

Abstract
Molecular characterizations of environmental microbial populations based on recovery and analysis of DNA generally assume efficient or unbiased extraction of DNA from different sample matrices and microbial groups. Appropriate controls to verify this basic assumption are rarely included. Here three different DNA extractions, performed with two commercial kits (FastDNA and UltraClean) and a standard phenol-chloroform method, and two alternative filtration methods (Sterivex and 25-mm-diameter polycarbonate filters) were evaluated, using the addition of Nitrosopumilus maritimus cells to track the recovery of DNA from marine Archaea. After the comparison, a simplified phenol-chloroform extraction method was developed and shown to be significantly superior, in terms of both the recovery and the purity of DNA, to other protocols now generally applied to environmental studies. The simplified and optimized method was used to quantify ammonia-oxidizing Archaea at different depth intervals in a fjord (Hood Canal) by quantitative PCR. The numbers of Archaea increased with depth, often constituting as much as 20% of the total bacterial community.
Efficient DNA extraction from environmental samples is fundamental to many culture-independent characterizations (10). Thus, there was an early and concerted effort to establish appropriate methods of DNA extraction from different types of environmental samples (14, 19, 25, 30, 34, 43, 47). DNA extraction efficiency is particularly important for quantitative PCR (qPCR), because poor DNA extraction efficiency results in the underestimation of gene copy numbers in the samples examined (6, 42).
Most methodological developments addressed DNA extraction from soil and sediment samples, with fewer comparative studies of the efficiency of collection and extraction from water samples (4, 13, 40). In part, a methodological focus on soils reflected the simplicity of filtration to collect aquatic populations and the generally good recovery of DNA from the Gram-negative bacteria making up a significant fraction of aquatic communities. However, small Archaea are now known to constitute a substantial fraction of the prokaryotic populations in marine and terrestrial systems (2, 7, 9, 20, 26, 31, 33, 45). Since the archaeal cell wall and membrane structures are distinct from those of bacteria, there is no assurance that commonly used extraction methods are adequate. With increasing reliance on commercially available bead-beating-type DNA extraction kits, these methods are now often used for different water samples (1, 5-7, 14, 19, 36). Although most protocols incorporate mechanical disruption to ensure more-uniform extraction than is possible by using methods that rely entirely on enzymatic digestion and/or chemical disruption (4, 13, 40), the suitability of these protocols for the concerted analysis of archaeal and bacterial populations has not been fully evaluated.
In the studies reported here, the recently isolated marine archaeon Nitrosopumilus maritimus strain SCM1 (22) was therefore used as a reference standard for evaluation of the commonly employed DNA extraction methods by using qPCR. This archaeon was then used as a reference for the development of a simple, rapid, and efficient method of extracting DNA from both archaeal and bacterial cells. The modified protocol was subsequently employed to characterize the vertical distribution of ammonia-oxidizing Archaea in a fjord (Hood Canal) in Puget Sound (Washington State), revealing a high fractional representation of Archaea relative to Bacteria not observed previously in coastal waters.
MATERIALS AND METHODS
Water sample collection and nutrient analysis.
Seawater samples were collected using a Niskin water sampler at an ocean remote chemical analyzer (ORCA) buoy in Hood Canal (Hoodsport; latitude, 47°0.43′N; longitude, 123°0.11′W; depth, 130 m) on 31 July 2008 and 28 April 2009. Vertical profiles of temperature, salinity, chlorophyll a, and dissolved oxygen were measured using the buoy-equipped sensors. The concentrations of nitrogen species (ammonia, nitrite, and nitrate) were determined as described previously (15, 16, 46). Cells for DNA extraction were collected on 0.22-μm-pore-size Sterivex cartridge filters (Millipore Corp., Bedford, MA) or on 0.22-μm-pore-size polycarbonate filters (diameter, 25 mm; GTTP; Millipore) by filtration of 50 to 500 ml seawater.
Quantification of DNA and gene copy numbers.
A modified fluorescence-based assay using SYBR green I dye (Molecular Probes, Eugene, OR) was used for DNA quantification. Environmental DNA or lambda DNA (1 μl) was added to 99 μl of Tris-EDTA (TE) buffer (pH 8.0) in each well of a 96-well flat-bottom black polystyrene microtiter plate (Corning Inc., Corning, NY). Following addition of 100 μl of SYBR green I diluted in TE buffer (5,000-fold dilution of the commercial product), the plate was incubated for 5 to 10 min at room temperature in the dark, and fluorescence was determined with a microplate reader (Tecan i-Control) using band-pass filters with an excitation wavelength of 485 nm and an emission wavelength of 520 nm. Concentrations were determined relative to a lambda DNA standard in the range of 0 to 500 ng ml−1. DNA quality was evaluated using the ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) by measuring absorptions at 260, 230, and 280 nm to estimate purity from solvents and protein contamination.
