

Summary
Background
Recent clinical studies suggest that the prophylactic use of recombinant factor VIIa (rFVIIa) markedly reduces the number of bleeding episodes in hemophilic patients with inhibitors. Given the short biological half-life of rFVIIa, it is unclear how rFVIIa could be effective in prophylactic treatment.
Objectives
To examine the extravascular distribution of pharmacologically administered rFVIIa to obtain clues on how rFVIIa could work in prophylaxis.
Methods
Recombinant mouse FVIIa tagged with AF488 fluorophore (AF488-FVIIa) was administered into mice via the tail vein. At different time intervals following the administration, mice were exsanguinated and various tissues were collected. The tissue sections were processed for immunohistochemistry to evaluate distribution of rFVIIa.
Results
rFVIIa, immediately following the administration, associated with the endothelium lining of large blood vessels. Within 1 h, rFVIIa bound to endothelial cells was transferred to the perivascular tissue surrounding the blood vessels and thereafter diffused throughout the tissue. In the liver, rFVIIa was localized to sinusoidal capillaries and accumulated in hepatocytes. In bone, rFVIIa was accumulated in the zone of calcified cartilage and some of it was retained there for a week. The common finding of the present study is that rFVIIa in extravascular spaces was mostly localized to regions that contain TF expressing cells.
Conclusions
The present study demonstrates that pharmacologically administered rFVIIa readily associates with the vascular endothelium and subsequently enters into extravascular spaces where it is likely to bind to TF and is retained for extended time periods. This may explain the prolonged pharmacological effect of rFVIIa.
Keywords: bio-distribution, endothelial cell protein C receptor, hemophilia, prophylaxis, rFVIIa, tissue factor
Introduction
Recombinant factor VIIa (rFVIIa) has been demonstrated to be an effective hemostatic agent for treatment of severe hemophilia patients with inhibitors against FVIII or FIX [1]. Recent reports suggest that regular administrations of rFVIIa may also prevent the development of mild–moderate joint bleeds [2–8]. In a randomized, multicenter, double-blinded prospective clinical trial involving 37 patients with severe hemophilia with inhibitors, it was found that daily injection of rFVIIa for 3 months decreased significantly the number of mild–moderate joint bleeds as compared with the 3 months of pretreatment observation time in the same patients [8]. Moreover, in a 3-month follow-up period, the number of hemorrhagic episodes was less than that observed in the pretreatment period [8]. The question arises as to why regular administration once daily of rFVIIa reduces the number of hemorrhagic events in the post-treatment period, given the short biological half-life of rFVIIa in the circulation. In this context it seemed of interest to study the bio-distribution of pharmacologically administered rFVIIa. By doing so it might be possible to identify a potential locale for retaining functional rFVIIa for a longer period of time than indicated by the plasma half-life. There is ample evidence for the presence of many coagulation proteins in the extravascular space [9,10]. Earlier studies demonstrated the presence of functional FVIIa-TF complexes in the extravascular tissue in an umbilical vein model system [11,12]. More recently, FVII-TF complexes were demonstrated around dermal vessels in the absence of injury [13]. A low recovery (approximately 50%) of intravenously injected rFVIIa also suggests a large extravascular distribution volume for rFVIIa [14].
Earlier studies of characterization of rFVIIa distribution, where 125I-rFVIIa was administered to rats and rFVIIa accumulation was examined by whole body autoradiography, revealed that the rFVIIa was associated with various tissues (e.g. lung, liver, kidney and spleen) at 30 min following the intravenous injection of 125I-rFVIIa. The concentration of 125I-rFVIIa declined sharply during 6 h after dosing and little radioactivity was detected at 24 h, suggesting that rFVIIa was rapidly excreted [15]. In contrast to other tissues, relatively large amounts of 125I-rFVIIa were sequestered in mineralized bone, where it released more slowly [15,16]. These earlier studies were limited mostly to 24 h following rFVIIa administration. Moreover, rFVIIa distribution was evaluated by whole-body autoradiography or counting the radioactivity, which provided no information on how rFVIIa was associated with specific tissues and cell types that are involved in rFVIIa metabolism. To understand potential mechanisms involved in rFVIIa-induced secondary prophylaxis, it is important to know the pharmacokinetics of rFVIIa at the cellular level. As a first step towards an increased understanding of potential mechanisms for a prolonged effect of intravenously injected rFVIIa, the present study has been carried out to characterize bio-distribution of rFVIIa following a single dose of rFVIIa.
