covered with a gas-permeable adhesive seal (Abgene) to prevent contamination between wells on the plate. A fresh gas-permeable seal was applied to each plate after the addition/transfer of reagents or culture. The strains were grown to mid-exponential phase (optical density at 600 nm [OD600] of 0.45 to 0.55) at 37°C and 150 rpm prior to the addition of norfloxacin (final concentration, 1 μg ml−1), followed by a further 1 h of incubation at 37°C to induce endogenous prophages.
FIG. 1.

Schematic workflow of the high-throughput 96-well plate-based method for the rapid induction of endogenous prophages from Shiga toxigenic Escherichia coli strains.
A 10-fold dilution of the culture liquor was performed by transferring 20 μl of each culture to a fresh 96-well plate containing 180 μl PB, including 5 μg ml−1 DNase I (Sigma), and the cultures were allowed to recover for 2 h at 37°C. Three identical recovery plates were produced for each plate of induced STEC isolates to provide a total volume of 600 μl of culture (three 200-μl wells) for the subsequent filtration of phages from each strain. Following recovery, a 1-ml syringe was used to remove the culture from the replicate wells for each strain (600 μl total), and the supernatant was filtered through a sterile 0.2-μm-pore-diameter filter into the appropriate well of a fresh 96-well PCR plate. Due to the retention of some culture supernatant in the filter, it was possible to retrieve only ca. 200 μl of filtered phage suspension for each strain, thus the requirement to perform the recovery plate step in triplicate. The recovered phage preparations were placed in an MJ research PTC-200 thermal cycler (Bio-Rad, Hertfordshire, United Kingdom) and incubated at 100°C for 10 min to inactivate the phages and DNase. For convenience, the samples were stored at −20°C in 96-well plate format prior to PCR analysis.
Bacterial genomes, and particularly those of E. coli strains, have been demonstrated to contain large numbers of remnant prophages (1), and as such, the removal of extraneous bacterial genomic DNA from induced phage suspensions was critical to ensure the subsequent PCR amplification of only the induced bacteriophage DNA. This was achieved by the inclusion of a DNase treatment step to destroy the genomic DNA of lysed bacterial cells, while bacteriophage DNA was protected from DNase by the viral capsid. We tested the addition of various concentrations of DNase I (Sigma) (0, 5, 10, and 15 μg ml−1) after the filtration of the culture liquor and also during the 2-h recovery incubation period. The data demonstrated that the addition of DNase I at a concentration of 5 μg ml−1 during the 2-h recovery period was sufficient to remove detectable levels of bacterial genomic DNA (Fig. 2), as determined by amplification with a primer set targeting the bacterial gapA (glyceraldehyde-3-phosphate dehydrogenase) gene (Table 1) during 35 amplification cycles. In contrast, phage suspensions obtained by flooding agar plates generated by plaque assay with PB (18) generated using the same STEC isolates produced strong amplification products for the gapA gene, demonstrating the requirement for a further DNase I step following phage diffusion from the top agar into PB (data not shown). The addition of 5 μg ml−1 DNase I directly into to the PB buffer used in the 96-well recovery plates for all inductions represents a further reduction in sample processing time, as it negates that need for a separate DNase treatment step following phage filtration. The removal of contaminating bacterial genomic DNA from phage preparations of all 188 STEC isolates used in this study was confirmed by the absence of PCR amplification with the E. coli gapA primer set (Table 1) (data not shown).
FIG. 2.