Nitrosopumilus maritimus strain SCM1, cultured as described previously (28), was used to evaluate the efficiency of cell breakage and DNA recovery. Cell breakage was evaluated microscopically following bead beating of 1 ml culture (1.1 × 107 ± 2.1 × 106 cells ml−1 [mean ± standard deviation {SD} for three replicates]) containing 0.9% sodium dodecyl sulfate (SDS), filtering onto a black polycarbonate filter, staining with Mowiol (Moviol)-SYBR green I, and counting as described previously (27). The efficiency of DNA recovery from marine Archaea was evaluated by filtering different volumes (10, 50, 100, and 200 ml) of SCM1 cultures (7.7 × 107 cells ml−1) onto Sterivex filters. Archaeal DNA recovery from seawater was evaluated by seeding two bottles containing 10 liters each of seawater collected from depths of 10 m (surface water) and 90 m (deep water) with SCM1 cells to a final concentration of 8.1 × 105 to 8.5 × 105 ml−1. Cells were immediately collected by filtration through Sterivex or polycarbonate filters and were stored at −80°C until processing.
Bacterial 16S rRNA and archaeal amoA (encoding the alpha subunit of crenarchaeal ammonia monooxygenase) gene copy numbers were determined using the LightCycler FastStart DNA Master SYBR green I kit (Roche Applied Science, Indianapolis, IN) and capillary system (LightCycler; Roche). The GM3-EUB338 primer set (22) was used to amplify 16S rRNA gene sequences in a 10-μl reaction volume containing 0.01 to 3 ng of the DNA template in the master mix (3.5 mM MgCl2 and 0.5 μM each primer). The amplification protocol was as follows: initial denaturation at 95.0°C for 5 min; 55 cycles of 95.0°C for 10 s, 55.0°C for 10 s, and 72.0°C for 20 s; and a melting curve analysis (65°C to 95°C) with a heating rate of 0.1°C/s. The standard curves were generated using a plasmid clone of the Streptococcus mutans 16S rRNA gene in a dilution series of 102 to 107 gene copies. Data were analyzed with the second derivative maximum method using LightCycler software (version 3.5.3; Roche). Archaeal amoA was quantified using the primer set consisting of CrenAmoAQ-F and CrenAmoAModR (33) with the same reaction chemistry used for the bacterial 16S rRNA gene amplification, but with the concentration of each primer adjusted to 1.0 μM. The amplification protocol was as follows: initial denaturation at 95°C for 5 min; 55 cycles of 95°C for 10 s, 54°C for 10 s, 72°C for 13 s, and a detection step at 80°C for 3 s; and a melting profile analysis (70°C to 95°C) with a heating rate of 0.1°C/s. The standard curve was generated using N. maritimus genomic DNA in a dilution series of 101 to 106 copies of the archaeal amoA gene, and data were analyzed as described for the 16S rRNA gene.
Standard DNA extraction.
The bottom of a Sterivex cartridge was broken down by compression with Vise Grip pliers; the filter was removed using sterilized tweezers and aseptically cut into two equal pieces; and each piece was inserted into a separate microcentrifuge tube. The 25-mm-diameter polycarbonate filter was folded and transferred to a microcentrifuge tube with tweezers so that the cell-coated surface faced the inside. Three standard methods were used to extract DNA from surface water samples: (i) the FastDNA SPIN kit for soil (MP Biomedicals, Solon, OH), (ii) the UltraClean soil DNA kit (MoBio Laboratories, Solana Beach, CA), and (iii) standard phenol-chloroform extraction. The commercial kits were used in accordance with the manufacturers' instructions. In order to normalize bead-beating efficiency, a FastPrep FP120 instrument (MP Biomedicals) was used at speed 6 for 40 s for all DNA extraction treatments.
For standard phenol-chloroform extraction, TE-saturated phenol at pH 8.0 (0.5 ml), 0.2 M sodium phosphate at pH 8.0 (0.5 ml), and 20% SDS (50 μl) were added to a lysing matrix E tube (MP Biomedicals) containing the collection filter. The tube was mechanically agitated, and DNA was extracted with sequential phenol (TE saturated; pH 8.0), phenol-chloroform-isoamyl alcohol (50:49:1) (pH 8.0), and chloroform-isoamyl alcohol (24:1) treatments, precipitated with ethanol using a general protocol, resuspended in 50 μl TE buffer, and stored at −20°C until further analysis.
Modified DNA extraction.
Two different coastal water samples were used to develop a modified phenol-chloroform extraction method, briefly described here and elaborated as a protocol in file S2 in the supplemental material. One piece of filter was placed in the bead-beating tube (lysing matrix E tube; MP Biomedicals) as described above, followed by addition of 0.35 ml phenol-chloroform-isoamyl alcohol (50:49:1) (TE saturated; pH 8.0) and 0.35 ml of 2× TENS buffer (100 mM Tris-HCl [pH 8.0], 40 mM EDTA, 200 mM NaCl, 2% SDS) (23). After bead beating, the tube was centrifuged at 16,000 × g for 5 min; the aqueous phase was transferred to a 2.0-ml Phase Lock Gel tube (Eppendorf, Westbury, NY); 0.3 ml of 7.5 M ammonium acetate was added; and the contents were mixed by repeated inversion. An equal volume of chloroform was then added; the contents were again mixed thoroughly by repeated inversion; and the tube was centrifuged at 16,000 × g for 5 min. The supernatant was transferred to the sample reservoir of a spin column (Microcon YM-50; Millipore), twice dialyzed against TE according to the manufacturer's instructions to a final volume of 50 μl, and stored at −20°C. For the qPCR assay, 10-fold or 100-fold-diluted samples in MilliQ water were used.