Materials and methods
Reagents
Mouse rFVIIa and polyclonal antibodies against mouse TF were obtained from Novo Nordisk, Denmark. Mouse rFIX was purchased from Hematological Technologies (Essex Junction, VT, USA). Antibodies against mouse endothelial cell protein C receptor (EPCR) were kindly provided by Charles Esmon (Oklahoma Medical Research Foundation, Oklahoma City, OK, USA). The Alexa Fluor-488 (AF488) microscale protein labeling kit and antibodies specific to AF488 were obtained from Molecular Probes (Invitrogen Corporation, Carlsbad, CA, USA). Antigen retrieval solution, secondary antibodies and substrates used for immunohistochemistry were obtained from Dako North America, Inc (Carpenteria, CA, USA).
Labeling of mouse rFVIIa and rFIX with AF488
Mouse rFVIIa was labeled with AF488 using the AF488 microscale protein labeling kit and following the manufacturer's instructions. Briefly, 100 μg of rFVIIa (at a concentration of 1 mg mL−1) were incubated with 50-fold molar excess of AF488 TFP ester for 15 min at room temperature. Free AF488 molecules were removed from the conjugate by passing it through a Bio-Gel P-6 fine resin column. The degree of labeling, as determined by following the manufacturer's instructional manual, is varied 3–5. Mouse rFIX was labeled with AF488 in an identical manner.
Administration of FVIIa to mice
The AF488-FVIIa (or AF488-FIX) was administered into anesthetized male C57BL/6 mice via the tail vein at a dose of 120 μg kg−1 bw in 100 μL of saline followed by flushing with 100 μL of saline. At different time intervals following AF488-FVIIa administration (from 10 min to 7 days), mice were exsanguinated by flushing saline through the heart and draining the blood by severing the renal artery. Various organs (e.g. the skin, lung, liver, kidney, brain, skeletal muscles and bone joints) were collected and fixed in Excel fixative (American Master*Tech Scientific Inc., Lodi, CA, USA). Bone joints, after the fixation, were decalcified in the EDF decal solution (Statlab Medical Products, McKinney, TX, USA). In selected experiments, blood (∼100 μL) was drawn into citrate anticoagulant from the orbital sinus before exsanguinating mice to measure AF488-FVIIa levels in circulation. Typically three to four mice were used for each time interval to evaluate FVIIa bio-distribution. For control studies, two to three mice were used for each time interval to evaluate FIX bio-distribution.
FVIIa activity and antigen assays
AF488-FVIIa and unmodified FVIIa enzymatic activity was measured in a factor X activation assay using WI-38 lung fibroblasts as a source for TF. To compare the binding of AF488-FVIIa and unmodified FVIIa to TF and EPCR, mouse lung embryonic fibroblasts and CHO cell stably transfected with EPCR, respectively, were cultured in a 96-well plate (1 × 104 cells/well). The confluent monolayers were incubated with varying concentrations of AF488-FVIIa or unmodified FVIIa for 3 h at 4 °C in calcium containing HEPES buffer (10 mm HEPES, 0.15 m NaCl, 4 mm KCl, 11 mm glucose, pH 7.5 buffer containing 5 mm CaCl2 and 1 mg ml−1 BSA). At the end of 3 h incubation, unbound FVIIa was removed, the cells were washed thrice with the buffer and the bound FVIIa was fixed with 4% paraformaldehyde for 30 min at 4 °C. The amount of FVIIa bound to the cells was determined in an ELISA using biotinylated rabbit anti-mouse FVIIa, followed by alkaline phosphatase-conjugated streptavidin and color development with BluePhos phosphatase substrate (KPL, Gaithersburg, MD, USA). To measure AF488-FVIIa levels, rabbit anti-AF488 IgG (10 μg ml−1) was coated onto a 96-well plate overnight at 4 °C. After blocking the wells with 0.1% gelatin, diluted plasma samples or AF488-FVIIa standards were added to the wells. AF488-FVIIa captured on the primary antibody was measured using biotinylated rabbit anti-mouse FVIIa as described the above. The lower detection limit of the assay was 1 ng ml−1 AF488-FVIIa.
