

Abstract
Xanthomonas citri subsp. citri is the causal agent of citrus canker, which has a significant impact on citrus production. In this study, we characterized the galU gene of X. citri subsp. citri. Two galU mutants (F6 and D12) were identified in an X. citri subsp. citri EZ-Tn5 <R6Kγori/KAN-2> Tnp transposon library. Rescue cloning, sequence analysis, and Southern blot analysis indicated that both of these mutants had a single copy of the EZ-Tn5 transposon inserted in galU in the chromosome. Further study showed that galU was required for biosynthesis of extracellular polysaccharides (EPS; xanthan gum) and capsular polysaccharide (CPS) and biofilm formation. Mutation of galU resulted in a loss of pathogenicity for grapefruit. The loss of pathogenicity of a galU mutant resulted from its inability to grow in planta rather than from the effect on virulence genes. Quantitative reverse transcription-PCR assays indicated that mutation of galU did not impair the expression of key virulence genes, such as pthA of X. citri subsp. citri. Although D12 had a growth rate similar to that of the wild-type strain in nutrient broth, no D12 population became established in the intercellular spaces of citrus leaves. Coinoculation of a galU mutant with the wild-type strain did not promote growth of the galU mutant in planta. Defects in EPS and CPS production, pathogenicity, and growth in planta of the galU mutant were complemented to the wild-type level using plasmid pCGU2.1 containing an intact galU gene. These data indicate that the galU gene contributes to X. citri subsp. citri growth in intercellular spaces and is involved in EPS and CPS synthesis and biofilm formation.
Citrus canker is a serious disease of most commercial citrus cultivars in subtropical citrus-producing areas of the world (12, 18). Citrus-producing areas without citrus canker consider the disease a quarantine pest due to the potential threat to citrus production. Thus, citrus canker has a significant impact on national and international citrus markets and trade (18). Citrus canker is a disease that is characterized by formation of necrotic raised lesions on leaves, stems, and fruits. On severely affected trees, citrus canker causes defoliation, twig dieback, general tree decline, blemished fruit, and premature fruit drop (19). Citrus canker is caused by the bacterial pathogen Xanthomonas citri subsp. citri (synonyms, Xanthomonas citri, Xanthomonas campestris pv. citri, and Xanthomonas axonopodis pv. citri) strain A (10, 44, 48). This bacterium is dispersed via windblown rain and enters the plant directly through stomata or through wounds, and then it grows in the intercellular spaces of the spongy mesophyll (18). The intercellular spaces are nutrient poor (1) and contain toxic substances (either preformed or induced) as part of the host defense response (35, 45). Bacteria have evolved complicated mechanisms to overcome the plant defense and acquire nutrients to colonize susceptible hosts.
Genome sequencing of X. citri subsp. citri has greatly increased our understanding of the interaction between citrus and X. citri subsp. citri. X. citri subsp. citri contains 4,313 genes, including 2,710 genes with assigned functions, 1,603 genes without known functions, and 54 RNA genes (13). About 6% of the X. citri subsp. citri genes are involved in pathogenicity, virulence, and adaptation. The major genes involved in pathogenicity and virulence are genes encoding secretion systems, including type III secretion system genes, effector genes, genes encoding cell wall-degrading enzymes, toxins, bacterial adhesins, and surface structural elements, and rpf (regulation of pathogenicity factors) genes, which are related to cell-cell signaling (13). Even though the genome of X. citri subsp. citri has been sequenced and 62.83% of the predicted open reading frames have been assigned functions, functional studies are necessary to experimentally characterize the genes related to X. citri subsp. citri pathogenesis and host adaptation. Transposon mutagenesis has been widely used for this purpose (23, 41).
GalU is a UTP-glucose-1-phosphate uridylyltransferase (synonym, UDP-glucose pyrophosphorylase), and it catalyzes the formation of UDP-glucose from glucose 1-phosphate and UTP. UDP-glucose is involved in synthesis of glucosylated surface structures as a substrate for glucosyltransferase, and it is a glycosyl donor in the enzymatic biosynthesis of carbohydrates (9, 46). Mutations in the galU gene have led to reduced virulence of a number of bacterial pathogens, including Escherichia coli (21, 28), Klebsiella pneumoniae (7), Shigella flexneri (43), Actinobacillus pleuropneumoniae (38), Vibrio cholerae (34), Pseudomonas aeruginosa (37), and Mesophilic aeromonas (49). The reduced virulence of galU mutants has been associated mainly with changes in lipopolysaccharides (LPS), capsular polysaccharides (CPS), or exopolysaccharides (EPS). GalU has also been reported to be involved in adhesion of E. coli (17). However, the role of the galU gene in the virulence of X. citri subsp. citri and other plant-pathogenic bacteria has not been studied. In this study, we characterized the galU gene of X. citri subsp. citri. This was part of our effort to characterize critical genes involved in the virulence of X. citri subsp. citri. To our knowledge, it is the first report of an analysis of galU for plant-pathogenic bacteria.
MATERIALS AND METHODS
Bacterial strains and growth media.
All of the strains used in this study are listed in Table 1. X. citri subsp. citri wild-type strain 306 (rifamycin resistant) (40) and mutant strains were grown in nutrient broth (NB), on nutrient agar (NA), or in NYG medium (11) at 28°C. E. coli strains were grown in Luria-Bertani (LB) medium at 37°C. Antibiotics were used at the following concentrations: rifamycin (Rif), 50 μg/ml; kanamycin (Kn), 50 μg/ml; ampicillin (Ap), 50 μg/ml; gentamicin (Gm), 5 μg/ml; and chloramphenicol (Cm), 35 μg/ml.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli strains | ||
| Mach1 T1R | F− φ80lacZΔM15 ΔlacX74 hsdR(rK− mK+) ΔrecA1398 endA1 tonA | Invitrogen |
| TransforMax EC100D pir+ | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara-leu)7697 galU galK λ−rpsL nupG pir+(DHFR) | Epicentre |
| HB101 | F−supE44 hsdS20(rB−mB−) recA13 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 leuB6 thi | 4 |
| X. citri subsp. citri strains | ||
| 306 | Rifr, causes citrus canker on citrus | 40 |
| D12 | galU::Tn5 derivative of strain 306, Rifr Knr | This study |
| F6 | galU::Tn5 derivative of strain 306, Rifr Knr | This study |
| CD | D12 containing pCGU2.1, Knr Cmr Gmr | This study |
| CF | F6 containing pCGU2.1, Knr Cmr Gmr | This study |
| Plasmids | ||
| pCR2.1 | PUC ori f1 ori lacZα+ Knr Apr | Invitrogen |
| pRK2013 | ColE1 Knr Tra+, conjugation helper plasmid | 14 |
| pUFR053 | IncW Mob+mob(P) lacZα+ Par+ Cmr Knr, shuttle vector | 15 |
| pCGU1.1 | galU gene from X. citri subsp. citri 306 cloned in pCR2.1 | This study |
| pCGU2.1 | galU gene on BamHI fragment from pCGU1.1 cloned in pUFR053 | This study |
Construction of X. citri subsp. citri mutant library.