As an alternative to the spin column method, DNA was recovered following chloroform extraction by alcohol precipitation using 0.6 volume (0.36 ml) of ice-cold isopropyl alcohol with the addition of 3 μl of GlycoBlue (Ambion, Austin, TX) by standard protocols. DNA was then resuspended in 50 μl TE buffer. DNA recovery for each method (precipitation with and without GlycoBlue versus spin column recovery) was evaluated using salmon sperm DNA (Ambion, Austin, TX) at final concentrations of 40, 100, 200, and 600 ng μl−1.
Three methods were used to calculate the gene copy number. In the first method, the Nvol method, the gene copy number was normalized per milliliter of filtered seawater. The gene copy number per milliliter was calculated using equation 1:
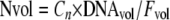 |
(1) |
where Nvol is the volume-based number, Fvol is the volume of filtered seawater (in milliliters), DNAvol is the volume of buffer in which the extracted DNA was dissolved (in microliters), and Cn is the copy number per microliter of DNA used for a single assay.
In the second method (the weight-based method), the gene copy number was normalized per nanogram of DNA using equation 2:
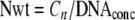 |
(2) |
where Nwt is the weight-based number and DNAconc is the DNA concentration (ng μl−1) used for a single assay.
In the third calculation, the gene copy number was estimated by the weight-volume-based (Nwt vol) method, which uses cell counts to estimate the total amount of DNA in a filtered sample based on the average weight of DNA in a single cell (equation 3). Seawater samples (2 ml) were fixed with 8% paraformaldehyde (final concentration, 0.8%), and cells were microscopically quantified as described previously (27).
 |
(3) |
where TCcount is the total cell count (expressed as cells per milliliter) and DNAsingle is the estimated weight of DNA per single cell (39).
RESULTS AND DISCUSSION
Method optimization.
Two filtration methods, followed by three different DNA extraction protocols, were evaluated by using surface water samples. There was no significant difference in DNA yields between the two filtration methods (Fig. 1). Thus, the selection of either filter type should be dictated primarily by the volume requirement (21). Bead beating was shown to be effective for disruption of N. maritimus cells, which was comparable to that observed for other cell types (4, 13, 41, 45), reducing cell numbers by 99.6% (4.0 × 104 ± 3.7 × 104 cells ml−1 [mean ± SD for three replicates]) versus 60.0% by use of SDS alone (9.6 × 106 ± 3.9 × 106 cells ml−1). However, it should be noted that 104 archaeal cells per milliliter still remained after the bead-beating treatment.
FIG. 1.

Effects of filtration and DNA extraction method on DNA yield. Data are means ± SDs for 6 to 12 replicates.
There were significant differences between the UV spectra of DNAs recovered by the different extraction methods (see Fig. S1 in the supplemental material). DNA isolated using the phenol-chloroform extraction method had a maximum absorbance between 260 and 275 nm and A260/280 and A260/230 ratios of 2.4 ± 1.0 (mean ± SD for 18 measurements) and 0.5 ± 0.2, respectively, indicating that the standard phenol-chloroform extraction method effectively eliminates UV radiation-absorbing materials and generates relatively clean DNA (see Fig. S1C in the supplemental material) (8, 32). On the other hand, no 260- to 275-nm maximum was observed for DNA isolated using the FastDNA or UltraClean kit (see Fig. S1A and B in the supplemental material). DNA extracted by the FastDNA kit had a peak around 237 to 254 nm, while DNA extracted by the UltraClean kit had a peak around 225 to 230 nm. A similar trend was also observed for the DNA isolated from freshwater biofilm samples using the UltraClean kit and for pure culture DNA extracted from Escherichia coli and N. maritimus using the FastDNA kit (data not shown). Therefore, direct spectrophotometric measurement may not provide accurate determination of the DNA concentration, presumably because commercial DNA extraction kits cannot sufficiently eliminate UV radiation-absorbing materials in the DNA samples.
An alternative fluorescence-based assay using SYBR green I dye—which is less affected by RNA and extracted cocontaminants than other commonly employed fluorescent dyes (29) and which has a linear relationship between 10 and 500 pg of DNA μl−1 (R2, 0.99)—was subsequently used to determine DNA concentrations. The DNA yield obtained with the UltraClean DNA extraction kit (0.52 ± 0.04 ng ml−1 [mean ± standard error {SE} for four replicates]) was significantly lower than those obtained with the FastDNA kit (1.40 ± 0.15 ng ml−1 [mean ± SE for four replicates]) and phenol-chloroform extraction (1.56 ± 0.22 ng ml−1 [mean ± SE for four replicates]) (P, <0.0001) (Table 1). The yields obtained by using the FastDNA kit and phenol-chloroform extraction were not significantly different. The recovery efficiency was calculated by the difference between realized and theoretical DNA yields; the latter was calculated from the total cell number (2.9 × 106 cells ml−1) and estimated cellular DNA mass (3 fg per cell) (39). Efficiencies were 52.4%, 18.7%, and 59.3% for FastDNA, UltraClean, and phenol-chloroform extraction, respectively. These values are all greater than those for the original enzymatic method for DNA extraction from Sterivex filters, which yielded roughly 10% of environmental DNA in aquatic samples (40).