Immunohistochemistry
After overnight fixation, the tissues were processed using graded alcohol and xylene and then embedded in paraffin using standard procedures. Thin sections (5 μm) were cut and deparaffinized. Rehydrated sections were processed for antigen retrieval using Dako Antigen Retrieval Solution. For bone joints, antigen retrieval was done by incubating the sections in 2 N HCl (pH 0.6–0.9) for 10 min. Tissue peroxidases were inactivated by treating the tissue sections with 3% H2O2 for 10 min. The sections were then blocked with antibody diluent solution containing 1% BSA. The sections were stained with control IgG, anti-AF488 IgG, anti-mouse TF IgG or anti-mouse EPCR IgG (diluted 1:250 to 1:1000 or 10 μg ml−1) in the blocking buffer. After rinsing the slides to remove excessive primary antibodies, tissue sections were labeled with biotinylated secondary antibodies followed by streptavidin-HRP using the Biotinylated Link Antibody Kit (Dako). Finally, the sections were stained with AEC substrate chromogen and counterstained with hematoxylin followed by mounting with aqueous mounting media. The stained sections were viewed under an Olympus BX50 microscope and photographed using an Olympus UCMAD3 camera and Picture Frame software. All sections were photographed using the same exposure settings.
Results
Our original objective was to investigate the in vivo distribution of rFVIIa administered to mice by monitoring the fluorescence of AF488 that was conjugated to rFVIIa. However, mouse tissue specimens exhibited high intrinsic fluorescence when excited at 488 nm, which made it difficult to evaluate objectively the distribution of AF488-FVIIa. Tissue sections also emitted intrinsic fluorescence when excited at other wavelengths, and therefore use of other fluorophores to tag rFVIIa was not considered. We then attempted to use frozen tissue sections to monitor the distribution of AF488-FVIIa by fluorescence microscopy. Although the intensity of intrinsic fluorescence of frozen tissues was lower than fixed tissues, there was still interference in the visualization of AF488-FVIIa distribution. Therefore, we have monitored the distribution of AF488-FVIIa by immunohistochemistry using antibodies that specifically detect AF488 of the AF488-FVIIa conjugate. The specificity of the antibodies to AF488 was confirmed in initial experiments by demonstrating the lack of staining in tissue sections from mice injected with saline or FVIIa, but not tagged with AF488. Immunostaining of AF488 in tissue sections reflects the staining of AF488-FVIIa protein conjugate and not free AF488, as free AF488 would be excreted from mice rapidly. Consistent with this, tissue sections from mice that received only free AF488 did not stain positively with AF488 antibodies (data not shown).
Characterization of AF488-FVIIa
To document whether AF488-modified rFVIIa behaves very similarly to unmodified rFVIIa, we evaluated the functionality of AF488-FVIIa in various assays. Analysis of FVIIa enzymatic activity in FX activation assay showed that AF488-FVIIa retained about 70–80% of the activity of unmodified rFVIIa. Cell binding assays, using mouse lung embryonic fibroblasts that constitutively express TF and CHO cells stably transfected with EPCR, showed that AF488-FVIIa bound to these cells in a similar profile and to a similar extent as that of unmodified rFVIIa (data not shown). Finally, AF488-FVIIa administered to mice was cleared from the circulation with t1/2 near to 29 min, which was very similar to that reported using 125I-labeled mouse or human rFVIIa (30 min) [17]. No AF488-FVIIa was detected in the plasma at 24 h following the administration of AF488-FVIIa.