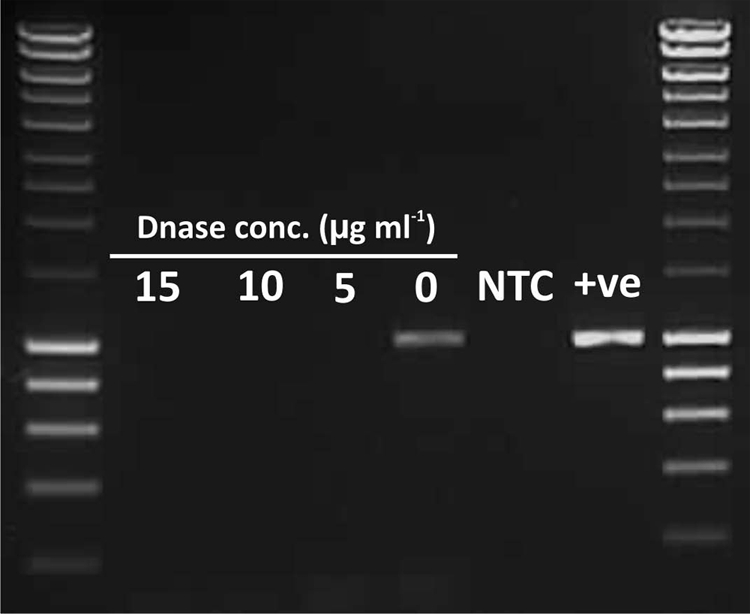
Agarose gel electrophoresis of PCR amplification products from induced Stx phage Φ24B filtrates with E. coli housekeeping gene gapA (glyceraldehyde-3-phosphate dehydrogenase) primers to determine the optimum DNase concentration for the removal of bacterial genomic DNA from the culture supernatant. NTC, no-template control; +ve, positive control (E. coli strain WG5 DNA). The molecular size marker is Hyperladder I (Bioline). The gapA gene amplicon size was ca. 1,000 bp and ran to a position similar to that of the 1,000-bp band of the molecular size marker (Hyperladder I) on the gel.
To determine whether phages induced from the putative STEC strains were capable of forming plaques (18), culture supernatant (10 μl) was spotted onto a lawn of E. coli WG5rif+ (19) indicator bacteria following the recovery step and prior to the filtration of the culture liquor. This was achieved using a Q tray low-profile square Bio assay dish (Genetix) containing Luria agar (200 ml) overlaid with 50 ml PB containing 0.4% (wt/vol) Difco agar, 300 μg ml−1 rifampin (rifampicin), and 1 ml (OD600 = 0.5) of the E. coli WG5rif+ indicator strain. All 94 inductions were spotted onto a single plate. The confluent or semiconfluent lysis of the bacterial indicator strain was observed for 70% of the 188 STEC strains (Fig. 3A). These data allowed comparisons to be drawn between the numbers of strains for which plaques were observed for each particular strain and the phage genes of interest that were amplified via PCR from filtered phages from the same induction experiment. However, the flexibility of this method to assess both the ability to infect an indicator strain and provide filtered supernatant for use in PCR studies from the same strain represents a significant benefit, making the method amenable to many applications in bacteriophage biology. A further advantage of this 96-well plate-based method compared to traditional plaque assay methods is the speed at which subsequent PCR amplifications can be performed, as phage suspensions are stored in 96-well plate format and are ready to be transferred directly into PCR plates using a multichannel pipette. This facilitates the rapid PCR typing of phage suspensions.
FIG. 3.

Plaque assay and PCR data for a total of 188 phage preparations induced from STEC strains. Numbers above the bars represent the number of strains out of the total of 188 tested in each category. (A) A comparison of the number of strains from which phages could be detected by plaque assay and a bacterial indicator strain and by the PCR amplification of the lambdoid bacteriophage-carried Q gene. (B) The proportion of STEC strains from which phages were observed via plaque assay in comparison to phage detection using Q gene and Stx1 and Stx2 gene-targeted PCR. (C) A comparison of the proportions of stx1 and stx2 gene variants detected in STEC isolates and detection of the lambdoid bacteriophage-encoded Q gene from the same strain.
Bacterial strains often contain more than one inducible bacteriophage type, and the filtered phage supernatants induced from each bacterial strain in this study therefore may represent a pool containing several different bacteriophages. Phage supernatants induced from STEC strains (2 μl) were added directly to 96-well plates containing PCR mastermix and Phusion High-Fidelity DNA polymerase (Finnzymes) and primers targeting the lambdoid Q gene and stx1 and stx2 genes (Table 1), as previously described (18). PCR amplification products of the Q, stx1, and stx2 genes were visualized using 1% agarose gel electrophoresis, and the amplification results for each strain were compared to the ability of each strain to direct the semiconfluent lysis of the bacterial indicator strain.