An EZ-Tn5 <R6Kγori/KAN-2> Tnp Transposome kit (Epicentre, Madison, WI) was used to make the X. citri subsp. citri mutant library by following the manufacturer's instructions. The cells recovered were diluted 500- to 1,000-fold, spread on NA plates containing Rif and Kn, and incubated at 28°C for 2 to 3 days. Mutants were kept at −80°C in 20% glycerol until they were used. The mutant library was screened by performing pathogenicity assays (see below) with a susceptible host, cultivar Duncan grapefruit (Citrus paradisi Macf. cv. Duncan). Mutants that caused no symptoms or reduced symptoms were selected for further study.
Rescue cloning of two nonmucoid mutants.
Two nonmucoid mutants, D12 and F6 (Table 1), were chosen from the mutant library for further analysis based on their nonpathogenic phenotypes in pathogenicity assays. To identify the insertion site in mutants D12 and F6, the rescue cloning method was used by following the manufacturer's instructions (Epicentre, Madison, WI). Briefly, genomic DNA (1 μg) of the D12 and F6 transformants were digested overnight with BamHI and SphI, end repaired (made blunt ended) using T4 DNA polymerase (New England Biolabs, Ipswich, MA), and 5′ phosphorylated so that they self-ligate. The digested DNA was purified using the Wizard SV gel and PCR clean-up system (Promega, Madison, WI) and allowed to self-ligate in the presence of T4 DNA ligase in a 10-μl mixture for 4 h at 16°C. The ligation mixture was electroporated into electrocompetent E. coli TransforMax EC100D pir+ (Epicentre, Madison, WI). Cells were immediately transferred to a 17- by 100-mm round-bottom polypropylene tube which contained 1 ml SOC (super optimal broth with catabolite repression) (20), and the preparation was gently mixed by pipetting. The cells were incubated for 1 h at 37°C with gentle shaking and then plated on LB agar containing Kn. A plasmid was isolated from Kn-resistant colonies, purified, and sequenced with primers R6KAN-2 RP-1 and KAN-2 FP-1 (Table 2). Sequencing was performed at the Interdisciplinary Center for Biotechnology Research sequencing facility at the University of Florida. Since the genome sequence of X. citri subsp. citri strain 306 has been published (13), transposon insertion sites in the F6 and D12 transformants were identified by aligning the corresponding X. citri subsp. citri 306 and mutated loci.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′ → 3′)a |
|---|---|
| galU-F1 | AGACAGTGCCGAAAGAAATGCTGC |
| galU-R1 | AACAGCGATCAGGCAACAATCAGC |
| Kan2-F1 | CGAGGCCGCGATTAAATTCCAACA |
| Kan2-R1 | AGGCAGTTCCATAGGATGGCAAGA |
| KAN-2 FP-1 | ACCTACAACAAAGCTCTCATCAACC |
| R6KAN-2 RP-1 | CTACCCTGTGGAACACCTACATCT |
| CGU-F | AATGATGGATCCCTGCCAAAGCCTTGACGC |
| CGU-R | AACAGAGGATCCAACATCACCACGCCCAAC |
Underlined nucleotides are not exact matches and were introduced to add a BamHI restriction enzyme site.
Nucleic acid isolation and PCR.
Genomic DNA was extracted using a Wizard genomic DNA purification kit (Promega, Madison, WI) by using the protocol for isolating genomic DNA from bacteria. After DNA precipitation, the pellet was dried in a Vacufuge (Eppendorf, Westbury, NY) for 5 min and dissolved in the DNA rehydration solution supplied with the kit. Bacterial plasmid DNA was isolated using the Wizard miniprep DNA purification system (Promega, Madison, WI). The concentration and purity of DNA were determined using an Agilent 8453 UV-visible spectrophotometer (Agilent Technologies, Santa Clara, CA).
All conventional PCRs were performed with a Bio-Rad DNAEngine Peltier thermal cycler (Bio-Rad, Hercules, CA). Amplification of the DNA was performed using 50-μl (total volume) mixtures with Taq DNA polymerase (Promega, Madison, WI). The PCR conditions were 95°C for 5 min, followed by 40 cycles of 30 s of denaturation at 95°C, 30 s of annealing at 52°C, and 1 to 3 min of extension (depending on the length of the amplicons) at 72°C.
Southern blot analysis.
For Southern blot hybridization, genomic DNA samples were purified (after isolation using a Wizard genomic DNA purification kit) using phenol-chloroform-isoamyl alcohol (25:24:1, vol/vol/vol) and chloroform-isoamyl alcohol (24:1, vol/vol) and a standard molecular biology protocol (42). The DNA was precipitated, washed with 70% (vol/vol) ethanol, and resuspended in RNase- and DNase-free water. Genomic DNA (3 μg per sample) was digested with BglII, subjected to electrophoresis on a 0.9% agarose gel, and transferred to a positively charged nylon membrane (Roche, Indianapolis, IN) using standard procedures (42). Probe generation, hybridization, and detection of chemiluminescence were performed using a digoxigenin (DIG)-High Prime II DNA labeling and detection starter kit as recommended by the manufacturer (Roche, Indianapolis, IN).
Complementation of the galU mutants.
The entire galU gene with 624 bp of upstream sequence and 388 bp of downstream sequence was amplified from genomic DNA of X. citri subsp. citri wild-type strain 306 by a PCR performed with primers CGU-F and CGU-R, which contained a BamHI restriction site (Table 2). The resulting 1.9-kb fragment was ligated to PCR 2.1-TOPO using the manufacturer's protocol (Invitrogen, Carlsbad, CA), resulting in pCGU1.1. A BamHI fragment containing the galU gene was isolated from pCGU1.1 and cloned into similarly digested pUFR053 that was treated with calf intestinal alkaline phosphatase (New England Biolabs, Ipswich, MA) (15), resulting in pCGU2.1 (Table 1). Plasmid pCGU2.1 was transferred into galU mutants D12 and F6 (galU::Tn5) by triparental mating with an E. coli helper strain containing pRK2013 (47). Transconjugants were selected on NA with Rif and Gm. The presence of pCGU2.1 was verified using PCR. Complementation assays were conducted to examine EPS and CPS production, the mucoid phenotype on NA medium, pathogenicity, and growth of the galU mutants in planta using plasmid pCGU2.1 containing an intact galU gene. Empty vector pUFR053 without the galU gene was used as a control.
Quantitative determination of EPS production.
To measure the quantity of EPS in culture supernatants, bacterial cells were grown in NB supplemented with 2% d-glucose at 28°C for 24 h with shaking at 200 rpm. Then 10-ml portions of the cultures were removed, and the cells were removed by centrifugation (5,000 × g for 20 min). Three volumes of 99% ethyl alcohol was added to the supernatants. The precipitated EPS was pelleted by centrifugation, dried, and weighed (51). Three independent replicates were used for each strain. The test was performed three times independently; only the results of one test are shown below.
Capsule assays.
Bacteria were grown at 28°C on NA with appropriate antibiotics. Samples were prepared by mixing and spreading a loop of bacteria with 1 drop of distilled water on a precleaned slide and air dried. Each sample was stained with 1% crystal violet and washed with 20% copper sulfate supplied with a capsule-staining kit (Eng Scientific Inc., Clifton, NJ) by following the manufacturer's instructions. The samples were photographed using a light microscope (Leica DMLB2; Leica Microsystems Wetzlar GmbH, Germany) with an oil immersion lens at a magnification of ×1,000.
LPS assays.