Distribution of rFVIIa in the skin
Marked differences in the distribution of rFVIIa were observed in the skin at different time intervals following the administration of AF488-FVIIa (Fig. 1A,B). Injected AF488-FVIIa conjugate initially (at 10 min) accumulated within the cytoplasm of cuboidal epithelial cells of the sebaceous glands beside hair follicles. Accumulation of AF488-FVIIa in the skin was more prominent between 30 min and 3 h following the administration of AF488-FVIIa (Fig. 1A,B). AEC staining, which represented the localization of injected AF488-FVIIa conjugate, was observed during these periods in the squamous epithelial cells of the epidermis (red arrows), as well as in the dermal region of the skin. Within the dermal region, the hair follicles and sebaceous glands were profusely stained. In addition to the bulbs of the hair follicle, collagen fibers (CF) in the dermal area were also stained. The connective tissue layers of the skin, including blood vessels in the hypodermal area, also demonstrated positive immunostaining for AF488 (Fig. 1B). Within 6–72 h after AF488-FVIIa administration, the accumulation of rFVIIa in the aforementioned regions was reduced (Fig. 1A). The accumulated rFVIIa disappeared faster from dermal connective tissue and hypodermal blood vessel areas than from the epidermal and hair follicle areas (Fig. 1B). By day 7, almost all of the rFVIIa accumulated in the skin was excreted/degraded as we observed no staining of AF488-FVIIa in the skin tissue at this time. Relatively light staining for AF488-FVIIa can be observed in epithelial cells of the hair follicle or epidermis 7 days after the administration of the drug (Fig. 1A). Comparable skin tissue sections were also immunostained with anti-TF antibodies. TF staining was observed in the epithelium, hair follicles and subcutaneous hypodermal region. No differences in the TF staining were observed following FVIIa administration (see Fig. S1).
Fig. 1.
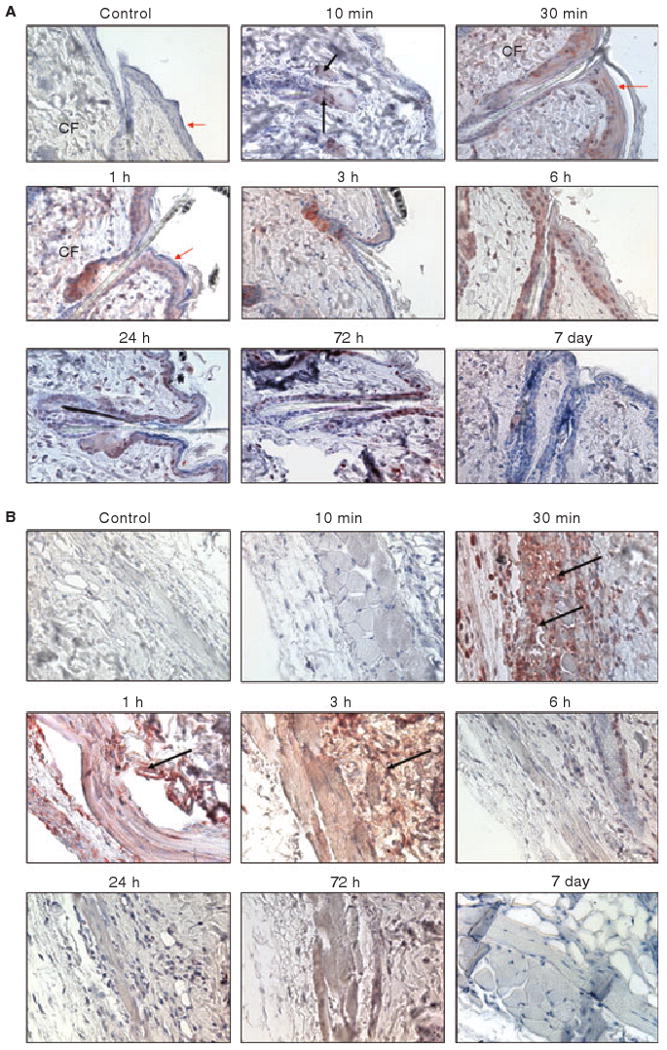
Immunostaining of skin tissues for rFVIIa in mice at different time intervals following the administration of a single pharmacological dose of rFVIIa (AF488-FVIIa). The sections were stained with anti-AF488 antibodies. Panel A, epidermis and upper dermis region; Panel B, lower dermis region. CF, collagen fibers. Red arrows point out squamous epithelial cells whereas black arrows point out hair follicles. Magnification, 400×. In this and other figures, control IgG staining was carried out with tissue sections of mice given AF488-FVIIa (30-min time period) or saline, where appropriate.