PCR amplification data demonstrated that of the 132 strains for which plaques were detected, the amplification of the lambdoid bacteriophage Q gene was possible for only 42 strains (Fig. 3B). Therefore, the endogenous prophages that produced plaques on the bacterial indicator strain where no Q gene amplification was identified may represent nonlambdoid phages that also can infect the indicator strain or novel lambdoid phages with a previously unidentified Q gene variant. However, Q gene amplification was observed in phage preparations induced from 28 bacterial strains (15% of total strains) for which no plaques were observed via plaque assay (Fig. 3B). Therefore, plaque assays did not detect all lambdoid phages from the bacterial phage preparations, and PCR provided a more sensitive method for their detection. These data support the use of the phage filtration and PCR-based approach described here in the rapid screening of phages, as we successfully identified more inducible lambdoid phage-containing strains than by plaque assay alone. Furthermore, of the 132 phage preparations for which plaques were observed, only 42 preparations contained lambdoid phages as detected by the PCR amplification of the Q gene. Therefore, the use of PCR to target and detect genes that are carried by phages of interest reduces the total number of samples for further characterization, i.e., only 42 of the 132 phage preparations were of interest here, and the PCR technique selected only those preparations that contained a lambdoid Q gene, whereas plaque assay could not discriminate between lambdoid and nonlambdoid phages, nor could it differentiate between lytic and temperate phages.
PCR data demonstrated that 37% of the 188 strains characterized in this study possessed inducible lambdoid phages, as detected by the amplification of the Q gene (Fig. 3A). The application of PCR primer sets targeting the stx gene demonstrated that 2% of the strains had inducible Stx1 phages and 14% carried inducible Stx2 phages (Fig. 3A). A surprising observation was the detection of phage preparations containing Stx1+ (two strains) and Stx2+ (four strains) phages for which no amplification product of the Q gene was observed (Fig. 3C). To date, all detected Stx phages are classed as lambdoid phages, and all contain the antiterminator gene, Q, for which a degenerate PCR primer set has been designed and applied as a genetic marker for lambdoid phages (14). The lack of detection of a Q gene in stx+ phage preparations therefore could be a result of unknown sequence diversity in the primer binding site of the Q gene or indicate the presence of stx genes in nonlambdoid phages. The amplification of the Q gene was observed for the majority of Stx1- and Stx2-containing phage preparations (67 and 85%, respectively) (Fig. 3C). However, it should be noted that phage preparations may contain more than one phage type, and therefore the amplification of the Q and stx genes in the same preparation does not confirm their presence in the same single-bacteriophage genome.
Using this method, it was possible for one person to induce four 96-well plates of strains (a total of 384 strains) in a single working day. This represents a significant reduction in the number of worker hours (9-fold reduction) and total number of days (20-fold reduction) involved in the induction of endogenous phages from strains described using previous methods (Table 2). Furthermore, reducing the culture volume and the use of 96-well plates significantly reduces the volume of reagents, solid and liquid culture media, and disposable plasticware required, and therefore the cost of the experiments is reduced compared to that of the current plaque assay-based methods (15).
TABLE 2.
Time required to induce endogenous prophages using traditional plaque assay methods and 96-well plate-based induction method described here
| Induction method | Protocol step timec (h) | Worker time (h) to process 384 strains (total no. of days) |
|---|---|---|
| Traditionala | Overnight culture setup (∼1) | 117e (39f) |
| Inoculate starter cultures (∼0.75) | ||
| Addition of norfloxacin (∼0.5) | ||
| Recovery setup (∼0.75) | ||
| Serial dilutions (∼1.5) | ||
| Plaque assay (∼1.5) | ||
| Diffusion step (∼1.5) | ||
| DNase treatment step (∼1.5) | ||
| 96-Well plateb | Overnight culture setup (∼4) | 12.75 (2g) |
| Inoculate starter cultures (∼0.25) | ||
| Addition of norfloxacin (∼0.25) | ||
| Recovery plate setup (∼0.25) | ||
| Culture liquor filtration (∼4 h × 2d) |
The traditional method included ten-milliliter culture volumes, norfloxacin induction, plaque assay, and diffusion of phages from top agar. For the induction of endogenous prophages from 30 bacterial strains. Due to the requirement to flood the top agar with buffer to allow phages to diffuse into the buffer for harvesting on serial dilutions of culture liquor for each strain, it is feasible to induce only 30 bacterial strains in a single experiment under containment level III conditions.