Bacteria were grown overnight at 28°C in NB, M9 glucose medium (8), or XVM2 medium (52). Ten-milliliter cultures were harvested at the exponential phase of growth and pelleted. LPS was isolated and treated with proteinase K (33), and then it was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Briefly, LPS samples were mixed with an equal volume of Laemmli sample buffer (pH 6.8) containing 62.5 mM Tris-HCl (pH 6.8), 2% SDS, 25% glycerol, and 0.01% bromophenol blue. The mixtures were boiled for 5 min, and 20-μl samples were loaded on precast Ready Gel Tris-HCl polyacrylamide gels (86 mm by 68 mm by 1.0 mm) containing 4 and 15% acrylamide in the stacking and separating gels, respectively (Bio-Rad Laboratories, Inc., Hercules, CA). Electrophoresis was performed at 12 mA for the stacking gels and at 25 mA for the separating gels until the bromophenol blue was 1 cm above the bottom of the gel. Immediately after electrophoresis each gel was stained using a silver stain kit (Bio-Rad Laboratories, Inc., Hercules, CA).
Biofilm assays.
Biofilm formation in borosilicate glass tubes was quantified as described previously (36). Bacteria were grown in NB with shaking to the mid-exponential growth phase and then diluted 1:10 in fresh NB containing appropriate antibiotics. One milliliter of a diluted bacterial suspension was placed in each borosilicate glass tube (Fisher Scientific, Pittsburgh, PA) and incubated without shaking for 48 h at 28°C. The culture medium was poured out, and attached bacterial cells were gently washed three times with distilled water, incubated at 60°C for 20 min, and then stained with 1.5 ml 0.1% crystal violet for 45 min. The unbound crystal violet was poured out, and the wells were washed with water. The crystal violet-stained cells were solubilized in 1.5 ml of ethanol-acetone (80:20, vol/vol). Biofilm formation was quantified by measuring the absorbance at 590 nm using an Agilent 8453 UV-visible spectrophotometer. Ten replicates were used for quantitative measurement.
Pathogenicity assays on plants.
Pathogenicity assays were conducted in a quarantine greenhouse facility at the Citrus Research and Education Center, Lake Alfred, FL. Assays were performed using fully expanded, immature leaves of cv. Duncan grapefruit. The X. citri subsp. citri wild-type and mutant strains used in this assay were grown with shaking at 28°C overnight in NB and were suspended in sterile tap water, and the concentration was adjusted to 108 CFU/ml. For the pathogenicity assays, bacterial suspensions containing both 108 and 105 CFU/ml were injection infiltrated into leaves with a needleless syringe (40, 50). The test was repeated three times, and similar results were obtained in all tests. Disease symptoms were photographed 5, 10, and 12 days postinoculation (DPI).
Bacterial growth assays in planta.
For assays of bacterial growth in the intercellular spaces of citrus leaves, the concentration of the starting inoculum used was 106 CFU/ml, and whole leaves were inoculated by infiltration of the abaxial leaf surface using a needleless syringe as described above. For coinoculation, wild-type strain 306 and the D12 mutant were mixed together (1:1) before inoculation. Briefly, bacterial cell counts were determined for four biological replicates at each sampling time point (0, 1, 2, 3, 4, 6, 8, or 10 DPI). Leaf disks from inoculated leaves were excised with a cork borer (leaf area, 1 cm2) and then ground in 1 ml sterile tap water. The samples were serially diluted and plated on NA plates with appropriate antibiotics. The colonies were counted 48 h after plating. The in planta growth assays were repeated three times independently with four replicates each time, but the results of only one experiment are described here.
RNA extraction and quantitative reverse transcription-PCR (QRT-PCR).
Bacteria were grown in XVM2 medium at 28°C with shaking at 200 rpm, and 1-ml samples of X. citri subsp. citri wild-type strain and D12 mutant cultures were collected at 13 h after inoculation. RNA was stabilized immediately by mixing it with 2 volumes of RNAprotect bacterial reagent (Qiagen, Valencia, CA) and was incubated at room temperature for 5 min. Bacterial cells were centrifuged at 5,000 × g for 10 min, and the cell pellets were stored at −80°C prior to RNA extraction.
Cell pellets were treated with lysozyme, and RNA extraction was performed using an RNeasy minikit (Qiagen, Valencia, CA). Contaminated genomic DNA was removed from RNA by treatment with a TURBO DNA-free kit (Ambion, Austin, TX). The concentration of RNA was determined with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and adjusted to 50 ng/μl for QRT-PCR.
Fifteen target genes were chosen for an expression study with 16S rRNA as the endogenous control, and the primers shown in Table 3 were designed using sequences of the X. citri subsp. citri genome and DNASTAR software (DNASTAR, Inc., Madison, WI). A one-step QRT-PCR was performed with a 7500 Fast real-time PCR system (Applied Biosystems, Inc., Foster City, CA) using a QuantiTect SYBR green RT-PCR kit (Qiagen, Valencia, CA). The total volume of the one-step QRT-PCR mixture was 25 μl, and this mixture contained 2× QuantiTect SYBR green RT-PCR master mixture (12.5 μl), 10 μM gene-specific primers (1.25 μl), QuantiTect RT mixture (0.5 μl), and 50 ng of RNA template (1 μl). The reaction mixture was incubated at 50°C for 30 min and at 95°C for 15 min and then was subjected to 40 cycles of 94°C for 15 s, 53°C for 30 s, and 72°C for 30 s. A melting curve analysis was performed to verify the specificity and identity of the QRT-PCR products. Three biological replicates were used for each strain. QRT-PCR was repeated once with another three independent biological replicates. A total of six independent biological replicates were used for each strain.
TABLE 3.
Genes and corresponding primers used for QRT-PCR analysis
| Gene | Product |
Primerb |
||
|---|---|---|---|---|
| Functiona | Component or protein | Directionc | Sequence (5′ → 3′) | |
| 16S rRNA | Ribosome component | F | CGCTTTCGTGCCTCAGTGTCAGTGTTGG | |
| R | GGCGTAAAGCGTGCGTAGGTGGTGGTT | |||
| hrpG | TTSS | TTSS regulator | F | GCCTTTCAATTCGCACGAGTTACACG |
| R | CACACGCCGGGGCTGGAAAAGA | |||
| hrpX | TTSS | TTSS regulator | F | AGCGATCTCTGCGTTGTCCTAC |
| R | ATACGCATCTTCGGCCTCTTCCTGA | |||
| hrcV | TTSS | TTSS component | F | GCGTTTGCGGCGTGCTTCATCT |
| R | CAATCTGGTGGTAGGCCTGGTCGTTTTCTT | |||
| pthA | TTSS | TTSS effector | F | TGGCGTCGGCAAACAGTGGTC |
| R | TGCTCCGGGGTCAGGTTCAGG | |||
| avrBs2 | TTSS | Avirulence protein | F | CGCGCCAATCACGACAAGGACTACTAC |
| R | CGGGCCAGCGTGCGGTTTTC | |||
| avrXacE1 | TTSS | Avirulence protein | F | TCGCGCTGGGCCGGAACATACC |
| R | GCGTCCGCGGCGATAACTCTTG | |||
| XAC0028 | CWDE | Cellulase | F | CGCTCCACGCTGCTTTTCAT |
| R | ATTCGGCACCGGACAGATTG | |||
| XAC0160 | CWDE | Xylanase | F | CATGGCCTGGCGGTCCTTGTGCT |
| R | GCGCGATCCGGCTGGCTTGTTC | |||
| XAC0165 | CWDE | Xylosidase | F | AGGGCGGGGCGTTGCTGTTCTAC |
| R | TCTTGCCGTCGGGACTGCTGTGA | |||
| peh-1 | CWDE | Endopolygalacturonase | F | AGTGGCAACGCGTTTCTGACC |
| R | CGCCTGCGTTGTTGCCCTTGAC | |||
| celD | CWDE | Glucan 1,4-beta-glucosidase | F | GATTTCGGCCGAGCGTCTGGA |
| R | GGATGCCGGCCTGGTTCTTCA | |||
| pglA | CWDE | Polygalacturonase | F | CAGCGCCGACGTCACCTTGTA |
| R | GTAGCCGGGACGCGAATAGACC | |||
| pelB | CWDE | Pectate lyase II | F | GAACTTCGGCGTGCGTGTGGTG |
| R | GTGAGCGAGGCGAAGATGGTGTTGTGGTC | |||
| gumB | EPS synthesis | Polysaccharide exporter | F | CTGACCGAAATCGAGAAGGGCACCAATC |
| R | GCGCCACACCATCACAAGAGGAGTCAGTTC | |||
| kpsF | CPS synthesis | Arabinose 5-phosphate isomerase | F | GCTTCACCGCCGACGACTTC |
| R | CGCTTGCGGCTCATTTCCATC | |||
TTSS, type III secretion system; CWDE, cell wall-degrading enzyme; EPS, extracellular polysaccharides; CPS, capsular polysaccharide.