At higher magnification (1000×), we observed differential staining in the blood vessels within the hypodermal region of the skin (Fig. 2). Ten minutes after the administration of AF488-FVIIa, faint staining of AF488 was noted on the endothelial cells lining blood vessels. Intense staining of AF488 was prominent at 30 min, at which time the staining was mostly limited to endothelial cells and the sub-endothelial cell matrix. At 3 h, the rFVIIa distribution was observed in the endothelium and also in the surrounding perivascular tissue. Thereafter, the intensity of rFVIIa staining was decreased and diffused throughout the connective tissue surrounding the blood vessel. No evidence of AF488-FVIIa was observed in the endothelium or perivascular tissues on day 7. Comparable sections were also immunostained with anti-mTF and anti-mEPCR antibodies. As expected, EPCR antibodies exclusively stained the endothelium lining blood vessels, whereas TF antibodies stained pericytes and smooth muscle cells surrounding the blood vessel but not the endothelium. We found no significant differences in the intensity and distribution of TF and EPCR in blood vessels following the administration of AF488-FVIIa (data not shown).
Fig. 2.
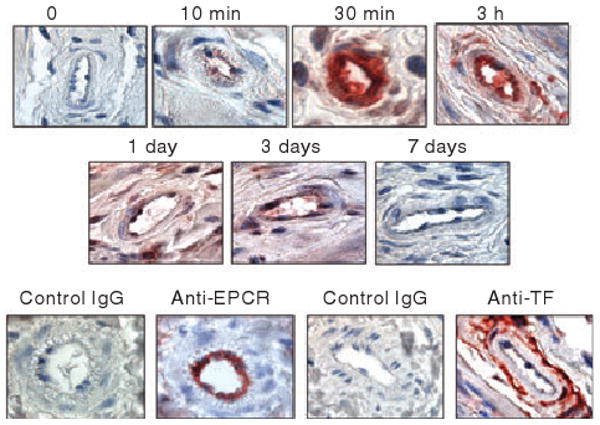
Endothelial and perivascular distribution of rFVIIa. Skin sections of mice administered with AF488-FVIIa were immunostained with AF488 antibodies and blood vessels were examined (1000×) to monitor the distribution of rFVIIa. In the bottom panel, the sections were stained with matching control IgGs, anti-EPCR mAb or anti-TF IgG.
Distribution of rFVIIa in the liver
The endothelial lining of the portal vein showed distinguishable staining of AF488-FVIIa at 10 min following the administration. Between 10 min and 6 h following the administration, sinusoids demonstrated significant staining of AF488-FVIIa (Fig. 3A). During this period, prominent and diffused cytoplasmic staining of AF488-FVIIa was also observed in hepatocytes. This staining was more intense in hepatocytes surrounding the portal vein compared with distal hepatocytes away from the portal vein. AF488-FVIIa staining was clearly visible in tissue sections from day 1 and very faint in sections obtained at 3 and 7 days post-administration of AF488-FVIIa.
Fig. 3.
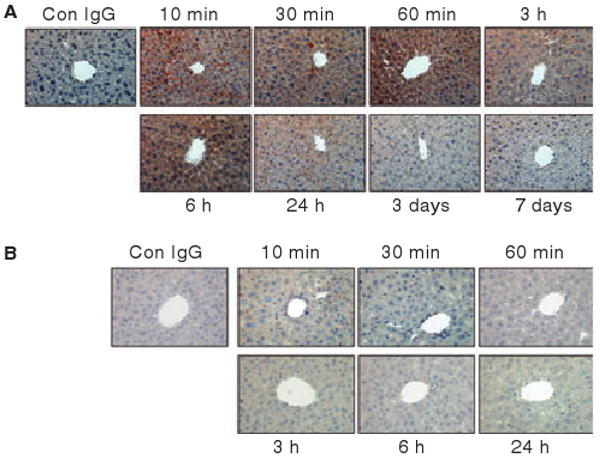
Immunostaining of liver sections with anti-AF488 antibodies of mice administered with AF488-FVIIa (panel A) or AF488-FIX (panel B). In panel A, the antibodies stained the portal vein, sinusoidal capillaries and hepatocytes. In panel B, only Kupffer cells were stained positively for AF488. Magnification, 400×.