The 96-well plate format included two hundred-microliter culture volumes, norfloxacin induction, and filtered phage preparations.
Time represents the actual manual labor time and does not include incubation periods between steps.
This step can be completed more rapidly if two people are present; one to perform culture filtration and one to remove plastic wrapping from sterile syringes and sterile filters and assist with transfer of plates and samples.
The traditional protocol requires 9 worker hours to induce 30 strains. Therefore, for comparison to the induction of 384 strains using the new method described here, it would require 117 h to perform 384 inductions using this method.
Based on 3 days per 30 strains: day 1, set up overnight cultures; day 2, induction and plaque assay; day 3, phage harvesting into buffer via diffusion and DNase I treatment. Thirteen 3-day experiments is a total of 39 days.
Based on 2 days per 384 strains: day 1, set up overnight cultures; day 2, induction, DNase step, and filtration of phage culture liquor.
The reported method now makes the screening of hundreds/thousands of bacterial strains for endogenous prophages feasible, reducing the processing time by up to 20-fold compared to that of traditional plaque assay-based methods (based on the induction of 384 strains) (Table 2). The method could easily be adapted to use any induction stimulus, as some bacterial hosts will be resistant/unresponsive to fluoroquinolone drugs. Moreover, the technique exhibits increased detection sensitivity and flexibility, enabling the addition of plaque assays, phage-spotting experiments, transmission electron microscopy analyses, qPCR, and supplementary assays to the standard protocol. The methodology also provides a prescreen of the sample set for downstream processing by PCR identification of only those phages containing genes of interest, in this case Q and stx, to address the dynamics of Stx phages in the environment or in the pathogenic profiles of outbreak-associated STEC strains. This is a versatile and inexpensive technique that is suitable for application in a wide range of studies of bacteriophage biology, including the rapidly emerging field of viral metagenomics. No specialized equipment is required, and the technique therefore has global utility to enable, for the first time, rapid analyses of significant numbers of bacteriophages to provide data on diversity, population structure, and gene carriage.
Acknowledgments
This research was funded by the Department for Environment, Food, and Rural Affairs (DEFRA) and the Biotechnology and Biological Sciences Research Council (BBSRC), both of the United Kingdom.
We are grateful to the owners of the dairy farm for allowing access to the sampling sites used in this study and David J. Rooks (University of Liverpool) for assistance with sampling.
Footnotes
Published ahead of print on 5 February 2010.