The primers were derived from the sequences of corresponding genes.
F, forward; R, reverse.
The amount of fluorescence was plotted as a function of the PCR cycle using the 7500 Fast system SDS software, and the threshold cycle (CT) values for each gene were determined. The ΔCT values for each target gene were calculated by subtracting the CT values for 16S rRNA (the endogenous control) from the CT values for the target gene. The ΔCT values for each gene for the D12 and wild-type strains were subjected to a simple t test (SAS), and the ΔΔCT values for target genes for D12 were obtained using the wild-type as a calibrator as described previously (53). The relative expression levels of target genes in XVM2 medium were obtained by using the  method, which calculated the fold change of the transcript levels of the target genes in D12 relative to the wild-type level.
method, which calculated the fold change of the transcript levels of the target genes in D12 relative to the wild-type level.
RESULTS
Generation of galU mutants of X. citri subsp. citri.
Two nonmucoid X. citri subsp. citri mutants, F6 and D12, were selected from the X. citri subsp. citri EZ-Tn5 library. Compared to wild-type strain 306 colonies, colonies of F6 and D12 on NA plates were smaller and less viscous. However, the growth rates of the mutants in NB broth were indistinguishable from that of the wild-type strain (data not shown).
The sites of transposon insertion in the F6 and D12 mutants were determined by rescue cloning. Sequencing results indicated that EZ-Tn5 was inserted between nucleotides 235 and 236 in D12 and between nucleotides 665 to 666 in F6 downstream of the translation start site (Fig. 1A). Insertion of EZ-Tn5 into the galU gene was confirmed by PCR analysis. PCR with gene-specific primers galU-F1 and galU-R1 targeted amplification of the interior region of galU (Table 2). This resulted in a 0.84-kb amplicon when X. citri subsp. citri 306 genomic DNA was the template but in approximately 2.8-kb amplicons when F6 and D12 were the templates due to insertion of the 1.938-kb EZ-Tn5 transposon (Fig. 1B). In addition, the F6 and D12 transformants were confirmed to have a single copy of EZ-Tn5 using Southern blot analysis (Fig. 1C). Southern blot analysis showed that a DIG-labeled 675-bp Kan2 DNA fragment hybridized to a 7.3-kb band of D12 and to a 2.8-kb band of F6. The difference in the sizes of the D12 and F6 bands that hybridized resulted from differences in the locations of the transposon insertion sites and their relative distances from the restriction site of BglII, which was used for digestion of the genomic DNA, in the galU gene. For D12, EZ-Tn5 (1.938 kb) was inserted into a 5.535-kb BglII fragment containing the 5′ end of the galU gene. For F6, EZ-Tn5 (1.938 kb) was inserted into a 0.9-kb BglII fragment containing the 3′ end of the galU gene. Consequently, a 7.3-kb band and a 2.8-kb band were hybridized for D12 and F6, respectively. This confirmed that a single copy of EZ-Tn5 was inserted into the genomes of F6 and D12 (Fig. 1C). No hybridization to the genomic DNA of X. citri subsp. citri wild-type strain 306 was observed when the Kan2 probe was used.
FIG. 1.

Sequence analysis of EZ-Tn5 insertion in the galU mutants. (A) Genomic location of galU on the X. citri subsp. citri chromosome and transposon insertion sites in the galU mutants. kefB encodes a transport protein, galU encodes a UTP-glucose-1-phosphate uridylyltransferase, and XCC2293 encodes a dehydratase protein. Bg, BglII restriction site. (B) PCR analysis confirming insertion of EZ-Tn5 into the galU gene: agarose gel electrophoresis of DNA amplified using primers galU-F1 and galU-R1 targeting the interior region of the galU gene from the X. citri subsp. citri wild-type 306, D12, and F6 strains. Lane 1, Invitrogen 1 Kb Plus DNA size marker; lane 2, D12, lane 3, F6, lane 4, X. citri subsp. citri 306. (C) Southern blot of DNA of X. citri subsp. citri wild-type strain 306 and galU mutants F6 and D12 digested with BglII. The membrane was probed with a 675-bp kan-2 gene fragment that was amplified using PCR with primers Kan-F1 and Kan-R1. Wt, wild type.
galU gene involvement in polysaccharide biosynthesis.
In order to study the role of the galU gene in polysaccharide biosynthesis, the major polysaccharides of X. citri subsp. citri, EPS, CPS, and LPS, were investigated. The results for only one galU mutant, D12, are described here since there were no differences between the results for galU mutants D12 and F6. There was a significant difference in EPS production between the X. citri subsp. citri wild-type strain (1.325 mg/ml) and D12 (0.012 mg/ml). Complementation with plasmid pCGU2.1 containing the entire galU gene restored EPS production in D12 to the wild-type level (1.725 mg/ml). The empty vector pUFR053 did not affect EPS production (0.0175 mg/ml). The D12 and wild-type strains were stained using a capsule-staining kit and observed with a microscope. The X. citri subsp. citri wild-type strain was covered with capsule, while there appeared to be no capsule in the galU mutant (Fig. 2A). Complementation with plasmid pCGU2.1 restored CPS production in D12 to the wild-type level. The empty vector pUFR053 did not complement CPS production in D12 (Fig. 2A). After proteinase K treatment, the LPS pattern of the galU mutant strain was indistinguishable from that of the wild-type strain when these strains were grown in NB, M9 glucose medium, or XVM2 medium (data not shown). The mucoid phenotype of strain D12 colonies was restored to that of the wild-type colonies on NA by complementation with plasmid pCGU2.1 but not by complementation with the empty vector pUFR053 (data not shown).
FIG. 2.