TABLE 1.
Comparative summary of DNA extraction methods and spiked experimentsa
| DNA extraction method | DNA yield (ng ml−1) | Total copy no. (103 ml−1) in: |
% Recovery in seeded samples | |
|---|---|---|---|---|
| Seeded samples | Nonseeded samples | |||
| FastDNA | 1.40 (0.15) | 88.8 (28.6) | 2.3 (0.65) | 10.7 (3.5) |
| UltraClean | 0.52 (0.04) | 60.5 (2.91) | 9.6 (2.68) | 6.3 (0.1) |
| Phenol-chloroform | 1.56 (0.22) | 180.4 (9.61) | 32.5 (8.43) | 18.7 (0.5) |
Data are means (SEs) for four replicates. Copy numbers were calculated by the volume-based method (equation 1).
To examine the quality of DNA, the PCR amplification efficiency of the bacterial 16S rRNA gene was examined (Table 2). The amplification efficiencies obtained using the phenol-chloroform method and the FastDNA kit were comparable, based on the eluted-volume-based method (Nvol) (Table 2), and were about two to three times greater than that with the UltraClean kit. This result suggested that the former two methods offer greater DNA yields than the UltraClean kit. In contrast, when the DNA weight-based estimation (Nwt) was used, there was no significant difference between the copy numbers obtained with the phenol-chloroform method and those obtained with the UltraClean kit, and both were about two times greater than that obtained using the FastDNA kit (Table 2). This result indicated that the quality of the DNA extracted by the former two methods was better than that of the DNA extracted with the FastDNA kit. To further evaluate the efficiency of DNA recovery and DNA quality from environmental samples, we used qPCR to quantify the recovery of archaeal amoA genes in surface water samples seeded with N. maritimus cells. The efficiency of recovery was highest for the phenol-chloroform method (18.7% ± 0.5% [mean ± SE for four replicates]) and lowest for the UltraClean kit (6.3% ± 0.1%) (Table 1). In consideration of DNA yield and purity (i.e., UV spectra) (see Fig. S1 in the supplemental material), the efficiency of amplification of the bacterial 16S rRNA gene (Table 2), and the efficiency of recovery of seeded N. maritimus cells (Table 1), the phenol-chloroform method was selected and used for further development of a DNA extraction protocol.
TABLE 2.
Amplification of 16S rRNA genes using real-time PCRa
| DNA extraction method | Bacterial 16S rRNA gene copy no. (106): |
|
|---|---|---|
| Per ml of seawater | Per ng of DNA | |
| FastDNA | 2.7 (0.099) | 0.79 (0.086) |
| UltraClean | 0.91 (0.025) | 1.4 (0.063) |
| Phenol-chloroform | 3.0 (0.053) | 1.5 (0.065) |
Purification steps were shortened to reduce the number of tube changes, which contribute to loss of DNA and increased processing time. We also observed that the Phase Lock Gel significantly reduced contamination and increased recovery (the level of recovery was previously reported to be as much as 30% greater [35]). In the downstream purification steps, ethanol precipitation, with or without a coprecipitate, was compared with a spin column (Table 3). The inclusion of GlycoBlue facilitated DNA handling and increased DNA recovery over that with general ethanol precipitation, most significantly when the amount of DNA was low (20 μg or less) (P < 0.05). This result was consistent with those of previous studies (4, 17). Alcohol precipitation with GlycoBlue and spin column purification methods were equivalent and not significantly different (Table 3).
TABLE 3.
Evaluation of downstream purification steps
| Treatment | Amt (%) of DNA recovereda with the following amt of DNA added per sample: |
|||
|---|---|---|---|---|
| 20 μg | 50 μg | 100 μg | 300 μg | |
| Ethanol | 7.5 ± 6.7 (37.4) | 32.6 ± 21.2 (65.1) | 67.1 ± 37.5 (67.1) | 197.5 ± 75.5 (65.8) |
| Ethanol + GlycoBlue | 17.5 ± 7.6 (87.4) | 38.8 ± 17.4 (77.5) | 72.0 ± 32.2 (72.0) | 198.3 ± 79.0 (66.1) |
| Spin column | 13.6 ± 0.5 (68.1) | 39.3 ± 1.1 (78.7) | 71.8 ± 2.3 (71.8) | 237.0 ± 5.4 (79.0) |
Amounts of DNA recovered are expressed as micrograms per sample and are means ± SEs for three replicates.
By use of the modified extraction method, DNA recovery from N. maritimus cells did not differ significantly for different filtration volumes or cell densities, showing good correspondence between yield and cell numbers (R2, 0.78; 16 measurements; P, <0.001) (Fig. 2). The efficiency of DNA extraction from strain SCM1 by the modified phenol-chloroform protocol (83.2% ± 7.4% [mean ± SE for 16 measurements]) was higher than that with the FastDNA kit (67.0% ± 6.8% [mean ± SE for 12 measurements]). The DNA yield and amplification efficiency of samples seeded with SCM1 cells were also greatly improved over those with the unmodified phenol-chloroform method; the percentage of recovery increased from 18.4% ± 0.5% (mean ± SE) to 79.4% ± 6.0% in samples from a depth of 10 m and from 28.9% ± 3.3% to 64.2% ± 2.0% in those from a depth of 90 m (Table 4).