To test whether the diffused staining of FVIIa observed reflects the specific binding and accumulation of FVIIa in the hepatocytes or a general catabolism of vitamin K-dependent clotting factors in the liver, mice were injected with AF488-FIX instead of AF488-FVIIa, and the liver sections were immunostained with AF488 antibodies. Although the sections were stained positively with AF488 antibodies, the pattern of AF488-FIX staining was completely different from that observed with liver sections of mice injected with AF488-FVIIa. The staining appeared punctuated and limited to the Kupffer cells (Fig. 3B). AF488-FIX staining was absent in sinusoids. Hepatocytes were mostly negative to AF488-FIX staining.
Distribution of rFVIIa in bone joints/bone marrow
At lower magnification (40×), synovial and the mineralized bone regions were stained for AF488-FVIIa in joint sections from mice administered rFVIIa (see Fig. S2). At higher magnification, it was found that rFVIIa staining was mostly localized to the zone of calcified cartilage within the growth plate region of the joints (Fig. 4A). The staining intensity of AF488-FVIIa was high and persisted until 24 h. Although the staining intensity of this region was decreased in tissue sections obtained on the 3rd and 7th day post AF488-FVIIa administration, rFVIIa staining was clearly visible even in tissue specimens collected on day 7 (Fig. 4A). Earlier studies in which rats were given 125I-labelled rFVIIa, Gla-domainless rFVIIa or FIX, suggested that accumulation of FVIIa in the bone was dependent on the Gla domain, and FIX, similar to rFVIIa, can also accumulate in the bone [16]. It is unclear from these studies whether any qualitative differences exist in the distribution of rFVIIa and FIX in mineralized bone. To investigate this, we immunostained the bone joint sections prepared from mice injected with AF488-FIX (Fig. 4B). Similar to FVIIa, FIX was found in the zone of calcified cartilage. Interestingly, FIX disappeared from the region by 24 h, whereas FVIIa persisted for a much longer time. We also immunostained tissue sections of bone joints collected from mice administered AF488-VIIa with anti-TF antibodies. The zone of calcified cartilage in the growth plate region stained intensely positive for anti-TF antibodies; no change in TF staining intensity or the pattern of staining was observed in mice that received rFVIIa (see Fig. S3).
Fig. 4.
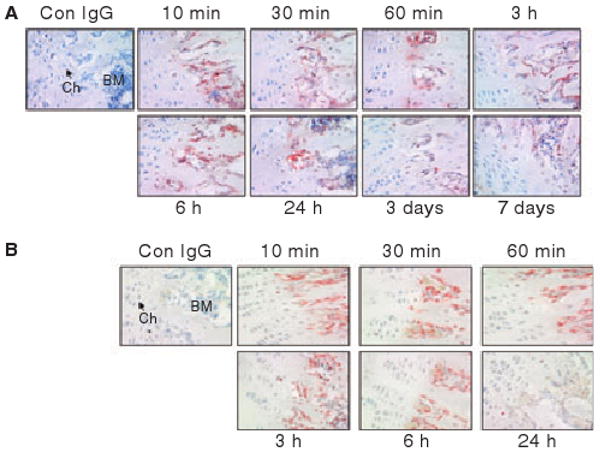
Immunostaining of bone joint sections with anti-AF488 antibodies of mice administered with AF488-FVIIa (panel A) or AF488-FIX (panel B). Ch, chondrocytes; BM, bone marrow. Magnification, 400×.
We also examined the association of AF488-FVIIa with blood vessels in bone joint regions. Blood vessels in the bone joint regions of mice collected 30 min post-FVIIa infusion stained intensely positive for rFVIIa (Fig. S4). FVIIa staining was limited to the endothelium and sub-endothelium. Tissue specimens from mice collected at 1 h and 6 h post-infusion also stained positively for AF488; however, the intensity of staining was very low. AF488-FVIIa was not detected in blood vessels of mice collected at day 7 after the administration. As expected, perivascular regions of blood vessels stained intensely positive for TF, and the intensity of TF staining was unaltered following FVIIa administration. In contrast to rFVIIa, rFIX administered to mice did not associate with endothelial cells lining blood vessels (see Fig. S5).