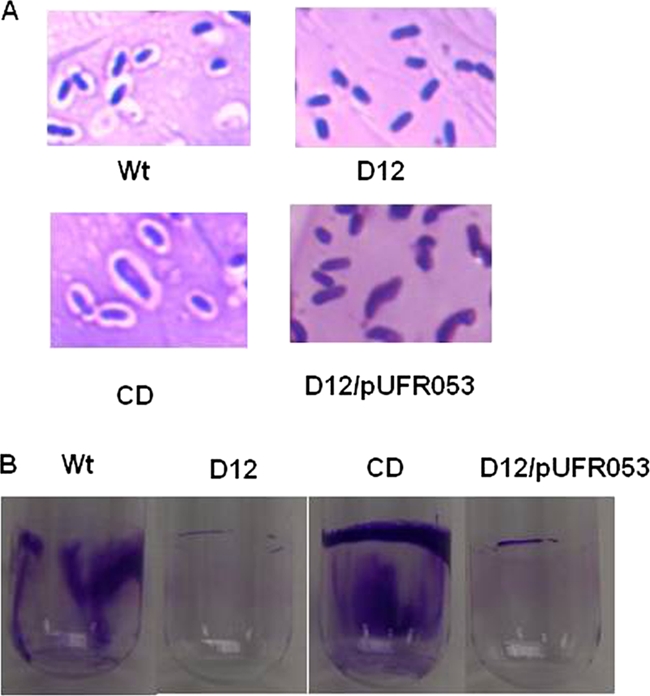
Effects of galU on the capsular polysaccharide and biofilm formation. (A) Capsule-stained X. citri subsp. citri strains observed with a light microscope (magnification, ×1,000). (B) Biofilm formation by X. citri subsp. citri strains as determined using crystal violet staining. Wt, X. citri subsp. citri wild-type strain 306; D12, galU mutant; CD, strain D12 complemented with pCGU2.1; D12/pUFR053, D12 complemented with empty vector pUFR053 without the galU gene.
galU gene involvement in biofilm formation.
Because of the significant effect of the galU gene on polysaccharide production, the galU gene was hypothesized to be involved in biofilm formation. As shown in Fig. 2B, less biofilm was observed for the galU mutant than for the wild-type strain. Ten replicates were used for quantitative measurement of biofilm. The absorbance of the crystal violet in the biofilm-staining assay for the wild-type strain (optical density at 590 nm [OD590], 0.811 ± 0.083) was 12.5 times greater than that for galU mutant strain D12 (OD590, 0.065 ± 0.011). Biofilm formation by the D12 mutant strain was restored by complementation with plasmid pCGU2.1 containing the intact galU gene (OD590, 1.753 ± 0.063) but not by complementation with the empty vector pUFR053 lacking the galU gene (OD590, 0.040 ± 0.013).
Pathogenicity assays.
Pathogenicity assays indicated that neither F6 nor D12 elicited a reaction on grapefruit, while the wild-type strain caused necrotic raised lesions typical of citrus canker on leaves when an inoculum containing a high concentration of bacteria (108 CFU/ml) was used (Fig. 3A). Similar results were obtained for an inoculum containing a lower concentration of bacteria (105 CFU/ml) (data not shown). Complementation with plasmid pCGU2.1 containing the entire galU gene restored the pathogenicity of the D12 and F6 mutants to the wild-type level and resulted in symptoms similar to those caused by the wild type on grapefruit (Fig. 3A). The empty vector pUFR053 without the galU gene did not complement the pathogenicity of either F6 and D12, and no symptoms were observed with the two mutants on grapefruit leaves (Fig. 3A).
FIG. 3.

Pathogenicity assays and growth of X. citri subsp. citri strains in planta. All strains were infiltrated into leaves with needleless syringes. (A) Host responses of cv. Duncan grapefruit inoculated with X. citri subsp. citri wild-type strain 306 (a), galU mutant D12 (b), CD (complemented mutant D12) (c), galU mutant F6 (d), CF (complemented mutant F6) (e), D12 with empty vector pUFR053 without the galU gene (f), F6 with pUFR053 without the galU gene (g), and water control (h). (B) Growth of X. citri subsp. citri wild-type strain 306, galU mutant D12, CD (complemented mutant D12), and D12 with empty vector pUFR053 in grapefruit leaves. (C) Growth of coinoculated X. citri subsp. citri wild-type strain 306 and galU mutant D12 in grapefruit leaves. □, wild-type strain 306; ▪, D12; ○, CD (complemented mutant D12); •, D12 with pUFR053. The in planta growth assays were repeated three times independently with four replicates each time, but the results of only one experiment are shown. The error bars indicate the standard errors of the means.
Effects of mutation of the galU gene on expression of key virulence genes.
Loss of pathogenicity could result from downregulation of key virulence genes. In order to test whether mutation of the galU gene affected the expression of key virulence genes, QRT-PCR assays were conducted. XVM2 medium has been reported to mimic the environment of plant intercellular spaces, and most virulence factors of Xanthomonas spp. can be induced in this medium (2, 52). Therefore, we grew D12 and the wild-type strain in XVM2 medium, removed samples at the exponential phase, and compared the levels of transcription of 15 genes using QRT-PCR. The 15 target genes included genes encoding enzymes involved in EPS and CPS biosynthesis and virulence factors, such as the type III secretion system (TTSS), effector proteins, and cell wall-degrading enzymes (CWDEs).
For QRT-PCR, the ΔCT and ΔΔCT values are appropriate subjects for statistical analysis (53). A t test was performed for the ΔCT values of the D12 and wild-type strains, and the ΔΔCT values of target genes for D12 were obtained using the wild type as a calibrator. Table 4 shows that five genes, including three CWDE genes (XAC0160, celD, and pglA), a CPS biosynthesis gene (kpsF), and a TTSS effector gene (pthA), were not expressed differently in the D12 and wild-type strains when they were grown in XVM2 medium. However, the expression of seven genes was significantly upregulated in D12 compared to the wild-type strain; these genes were two avirulence genes (avrXacE1and avrBs2), one TTSS component gene (hrcV), two TTSS regulator genes (hrpG and hrpX), one CWDE gene (peh-1), and one EPS synthesis gene (gumB). Three genes encoding CWDEs (XAC0028, pelB, and XAC0165) were significantly downregulated at the transcription level in D12 compared to the wild-type strain.
TABLE 4.
Comparison of expression of key virulence genes in the wild type and the galU mutant grown in XVM2 medium using QRT-PCR
| Gene | Mean ΔΔCTa | SD | 95% Confidence interval | P | Change (fold)b |
|---|---|---|---|---|---|
| hrpG | −1.6804 | 0.5275 | −2.3590, −1.0018 | 0.0003c | 3.2052 |
| hrpX | −2.1666 | 0.5206 | −2.8363, −1.4968 | <0.0001c | 4.4896 |
| hrcV | −2.0417 | 0.4961 | −2.6799, −1.4035 | <0.0001c | 4.1173 |
| pthA | −0.2077 | 0.3894 | −0.7086, 0.2933 | 0.3774 | 1.1548 |
| avrBs2 | −2.1722 | 0.6046 | −2.9499, −1.3944 | <0.0001c | 4.5071 |
| avrXacE1 | −2.4682 | 1.0118 | −3.7699, −1.1665 | 0.0018c | 5.5335 |
| XAC0028 | 2.2138 | 0.7257 | 1.2802, 3.1474 | 0.0004c | 0.2156 |
| XAC0160 | 0.5413 | 1.3596 | −1.2078, 1.2903 | 0.5062 | 0.6872 |
| XAC0165 | 1.2379 | 0.3362 | 0.8054, 1.6705 | <0.0001c | 0.4240 |
| peh-1 | −2.8464 | 0.9095 | −4.0164, −1.6764 | 0.0003c | 7.1920 |
| celD | 0.4748 | 1.0723 | −0.9047, 1.8542 | 0.4609 | 0.7196 |
| pglA | −0.5460 | 0.8444 | −1.6322, 0.5402 | 0.2889 | 1.4600 |
| pelB | 1.2397 | 0.2500 | 0.9182, 1.5613 | <0.0001c | 0.4235 |
| gumB | −0.9043 | 0.5975 | −1.6730, −0.1356 | 0.0255c | 1.8716 |
| kpsF | −0.3704 | 0.9612 | −1.6069, 0.8661 | 0.5196 | 1.2927 |
The mean ΔΔCT was determined using six biological replicates.