FIG. 2.

Relationship between filtration volume and DNA recovery of Nitrosopumilus maritimus cells. Data are means ± SDs for four measurements each.
TABLE 4.
DNA yield and qPCR-based enumeration of the amoA gene before and after modification of the extraction methoda
| DNA extraction method and sampling depth (m) | DNA yield (ng ml−1) | Total copy no. (103 ml−1) in: |
% Recovery in seeded samples | |
|---|---|---|---|---|
| Seeded samples | Nonseeded samples | |||
| Phenol-chloroform | ||||
| 10 | 1.64 (0.06) | 150 (6.4) | 1.05 (0.12) | 18.4 (0.5) |
| 90 | 0.21 (0.02) | 313 (0.09) | 36.3 (1.05) | 28.9 (3.3) |
| New method | ||||
| 10 | 2.01 (0.18) | 664 (72.1) | 18.2 (2.8) | 79.4 (6.0) |
| 90 | 0.90 (0.08) | 690 (14.9) | 172 (1.58) | 64.2 (2.0) |
Data are calculated by the volume-based method (equation 1) and are means (SEs) for six (10-m samples) or four (90-m samples) replicates.
Population structure of the Hood Canal water column.
The modified DNA extraction method was used to characterize seawater samples collected from 11 depths (between 5 and 110 m) at a well-monitored field site in Hood Canal. This long, narrow fjord connected to Puget Sound has a shallow sill near its mouth (∼55 m) and depths exceeding 150 m. It is a highly dynamic system, due to seasonal shifts in freshwater input and illumination, as well as the replacement of bottom waters near the end of summer with dense (salty) waters that have upwelled along the coast, entering through the Strait of Juan de Fuca to Admiralty Inlet. High productivity during summer (∼3,000 mg of C m−2 day−1) is driven by the intrusion of the nitrate-rich (∼25 μM) Puget Sound water and is associated with an appearance of an oxygen-depleted water mass and resuspension of ammonia from the sediment to the water column (37). We anticipated that these features could make this fjord a valuable model system in which to explore the relative contributions of Archaea and Bacteria to the nitrogen cycle, with a specific focus on nitrification. Temperature and salinity profiles indicated that there was relatively little impact of freshwater in the near surface (Fig. 3A). Synechococcus numbers, chlorophyll a, and oxygen concentrations were high near the surface and decreased with depth (Fig. 3B, C, and F). A nitrite maximum was detected at a 10- to 30-m depth and was nearly coincident with an ammonium peak (Fig. 3D). The nitrate concentration increased with depth, reaching 34.7 μM at 105 m (Fig. 3E). Total cell numbers were in the range of 2.2 × 105 to 1.1 × 106 ml−1 (Fig. 3F), and there was good correspondence between the DNA yield per milliliter and the 16S rRNA copy number estimated by the volume-based (Nvol; equation 1) method (R2, 0.70; P, <0.01).
FIG. 3.

Vertical profile of ammonia-oxidizing Archaea determined by the modified DNA extraction method. (A) Temperature and salinity; (B) chlorophyll a; (C) dissolved oxygen; (D) ammonium and nitrite concentrations; (E) nitrate concentration; (F) total bacterial and Synechococcus cell numbers; (G) archaeal amoA and bacterial 16S rRNA gene copy numbers based on volume estimation (Nvol); (H) archaeal amoA and bacterial 16S rRNA gene copy numbers based on weight-volume estimation (Nwt vol); (I) copy number ratio between archaeal amoA and bacterial 16S rRNA genes. Quantitative PCR data are means ± SEs for four (archaeal amoA gene) and eight (bacterial 16S rRNA gene) replicates. Coccoid cells autofluorescing yellow under green-light excitation were counted as Synechococcus (38).
An extraction efficiency of 3.5 ± 0.8 fg (mean ± SE for 11 measurements) of DNA per bacterial cell was calculated using the SYBR green I counts shown in Fig. 3F. Assuming that most cells were bacterial, this corresponded to a 16S rRNA gene copy number in the range of 4.3 ± 0.6 (mean ± SE) per cell, similar to average 16S rRNA gene copy numbers (e.g., 3.8 per cell) (11) and per-cell DNA contents (e.g., 2.6 to 5.75 fg cell−1) among Bacteria (12, 18, 39). All environmental data sets revealed a transition between surface and deeper waters between 10 and 20 m, which also marked a transition in the abundance of amoA-containing Archaea (Fig. 3G and H). A peak of the amoA gene copy number (3.2 × 105 copies ml−1) was detected at 20 to 25 m, which corresponded with the bottom of the nitrite maximum and an apparent nitracline (Fig. 3D, E, and H). The copy number ratio between archaeal amoA and bacterial 16S rRNA increased with depth from 5 to 60 m, reaching a maximum of approximately 20%, with the assumption that the average 16S rRNA copy number per cell does not vary significantly with depth (Fig. 3I). This depth-related pattern of abundance is similar to the findings of previous reports in open marine regions, though spanning a much narrower depth interval (2, 3, 9, 20, 33). There was reasonably good correlation between the copy number ratio and the oxygen concentration (R2, 0.86; 11 measurements; P < 0.001), supporting previous reports that high numbers of Crenarchaeota are often found in low-oxygen water masses (2, 6, 24, 44).