REFERENCES
- 1.Allison, H. E. 2007. Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol. 2:165-174. [DOI] [PubMed] [Google Scholar]
- 2.Herold, S., J. Siebert, A. Huber, and H. Schmidt. 2005. Global expression of prophage genes in Escherichia coli O157: H7 strain EDL933 in response to norfloxacin. Antimicrob. Agents Chemother. 49:931-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James, C. E., K. N. Stanley, H. E. Allison, H. J. Flint, C. S. Stewart, R. J. Sharp, J. R. Saunders, and A. J. McCarthy. 2001. Lytic and lysogenic infection of diverse Escherichia coli and Shigella strains with a verocytotoxigenic bacteriophage. Appl. Environ. Microbiol. 67:4335-4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.La Ragione, R. M., I. M. McLaren, G. Foster, W. A. Cooley, and M. J. Woodward. 2002. Phenotypic and genotypic characterization of avian Escherichia coli O86: K61 isolates possessing a gamma-like intimin. Appl. Environ. Microbiol. 68:4932-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loœ, J. M., P. Golec, G. Wegrzyn, A. Wegrzyn, and M. Los. 2008. Simple method for plating Escherichia coli bacteriophages forming very small plaques or no plaques under standard conditions. Appl. Environ. Microbiol. 74:5113-5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsushiro, A., K. Sato, H. Miyamoto, T. Yamamura, and T. Honda. 1999. Induction of prophages of enterohemorrhagic Escherichia coli O157:H7 with norfloxacin. J. Bacteriol. 181:2257-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Brien, A. D., T. A. Lively, T. W. Chang, and S. L. Gorbach. 1983. Purification of Shigella dysenteriae 1 (Shiga)-like toxin from Escherichia coli O157:H7 strain associated with haemorrhagic colitis. Lancet ii:573. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien, A. D., J. W. Newland, S. F. Miller, R. K. Holmes, H. W. Smith, and S. B. Formal. 1984. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 226:694-696. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien, A. O., T. A. Lively, M. E. Chen, S. W. Rothman, and S. B. Formal. 1983. Escherichia coli O157:H7 strains associated with haemorrhagic colitis in the United States produce a Shigella dysenteriae 1 (SHIGA) like cytotoxin. Lancet i:702. [DOI] [PubMed] [Google Scholar]
- 10.Paton, A. W., J. C. Paton, P. N. Goldwater, M. W. Heuzenroeder, and P. A. Manning. 1993. Sequence of a variant Shiga-like toxin type I operon of Escherichia coli O111H. Gene 129:87-92. [DOI] [PubMed] [Google Scholar]
- 11.Riley, L. W., R. S. Remis, S. D. Helgerson, H. B. McGee, J. G. Wells, B. R. Davis, R. J. Hebert, E. S. Olcott, L. M. Johnson, N. T. Hargrett, P. A. Blake, and M. L. Cohen. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308:681-685. [DOI] [PubMed] [Google Scholar]
- 12.Robinson, S. E., P. E. Brown, E. John Wright, M. Bennett, C. A. Hart, and N. P. French. 2005. Heterogeneous distributions of Escherichia coli O157 within naturally infected bovine faecal pats. FEMS Microbiol. Lett. 244:291-296. [DOI] [PubMed] [Google Scholar]
- 13.Robinson, S. E., E. J. Wright, C. A. Hart, M. Bennett, and N. P. French. 2004. Intermittent and persistent shedding of Escherichia coli O157 in cohorts of naturally infected calves. J. Appl. Microbiol. 97:1045-1053. [DOI] [PubMed] [Google Scholar]
- 14.Rooks, D. J., Y. Yan, J. E. McDonald, M. J. Woodward, A. J. McCarthy, and H. E. Allison. 2009. Development and validation of a qPCR-based method for quantifying Shiga toxin-encdoing and other lambdoid phages. Environ. Microbiol. doi: 10.1111/j.1462-2920.2010.02162.x. [DOI] [PubMed]
- 15.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, vol. 1, 3rd ed., p. 2.25-2.32. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 16.Schmidt, H., M. Bielaszewska, and H. Karch. 1999. Transduction of enteric Escherichia coli isolates with a derivative of Shiga toxin 2-encoding bacteriophage phi 3538 isolated from Escherichia coli O157:H7. Appl. Environ. Microbiol. 65:3855-3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith, D. L., C. E. James, M. J. Sergeant, Y. Yaxian, J. R. Saunders, A. J. McCarthy, and H. E. Allison. 2007. Short-tailed Stx phages exploit the conserved YaeT protein to disseminate Shiga toxin genes among enterobacteria. J. Bacteriol. 189:7223-7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith, D. L., B. M. Wareing, P. C. Fogg, L. M. Riley, M. Spencer, M. J. Cox, J. R. Saunders, A. J. McCarthy, and H. E. Allison. 2007. Multilocus characterization scheme for Shiga toxin-encoding bacteriophages. Appl. Environ. Microbiol. 73:8032-8040. [DOI] [PMC free article] [PubMed] [Google Scholar]