In addition to the zone of calcified cartilage, FVIIa also accumulated in the bone marrow. Bone marrow in mice collected at 10 min to 24 h post AF488-FVIIa administration stained intensely positive for AF488-FVIIa (Fig. 5A). Although the staining intensity was somewhat decreased in bone marrow tissue specimens from day 3 and day 7, AF488-FVIIa staining in the bone marrow was clearly evident even on day 7, suggesting that FVIIa accumulated in the bone marrow was retained for longer time periods. Within the bone marrow, megakaryocytes stained intensely positive for FVIIa. Factor IX given to mice also accumulated in the bone marrow; however, FIX appeared to be cleared from bone marrow much faster then FVIIa as we found little AF488-FIX staining in bone marrow sections of post day 1 samples (Fig. 5B). The bone marrow consistently stained positive to TF. Megakaryocytes in the bone marrow were stained intensely positive to TF. No detectable changes were found in TF expression levels following rFVIIa administration (see Fig. S6). Certain cell populations in bone marrow were also stained positive for EPCR (data not shown). We attempted to immunostain the serially sectioned specimens of bone marrow for FVIIa, TF and EPCR in order to determine whether FVIIa in the bone marrow was associated with TF and/or EPCR expressing cells. However, we were unable to obtain serial sections in a reproducible manner as a result of the lack of uniformity in bone marrow cells in serial sections and detachment of samples from the slide during processing.
Fig. 5.
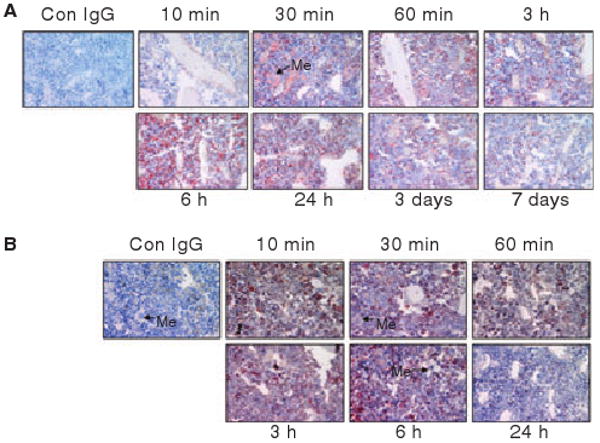
Accumulation of rFVIIa in bone marrow at different time points after a single dose of rFVIIa. Bone sections (from the joint region) of mice administered AF488-FVIIa (panel A) or AF488-FIX (panel B) were stained with anti-AF488 antibodies. Me, megakaryocyte. Magnification, 400×.
Distribution of rFVIIa in the kidney
In the kidney, differential detection of AF488-FVIIa was observed at different time intervals following AF488-FVIIa administration. At 10 min after the injection, rFVIIa was found to localize within the glomerular capillaries consisting of endothelial cells from afferent and efferent arterioles and podocytes (Fig. 6). Intense FVIIa distribution was also noticed in the cross-sections of proximal and distal convoluted tubules, which are lined by cuboidal epithelial cells. Similar staining was observed after 30 min and 1 h following rFVIIa injection. The staining intensity was reduced or negative in glomerular capillaries between 3 h and 72 h post-injection; however, AF488-FVIIa staining in the kidney tubules remained positive during this time frame. After the 7th day of FVIIa administration, the kidney sections showed no evidence for the presence of FVIIa. The medullary rays (collecting tubes containing epithelial cells) showed similar trends of intense staining for AF488 at earlier time points (10 min to 3 h) and reduced or no staining thereafter (data not shown). Staining of kidney tissue sections for TF revealed the staining in the glomerular capillaries, the parietal epithelial layer of the Bowman's capsule, and on the walls of the proximal and distal convoluted tubules. The intensity of TF staining remained unchanged at different time points after rFVIIa injection (data not shown).
Fig. 6.
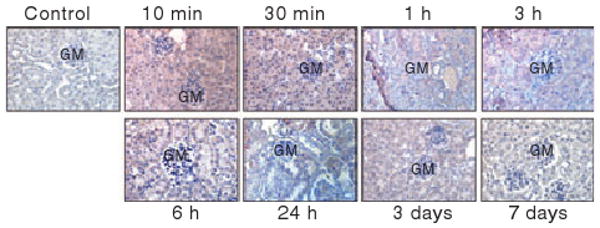
Distribution of rFVIIa in the kidney at different time intervals following a single dose of rFVIIa. Kidney sections of mice that received a single dose of AF488-FVIIa were immunostained with AF488 antibodies to evaluate rFVIIa distribution. GM, glomerulus. Magnification, 400×.