Summary.
Achieving uniform extraction, high yield, and purity of DNA is essential for unbiased molecular analyses of environmental samples, although it is generally considered difficult to achieve all objectives at the same time (25). As a result of this uncertainty, two alternative calculations of gene copy number have frequently been used to express qPCR results: the volume-based (Nvol; copies per milliliter of seawater) and DNA weight-based (Nwt; copies per nanogram DNA) methods. Both expressions of abundance have advantages with respect to recognized problems of DNA extraction and quantification (3). The eluted-volume-based method is designed to express absolute abundance without the requirement for a DNA concentration measurement (1, 24, 33). However, the great drawbacks of eluted-volume-based calculation are variable DNA recovery (i.e., DNA loss) among different samples and filtration methods and uncertainty about the effects of contaminants that may differ among samples and interfere with PCR-based methods of quantification (3, 24). In the present study, the recovery of seeded cells ranged from 6.3 to 79.4% among different DNA extraction methods and samples. An alternative method, the DNA weight-based calculation, expresses abundance as a fraction of total DNA and therefore serves to normalize differences in DNA recovery among samples (Nwt; equation 2). However, weight-based copy numbers do not provide a direct measure of abundance. Thus, we incorporated cell counts to provide additional metrics to evaluate DNA recovery. Total cell counts and the average DNA content per cell (39) were used to estimate the amount of DNA present in a known sample volume (Nwt vol; equation 3). This estimate, compared to the volume-based recovery values, showed a smooth vertical profile, independent of the noise caused by differences in DNA extraction efficiency between samples, and could possibly be used in future studies (Fig. 3G to H).
Among the various methods evaluated, the modified phenol-chloroform method offered superior recovery of total bacterial and archaeal DNA of high quality (Table 3). Recovery (total DNA) and quality (16S rRNA gene copy number) compared to direct cell counts were fully consistent with the predicted range of values for DNA content cell−1 and 16S rRNA gene copy numbers. The maximum efficiency of extraction of archaeal DNA achieved using the modified method (64.2 to 79.4%) may be attributed in part to loss of small archaeal cells during filtration (22).
Our initial inspection of population profiles using this method revealed that ammonia-oxidizing Archaea were a significant population at the time of sampling, increasing with depth and reaching a maximum of 3.2 × 105 cells ml−1 as inferred from amoA gene copy numbers (Fig. 3H). This is a much greater abundance of ammonia-oxidizing Archaea than that determined by previous studies of other marine systems, reflecting the improvements in extraction and quantification established in this study and possibly also features unique to the Hood Canal fjord. Thus, we anticipate that this relatively simple protocol will provide a better analytical foundation for establishing the contribution of Archaea and other microorganisms to key biogeochemical processes in the ocean.
Supplementary Material
Acknowledgments
We thank José R. de la Torre, Florian U. Moeller, and Kyle C. Kosta for early contributions to this study. We also thank Colin A Smith for helping with sample collection and Allen H. Devol for providing buoy data. We thank E. Michelle L. Starke for initial assistance with the qPCR assay. We acknowledge Peter F. Andeer for critical reading of the manuscript.
This work was supported by National Science Foundation (NSF) Microbial Interactions Program grant MCB-0604448 to D.A.S. and José R. de la Torre and by NSF Biological Oceanography grant OCE-0623174 to A. E. Ingalls, D.A.S., and A. H. Devol.
Footnotes
Published ahead of print on 29 January 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Agogué, H., M. Brink, J. Dinasquet, and G. J. Herndl. 2008. Major gradients in putatively nitrifying and non-nitrifying Archaea in the deep North Atlantic. Nature 456:788-791. [DOI] [PubMed] [Google Scholar]
- 2.Baltar, F., J. Arístegui, J. M. Gasol, S. Hernández-León, and G. J. Herndl. 2007. Strong coast-ocean and surface-depth gradients in prokaryotic assemblage structure and activity in a coastal transition zone region. Aquat. Microb. Ecol. 50:63-74. [Google Scholar]
- 3.Beman, J. M., B. N. Popp, and C. A. Francis. 2008. Molecular and biogeochemical evidence for ammonia oxidation by marine Crenarchaeota in the Gulf of California. ISME J. 2:429-441. [DOI] [PubMed] [Google Scholar]
- 4.Boström, K. H., K. Simu, A. Hagström, and L. Riemann. 2004. Optimization of DNA extraction for quantitative marine bacterioplankton community analysis. Limnol. Oceanogr. Methods 2:365-373. [Google Scholar]