The change in expression in D12 was calculated using  .
.
Values are significantly different when P is <0.05.
In planta growth of the galU mutant.
Since mutation of the galU gene did not impair most key virulence genes of X. citri subsp. citri, the loss of pathogenicity of the galU mutant was hypothesized to be due to the lack of growth in planta. To test this hypothesis, the wild-type and D12 mutant strains were tested to determine their growth in grapefruit leaves. Although there were no differences between the wild type and the galU mutants in the ability to grow in NB (data not shown), the growth of galU mutant D12 was significantly reduced in planta compared to the growth of the wild-type strain. The concentration of the wild-type strain in planta was 4 × 108 CFU/cm2 at 10 DPI, compared to 6 × 102 CFU/cm2 for galU mutant D12 (Fig. 3B). Strain D12 with complementation plasmid pCGU2.1 containing the entire galU gene grew to the same concentration as the wild type in planta (Fig. 3B). Addition of he empty vector pUFR053 without the galU gene did not restore the growth of D12 in planta.
To determine whether the X. citri subsp. citri wild-type strain affected the growth of the galU mutant in planta, the wild-type and D12 strains were coinoculated into grapefruit leaves as described above. Coinoculation of the wild-type strain and the D12 strain did not affect the growth of D12 in planta compared to the growth of D12 alone (Fig. 3C).
DISCUSSION
Multiple genes, including galETKM, galU, and galR, are involved in formation of the sugar nucleotide precursors UDP-galactose and UDP-glucose for polysaccharide synthesis. The galETKM genes normally form an operon, while the galU and galR genes are not clustered in the genome (17). Knockout of these genes affects the outer membrane properties of bacteria and the virulence of Azospirullum brasilense (25), Klebsiella pneumoniae (7), Streptococcus pneumoniae (32), and E. coli O157:H7 (21) in hosts. In this study, a galU mutant of X. citri subsp. citri was characterized to determine the effects of a mutation on the synthesis of major polysaccharides, pathogenicity, and growth in intercellular spaces. Apparently, the galU gene in X. citri subsp. citri is involved in EPS and CPS production. This is consistent with the critical role of the UTP-glucose-1-phosphate uridylyltransferase, which is responsible for synthesis of UDP-glucose from glucose 1-phosphate and UTP and for galactose and glucose interconversion through the Leloir pathway and plays a pivotal role in carbohydrate metabolism in different organisms (16). The phenotype of the mutant apparently resulted from mutation of the galU gene rather than from malfunction of downstream genes. The galU gene is the last gene of the operon containing XAC2295 encoding one hypothetical protein, XAC2294 encoding a lipopolysaccharide core biosynthesis protein, XAC2293 encoding a dehydratase, and galU (13). The intergenic distance between the galU gene and the downstream gene kefB is 174 bp. The galU gene and the downstream kefB gene were predicted to belong to different operons based on operon prediction using SOFTBERRY (Softberry, Inc.). Thus, transposon mutation of the galU gene should not affect the function of the kefB gene (13). In addition, defects of the galU mutant in EPS production, the mucoid phenotype, pathogenicity, and growth in planta were complemented so that the levels were the same as the wild-type levels when plasmid pCGU2.1 containing an intact galU gene was used but not when the pUFR053 vector without the galU gene was used.
Mutation of the galU gene blocks EPS and CPS biosynthesis in X. citri subsp. citri. Both EPS and CPS are important components of the bacterial outer surface. Capsular polysaccharides are linked to the cell surface, while EPS molecules appear to be released onto the cell surface with no visible means of attachment and form an amorphous layer on the outer surface (39). The EPS produced by xanthomonads, xanthan, consists of repeating pentasaccharide units with the structure mannose-(β1,4)-glucuronic acid-(β-1,2)-mannose-(α-1,3)-cellobiose (24). Three sugar nucleotides, UDP-glucose, UDP-glucuronic acid, and GDP-mannose, are required precursors for EPS synthesis. UDP-glucose is the substrate for UDP-glucuronic acid synthesis and also affects the intercellular concentration of UDP-galactose (3). Mutation of the galU gene eliminated synthesis of one of the major precursors, UDP-glucose, and may also affect UDP-glucuronic acid for EPS synthesis in X. citri subsp. citri. Therefore, EPS production is deficient in the galU mutant of X. citri subsp. citri. Likewise, a deficiency of the major sugar nucleotide precursor, UDP-glucose, and possibly UDP-galactose and UDP-glucuronic acid in the galU mutant also accounts for the defect in CPS synthesis in the galU mutant. Previous studies also indicated that GalU is involved in EPS and CPS synthesis (3, 29, 31, 32).
The lack of pathogenicity of the galU mutant results from its inability to grow in planta rather than from an effect on the virulence genes. In order to determine why the galU mutant was not pathogenic, we first investigated the effect of the galU gene on the expression of genes encoding EPS and CPS biosynthesis proteins and key virulence factors, such as the type III secretion system (TTSS), effector proteins, and cell wall-degrading enzymes (CWDEs). The TTSS effector PthA is the pathogenicity determinant of X. citri subsp. citri that is required to cause canker symptoms on hosts (5). The level of expression of the pthA gene was the same in the galU mutant as it was in the wild-type strain. Overall, none of the virulence genes tested in this study showed reduced expression in the galU mutant, except for three CWDE genes whose involvement in X. citri subsp. citri virulence is still not known. Growth assays indicated that the galU mutant grew poorly in the intercellular environment (Fig. 3). This was probably due to a defect in synthesis of EPS and CPS, which have been shown to act as a barrier under stress conditions (39). Capsular polysaccharides have been reported to be essential for growth of Erwinia amylovora in planta (6). Capsule-like structures around X. citri subsp. citri were observed in infected Mexican lime and Yuzu leaves by transmission electron microscopy (30). Interestingly, coinoculation of the wild-type strain and the D12 strain did not rescue the growth of D12 in planta. E. amylovora exopolysaccharide amylovoran mutants can be rescued by wild-type strains, which presumably envelope the mutants in a biofilm (26, 27). Apparently, the mechanism by which the growth of the galU mutant was impaired in planta was different from the mechanism by which the growth of the amylovoran mutants of E. amylovora was impaired and remains to be explored.