- 5.Bower, P. A., C. O. Scopel, E. T. Jensen, M. M. Depas, and S. L. McLellan. 2005. Detection of genetic markers of fecal indicator bacteria in Lake Michigan and determination of their relationship to Escherichia coli densities using standard microbiological methods. Appl. Environ. Microbiol. 71:8305-8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coolen, M. J., B. Abbas, J. van Bleijswijk, E. C. Hopmans, M. M. Kuypers, S. G. Wakeham, and J. S. Sinninghe Damste. 2007. Putative ammonia-oxidizing Crenarchaeota in suboxic waters of the Black Sea: a basin-wide ecological study using 16S ribosomal and functional genes and membrane lipids. Environ. Microbiol. 9:1001-1016. [DOI] [PubMed] [Google Scholar]
- 7.De Corte, D., T. Yokokawa, M. M. Varela, H. Agogué, and G. J. Herndl. 2009. Spatial distribution of Bacteria and Archaea and amoA gene copy numbers throughout the water column of the Eastern Mediterranean Sea. ISME J. 3:147-158. [DOI] [PubMed] [Google Scholar]
- 8.Dell'Anno, A., M. Fabiano, G. C. A. Duineveld, A. Kok, and R. Danovaro. 1998. Nucleic acid (DNA, RNA) quantification and RNA/DNA ratio determination in marine sediments: comparison of spectrophotometric, fluorometric, and high-performance liquid chromatography methods and estimation of detrital DNA. Appl. Environ. Microbiol. 64:3238-3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLong, E. F., L. T. Taylor, T. L. Marsh, and C. M. Preston. 1999. Visualization and enumeration of marine planktonic archaea and bacteria by using polyribonucleotide probes and fluorescent in situ hybridization. Appl. Environ. Microbiol. 65:5554-5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinstein, L. M., W. J. Sul, and C. B. Blackwood. 2009. Assessment of bias associated with incomplete extraction of microbial DNA from soil. Appl. Environ. Microbiol. 75:5428-5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fogel, G. B., C. R. Collins, J. Li, and C. F. Brunk. 1999. Prokaryotic genome size and SSU rDNA copy number: estimation of microbial relative abundance from a mixed population. Microb. Ecol. 38:93-113. [DOI] [PubMed] [Google Scholar]
- 12.Fuhrman, J. A., and F. Azam. 1982. Thymidine incorporation as a measure of heterotrophic bacterioplankton production in marine surface waters: evaluation and field results. Mar. Biol. 66:109-120. [Google Scholar]
- 13.Fuhrman, J. A., D. E. Comeau, A. Hagström, and A. M. Chan. 1988. Extraction from natural planktonic microorganisms of DNA suitable for molecular biological studies. Appl. Environ. Microbiol. 54:1426-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon, K. V., M. C. Vickery, A. DePaola, C. Staley, and V. J. Harwood. 2008. Real-time PCR assays for quantification and differentiation of Vibrio vulnificus strains in oysters and water. Appl. Environ. Microbiol. 74:1704-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grasshoff, K., M. Ehrhardt, and K. Kremling. 1999. Methods of seawater analysis, 3rd ed. Wiley-VCH Verlag GmbH, Weinheim, Germany.
- 16.Holmes, R. M., A. Aminot, R. Kérouel, B. A. Hooker, and B. J. Peterson. 1999. A simple and precise method for measuring ammonium in marine and freshwater ecosystems. Can. J. Fish. Aquat. Sci. 56:1801-1808. [Google Scholar]
- 17.Jeffrey, W. H., S. Nazaret, and R. Von Haven. 1994. Improved method for recovery of mRNA from aquatic samples and its application to detection of mer expression. Appl. Environ. Microbiol. 60:1814-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeffrey, W. H., R. Von Haven, M. P. Hoch, and R. B. Coffin. 1996. Bacterioplankton RNA, DNA, protein content and relationships to rates of thymidine and leucine incorporation. Aquat. Microb. Ecol. 10:87-95. [Google Scholar]
- 19.Jiang, J. L., K. A. Alderisio, A. Singh, and L. H. Xiao. 2005. Development of procedures for direct extraction of Cryptosporidium DNA from water concentrates and for relief of PCR inhibitors. Appl. Environ. Microbiol. 71:1135-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karner, M. B., E. F. DeLong, and D. M. Karl. 2001. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409:507-510. [DOI] [PubMed] [Google Scholar]
- 21.Kirchman, D. L., L. Y. Yu, B. M. Fuchs, and R. Amann. 2001. Structure of bacterial communities in aquatic systems as revealed by filter PCR. Aquat. Microb. Ecol. 26:13-22. [Google Scholar]
- 22.Könneke, M., A. E. Bernhard, J. R. de la Torre, C. B. Walker, J. B. Waterbury, and D. A. Stahl. 2005. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437:543-546. [DOI] [PubMed] [Google Scholar]
- 23.Kuske, C. R., K. L. Banton, D. L. Adorada, P. C. Stark, K. K. Hill, and P. J. Jackson. 1998. Small-scale DNA sample preparation method for field PCR detection of microbial cells and spores in soil. Appl. Environ. Microbiol. 64:2463-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam, P., M. M. Jensen, G. Lavik, D. F. McGinnis, B. Muller, C. J. Schubert, R. Amann, B. Thamdrup, and M. M. Kuypers. 2007. Linking crenarchaeal and bacterial nitrification to anammox in the Black Sea. Proc. Natl. Acad. Sci. U. S. A. 104:7104-7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leff, L. G., J. R. Dana, J. V. McArthur, and L. J. Shimkets. 1995. Comparison of methods of DNA extraction from stream sediments. Appl. Environ. Microbiol. 61:1141-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leininger, S., T. Urich, M. Schloter, L. Schwark, J. Qi, G. W. Nicol, J. I. Prosser, S. C. Schuster, and C. Schleper. 2006. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442:806-809. [DOI] [PubMed] [Google Scholar]
- 27.Lunau, M., A. Lemke, K. Walther, W. Martens-Habbena, and M. Simon. 2005. An improved method for counting bacteria from sediments and turbid environments by epifluorescence microscopy. Environ. Microbiol. 7:961-968. [DOI] [PubMed] [Google Scholar]
- 28.Martens-Habbena, W., P. M. Berube, H. Urakawa, J. de la Torre, and D. A. Stahl. 2009. Ammonia oxidation kinetics determines niche separation of nitrifying Archaea and Bacteria. Nature 461:976-979. [DOI] [PubMed] [Google Scholar]
- 29.Martens-Habbena, W., and H. Sass. 2006. Sensitive determination of microbial growth by nucleic acid staining in aqueous suspension. Appl. Environ. Microbiol. 72:87-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin-Laurent, F., L. Philippot, S. Hallet, R. Chaussod, J. C. Germon, G. Soulas, and G. Catroux. 2001. DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl. Environ. Microbiol. 67:2354-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massana, R., A. E. Murray, C. M. Preston, and E. F. DeLong. 1997. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl. Environ. Microbiol. 63:50-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller, D. N., J. E. Bryant, E. L. Madsen, and W. C. Ghiorse. 1999. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 65:4715-4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mincer, T. J., M. J. Church, L. T. Taylor, C. Preston, D. M. Karl, and E. F. DeLong. 2007. Quantitative distribution of presumptive archaeal and bacterial nitrifiers in Monterey Bay and the North Pacific Subtropical Gyre. Environ. Microbiol. 9:1162-1175. [DOI] [PubMed] [Google Scholar]
- 34.Mumy, K. L., and R. H. Findlay. 2004. Convenient determination of DNA extraction efficiency using an external DNA recovery standard and quantitative-competitive PCR. J. Microbiol. Methods 57:259-268. [DOI] [PubMed] [Google Scholar]
- 35.Murphy, N. R., and R. J. Hellwig. 1996. Improved nucleic acid organic extraction through use of a unique gel barrier material. Biotechniques 21:934-939. [DOI] [PubMed] [Google Scholar]
- 36.Myers, M. L., G. Panicker, and A. K. Bej. 2003. PCR detection of a newly emerged pandemic Vibrio parahaemolyticus O3:K6 pathogen in pure cultures and seeded waters from the Gulf of Mexico. Appl. Environ. Microbiol. 69:2194-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newton, J. A., S. L. Albertson, K. V. Voorhis, C. Maloy, and E. Siegel. 2002. Washington State marine water quality in 1998 through 2000. Publication 02-03-056. Washington State Department of Ecology, Environmental Assessment Program, Olympia, WA.
- 38.Putland, J. N., and R. B. Rivkin. 1999. Influence of storage mode and duration on the microscopic enumeration of Synechococcus from a cold coastal ocean environment. Aquat. Microb. Ecol. 17:191-199. [Google Scholar]
- 39.Simon, M., and F. Azam. 1989. Protein content and protein synthesis rates of planktonic marine bacteria. Mar. Ecol. Prog. Ser. 51:201-213. [Google Scholar]
- 40.Somerville, C. C., I. T. Knight, W. L. Straube, and R. R. Colwell. 1989. Simple, rapid method for direct isolation of nucleic acids from aquatic environments. Appl. Environ. Microbiol. 55:548-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steffan, R. J., J. Goksoyr, A. K. Bej, and R. M. Atlas. 1988. Recovery of DNA from soils and sediments. Appl. Environ. Microbiol. 54:2908-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoeckel, D. M., E. A. Stelzer, and L. K. Dick. 2009. Evaluation of two spike-and-recovery controls for assessment of extraction efficiency in microbial source tracking studies. Water Res. 43:4820-4827. [DOI] [PubMed] [Google Scholar]
- 43.Tsai, Y. L., and B. H. Olson. 1991. Rapid method for direct extraction of DNA from soil and sediments. Appl. Environ. Microbiol. 57:1070-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varela, M. M., H. M. van Aken, E. Sintes, and G. J. Herndl. 2008. Latitudinal trends of Crenarchaeota and Bacteria in the meso- and bathypelagic water masses of the Eastern North Atlantic. Environ. Microbiol. 10:110-124. [DOI] [PubMed] [Google Scholar]
- 45.Wuchter, C., B. Abbas, M. J. L. Coolen, L. Herfort, J. van Bleijswijk, P. Timmers, M. Strous, E. Teira, G. J. Herndl, J. J. Middelburg, S. Schouten, and J. S. Sinninghe Damsté. 2006. Archaeal nitrification in the ocean. Proc. Natl. Acad. Sci. U. S. A. 103:12317-12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang, J.-Z., and C. J. Fischer. 2006. A simplified resorcinol method for direct spectrophotometric determination of nitrate in seawater. Mar. Chem. 99:220-226. [Google Scholar]
- 47.Zhou, J., M. A. Bruns, and J. M. Tiedje. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.