Interestingly, GalU represents a potential target for screening antimicrobial compounds for control of citrus canker disease. Since GalU is required for X. citri subsp. citri growth in planta, antimicrobial compounds that inhibit GalU activity could potentially render X. citri subsp. citri virtually avirulent. In addition, prokaryotic UTP-glucose-1-phosphate uridylyltransferases appear to be completely unrelated to their eukaryotic counterparts and have totally different structures even though they have almost identical catalytic properties (22). This interesting finding suggests that putative antimicrobial inhibitors of GalU might not be harmful for humans and the citrus hosts.
In summary, our data provide insights into the roles of the galU gene in EPS and CPS production, as well as biofilm formation, in X. citri subsp. citri. Our results also suggest that the galU gene plays a pivotal role in X. citri subsp. citri growth in the intercellular spaces of citrus leaves.
Acknowledgments
This work was supported by USDA-CSREES Special Citrus Canker Grant Project 73402.
We thank James Graham and Jeffrey Jones for critical reading of the manuscript. We thank James Graham for help with greenhouse work.
Footnotes
Published ahead of print on 29 January 2010.
REFERENCES
- 1.Alfano, J. R., and A. Collmer. 1996. Bacterial pathogens in plants: life up against the wall. Plant Cell 8:1683-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Astua-Monge, G., J. Freitas-Astua, G. Bacocina, J. Roncoletta, S. Carvalho, and M. Machado. 2005. Expression profiling of virulence and pathogenicity genes of Xanthomonas axonopodis pv. citri. J. Bacteriol. 187:1201-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boels, I., A. Ramos, M. Kleerebezem, and W. de Vos. 2001. Functional analysis of the Lactococcus lactis galU and galE genes and their impact on sugar nucleotide and exopolysaccharide biosynthesis. Appl. Environ. Microbiol. 67:3033-3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyer, H., and D. Roulland-Dussoix. 1969. A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41:459-472. [DOI] [PubMed] [Google Scholar]
- 5.Brunings, A. M., and D. W. Gabriel. 2003. Xanthomonas citri: breaking the surface. Mol. Plant Pathol. 4:141-157. [DOI] [PubMed] [Google Scholar]
- 6.Bugert, P., and K. Geider. 1995. Molecular analysis of the ams operon required for exopolysaccharide synthesis of Erwinia amylovora. Mol. Microbiol. 15:917-933. [DOI] [PubMed] [Google Scholar]
- 7.Chang, H. Y., J. H. Lee, W. L. Deng, T. F. Fu, and H. L. Peng. 1996. Virulence and outer membrane properties of a galU mutant of Klebsiella pneumoniae CG43. Microb. Pathog. 20:255-261. [DOI] [PubMed] [Google Scholar]
- 8.Clowes, R. C., and W. Hayes. 1968. Experiments in microbial genetics. Blackwell Scientific Publications, Oxford, United Kingdom.
- 9.Csonka, L. N., and W. Epstein. 1996. Osmoregulation, p. 1210-1223. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 10.Cubero, J., and J. H. Graham. 2002. Genetic relationship among worldwide strains of Xanthomonas causing canker in citrus species and design of new primers for their identification by PCR. Appl. Environ. Microbiol. 68:1257-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniels, M., C. Barber, P. Turner, M. Sawczyc, R. Byrde, and A. Fielding. 1984. Cloning of genes involved in pathogenicity of Xanthomonas campestris pv. campestris using the broad host range cosmid pLAFR1. EMBO J. 3:3323-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das, A. K. 2003. Citrus canker—a review. J. Appl. Hortic. 5:52-60. [Google Scholar]
- 13.da Silva, A. C., J. A. Ferro, F. C. Reinach, C. S. Farah, L. R. Furlan, R. B. Quaggio, C. B. Monteiro-Vitorello, M. A. Van Sluys, N. F. Almeida, L. M. Alves, A. M. do Amaral, M. C. Bertolini, L. E. Camargo, G. Camarotte, F. Cannavan, J. Cardozo, F. Chambergo, L. P. Ciapina, R. M. Cicarelli, L. L. Coutinho, J. R. Cursino-Santos, H. El-Dorry, J. B. Faria, A. J. Ferreira, R. C. Ferreira, M. I. Ferro, E. F. Formighieri, M. C. Franco, C. C. Greggio, A. Gruber, A. M. Katsuyama, L. T. Kishi, R. P. Leite, E. G. Lemos, M. V. Lemos, E. C. Locali, M. A. Machado, A. M. Madeira, N. M. Martinez-Rossi, E. C. Martins, J. Meidanis, C. F. Menck, C. Y. Miyaki, D. H. Moon, L. M. Moreira, M. T. Novo, V. K. Okura, M. C. Oliveira, V. R. Oliveira, H. A. Pereira, A. Rossi, J. A. Sena, C. Silva, R. F. de Souza, L. A. Spinola, M. A. Takita, R. E. Tamura, E. C. Teixeira, R. I. Tezza, M. Trindade dos Santos, D. Truffi, S. M. Tsai, F. F. White, J. C. Setubal, and J. P. Kitajima. 2002. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417:459-463. [DOI] [PubMed] [Google Scholar]
- 14.Ditta, G., S. Stanfield, D. Corbin, and D. Helinski. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. U. S. A. 77:7347-7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Yacoubi, B., A. Brunings, Q. Yuan, S. Shankar, and D. Gabriel. 2007. In planta horizontal transfer of a major pathogenicity effector gene. Appl. Environ. Microbiol. 73:1612-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frey, P. A. 1996. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 10:461-470. [PubMed] [Google Scholar]
- 17.Genevaux, P., P. Bauda, M. DuBow, and B. Oudega. 1999. Identification of Tn10 insertions in the rfaG, rfaP, and galU genes involved in lipopolysaccharide core biosynthesis that affect Escherichia coli adhesion. Arch. Microbiol. 172:1-8. [DOI] [PubMed] [Google Scholar]
- 18.Gottwald, T. R., J. H. Graham, and T. S. Schubert. 2002. Citrus canker: the pathogen and its impact. Plant Health Prog. doi: 10.1094/PHP-2002-0812-01-RV. [DOI]
- 19.Graham, J. H., T. R. Gottwald, J. Cubero, and D. S. Achor. 2004. Xanthomonas axonopodis pv. citri: factors affecting successful eradication of citrus canker. Mol. Plant Pathol. 5:1-15. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 21.Ho, T. D., and M. K. Waldor. 2007. Enterohemorrhagic Escherichia coli O157:H7 gal mutants are sensitive to bacteriophage P1 and defective in intestinal colonization. Infect. Immun. 75:1661-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hossain, S., K. Tanizawa, Y. Kazuta, and T. Fukui. 1994. Overproduction and characterization of recombinant UDP-glucose pyrophosphorylase from Escherichia coli K-12. J. Biochem. 115:965-972. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs, M., A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond, R. Levy, L. Chun-Rong, D. Guenthner, D. Bovee, M. Olson, and C. Manoil. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:14339-14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jansson, P., L. Kenne, and B. Lindberg. 1975. Structure of extracellular polysaccharide from Xanthomonas campestris. Carbohydr. Res. 45:275-282. [DOI] [PubMed] [Google Scholar]
- 25.Jofre, E., A. Lagares, and G. Mori. 2004. Disruption of dTDP-rhamnose biosynthesis modifies lipopolysaccharide core, exopolysaccharide production, and root colonization in Azospirillum brasilense. FEMS Microbiol. Lett. 231:267-275. [DOI] [PubMed] [Google Scholar]
- 26.Koczan, J., M. McGrath, G. W. Sundin, and Y. Zhao. 2008. Biofilm formation in Erwinia amylovora: implications in pathogenicity, p. 67-71. In K. B. Johnson and V. O. Stockwell (ed.), ISHS Acta Horticulturae 793. XI International Workshop on Fire Blight. ISHS, Leuven, Belgium.
- 27.Koczan, J., M. McGrath, Y. Zhao, and G. W. Sundin. 2009. Contribution of Erwinia amylovora exopolysaccharides amylovoran and levan to biofilm formation: implications in pathogenicity Phytopathology 99:1237-1244. [DOI] [PubMed] [Google Scholar]
- 28.Komeda, Y., T. Icho, and T. Iino. 1977. Effects of galU mutation on flagellar formation in Escherichia coli. J. Bacteriol. 129:908-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai, Y., H. Peng, and H. Chang. 2001. Identification of genes induced in vivo during Klebsiella pneumoniae CG43 infection. Infect. Immun. 69:7140-7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, I., K. Kim, J. Hyun, Y. Lee, and E. Park. 2009. Comparative ultrastructure of nonwounded Mexican lime and Yuzu leaves infected with the citrus canker bacterium Xanthomonas citri pv. citri. Microsc. Res. Tech. 72:507-516. [DOI] [PubMed] [Google Scholar]
- 31.Mollerach, M., and E. García. 2000. The galU gene of Streptococcus pneumoniae that codes for a UDP-glucose pyrophosphorylase is highly polymorphic and suitable for molecular typing and phylogenetic studies. Gene 260:77-86. [DOI] [PubMed] [Google Scholar]
- 32.Mollerach, M., R. Lopez, and E. Garcia. 1998. Characterization of the galU gene of Streptococcus pneumoniae encoding a uridine diphosphoglucose pyrophosphorylase: a gene essential for capsular polysaccharide biosynthesis. J. Exp. Med. 188:2047-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesper, J., D. Kapfhammer, K. Klose, H. Merkert, and J. Reidl. 2000. Characterization of Vibrio cholerae O1 antigen as the bacteriophage K139 receptor and identification of IS1004 insertions aborting O1 antigen biosynthesis. J. Bacteriol. 182:5097-5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nesper, J., C. Lauriano, K. Klose, D. Kapfhammer, A. Kraiss, and J. Reidl. 2001. Characterization of Vibrio cholerae O1 El tor galU and galE mutants: influence on lipopolysaccharide structure, colonization, and biofilm formation. Infect. Immun. 69:435-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osbourn, A. E. 1996. Preformed antimicrobial compounds and plant defense against fungal attack. Plant Cell 8:1821-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pratt, L., and R. Kolter. 1998. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol. Microbiol. 30:285-293. [DOI] [PubMed] [Google Scholar]
- 37.Priebe, G., C. Dean, T. Zaidi, G. Meluleni, F. Coleman, Y. Coutinho, M. Noto, T. Urban, G. Pier, and J. Goldberg. 2004. The galU gene of Pseudomonas aeruginosa is required for corneal infection and efficient systemic spread following pneumonia but not for infection confined to the lung. Infect. Immun. 72:4224-4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rioux, S., C. Galarneau, J. Harel, J. Frey, J. Nicolet, M. Kobisch, J. Dubreuil, and M. Jacques. 1999. Isolation and characterization of mini-Tn10 lipopolysaccharide mutants of Actinobacillus pleuropneumoniae serotype 1. Can. J. Microbiol. 45:1017-1026. [DOI] [PubMed] [Google Scholar]
- 39.Roberts, I. 1996. The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu. Rev. Microbiol. 50:285-315. [DOI] [PubMed] [Google Scholar]
- 40.Rybak, M., G. V. Minsavage, R. E. Stall, and J. B. Jones. 2009. Identification of Xanthomonas citri ssp. citri host specificity genes in a heterologous expression host. Mol. Plant Pathol. 10:249-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salama, N. R., and C. Manoil. 2006. Seeking completeness in bacterial mutant hunts. Curr. Opin. Microbiol. 9:307-311. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 43.Sandlin, R., K. Lampel, S. Keasler, M. Goldberg, A. Stolzer, and A. Maurelli. 1995. Avirulence of rough mutants of Shigella flexneri: requirement of O antigen for correct unipolar localization of IcsA in the bacterial outer membrane. Infect. Immun. 63:229-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaad, N., E. Postnikova, G. Lacy, A. Sechler, I. Agarkova, P. Stromberg, V. Stromberg, and A. Vidaver. 2006. Emended classification of xanthomonad pathogens on citrus. Syst. Appl. Microbiol. 29:690-695. [DOI] [PubMed] [Google Scholar]
- 45.Segura, A., M. Moreno, F. Madueno, A. Molina, and F. Garcia-Olmedo. 1999. Snakin-1, a peptide from potato that is active against plant pathogens. Mol. Plant-Microbe Interact. 12:16-23. [DOI] [PubMed] [Google Scholar]
- 46.Stimson, E., M. Virji, K. Makepeace, A. Dell, H. Morris, G. Payne, J. Saunders, M. Jennings, S. Barker, and M. Panico. 1995. Meningococcal pilin: a glycoprotein substituted with digalactosyl 2,4-diacetamido-2,4,6-trideoxyhexose. Mol. Microbiol. 17:1201-1214. [DOI] [PubMed] [Google Scholar]
- 47.Swarup, S., R. De Feyter, R. H. Brlansky, and D. W. Gabriel. 1991. A pathogenicity locus from Xanthomonas citri enables strains from several pathovars of Xanthomonas campestris to elicit canker-like lesions on citrus. Phytopathology 81:802-809. [Google Scholar]
- 48.Vauterin, L., B. Hoste, K. Kersters, and J. Swings. 1995. Reclassification of Xanthomonas. Int. J. Syst. Bacteriol. 45:472-489. [Google Scholar]
- 49.Vilches, S., R. Canals, M. Wilhelms, M. T. Salo, Y. A. Knirel, E. Vinogradov, S. Merino, and J. M. Tomas. 2007. Mesophilic Aeromonas UDP-glucose pyrophosphorylase (GalU) mutants show two types of lipopolysaccharide structures and reduced virulence. Microbiology 153:2393-2404. [DOI] [PubMed] [Google Scholar]
- 50.Viloria, Z., D. L. Drouillard, J. H. Graham, and J. W. Grosser. 2004. Screening triploid hybrids of ‘Lakeland’ limequat for resistance to citrus canker. Plant Dis. 88:1056-1060. [DOI] [PubMed] [Google Scholar]
- 51.Vojnov, A., A. Zorreguieta, J. Dow, M. Daniels, and M. Dankert. 1998. Evidence for a role for the gumB and gumC gene products in the formation of xanthan from its pentasaccharide repeating unit by Xanthomonas campestris. Microbiology 144:1487-1493. [DOI] [PubMed] [Google Scholar]
- 52.Wengelnik, K., C. Marie, M. Russel, and U. Bonas. 1996. Expression and localization of HrpA1, a protein of Xanthomonas campestris pv. vesicatoria essential for pathogenicity and induction of the hypersensitive reaction. J. Bacteriol. 178:1061-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan, J., A. Reed, F. Chen, and C. J. Stewart. 2006. Statistical analysis of real-time PCR data. BMC Bioinformatics 7:85. [DOI] [PMC free article] [PubMed] [Google Scholar]