Kidney tissues of AF488-FIX injected mice stained very weakly for AF488 even at early time points (data not shown). We found little AF488-FIX in kidney tissues collected 6 h post-administration. In contrast to rFVIIa staining, the glomeruli were negative for FIX. The Bowman's capsule and the proximal and distal convoluted tubules were stained weakly for FIX in time points of 10 min to 3 h.
Distribution of rFVIIa in the heart, brain, lung and skeletal muscles
AF488-FVIIa administered to mice was localized primarily to blood vessels in the heart immediately following the administration (Fig. 7). We observed intense staining of AF488-FVIIa on the endothelium and sub-endothelium at 10 min and a weak staining thereafter. At 10 min, not only endothelial cells but also perivascular supportive cells, namely fibroblasts and pericytes, were also stained positively for FVIIa. FVIIa bound to blood vessels appeared to enter deep into the heart tissue as we observed diffused, albeit weak, staining of AF488 at later time intervals. This weak staining of FVIIa persisted even 7 days after FVIIa administration. When heart tissue specimens from AF488-FVIIa injected mice were stained for TF, we observed TF in the supportive fibroblasts around the blood vessels but not in other cells (data not shown). No changes in TF expression were observed in the heart following FVIIa administration.
Fig. 7.
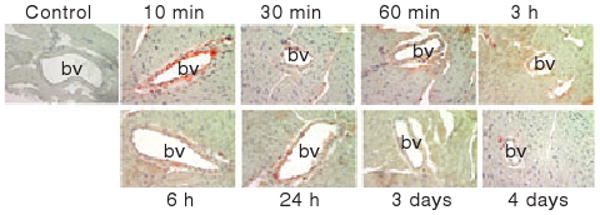
Distribution of rFVIIa in the heart at different time intervals following a single dose of rFVIIa. Heart sections of mice that received a single dose of AF488-FVIIa were immunostained with AF488 antibodies to evaluate rFVIIa distribution. bv, blood vessel. Magnification, 400×.
Consistent with the well-appreciated function of the blood–brain barrier, no immunostaining of AF488-FVIIa was observed in brain tissue following the administration of AF488-FVIIa (see Fig. S7). As expected, brain tissue stained positively for TF. The white matter of brain and the perivascular region of the blood vessels stained intensely positive for TF (see Fig. S8).
Unlike other organs (e.g. the skin, liver and kidney), lung sections from mice administered AF488-FVIIa showed no evidence for accumulation of FVIIa. None of the alveoli, bronchioles or blood vessels demonstrated positive immunostaining with AF488 (see Fig. S9). Similar data were obtained in mice administered with AF488-FIX (data not shown). In contrast to FVIIa immunostaining, the lung sections showed distinct TF staining on the alveolar wall, perivascular area of the blood vessels, and inner epithelial lining of the bronchioles (see Fig. S10).
Similar to lung tissue, the skeletal muscle fiber (multinucleated syncytium) did not exhibit the accumulation of rFVIIa following the administration of pharmacological doses of rFVIIa (data not shown). The skeletal muscle fibers were also found to be negative for TF expression (data not shown).
Discussion
Although there are a number of investigations studying the pharmacokinetics of rFVIIa [15,17–20], little information is available on the extravascular distribution of pharmacologically administered rFVIIa. The data presented herein provide evidence that rFVIIa, given at a pharmacological dose, enters into the extravascular compartment and accumulates differentially in various tissues. rFVIIa sequestered in some of the tissues, particularly in the calcified cartilage and liver, is retained for longer time periods. A slow release of rFVIIa from these storage sites may be at least partly responsible for the prolonged effect of rFVIIa observed in recent clinical studies [8].
In agreement with earlier studies [16], the present study showed that the pharmacologically administered rFVIIa accumulated in the bone, particularly in the zone of calcified cartilage. From all the tissues we have examined, the mineralized bone retained rFVIIa for the longest time period. We found large amounts of rFVIIa associated with the mineralized bone immediately following the administration of rFVIIa and the levels remained constant for the next 6 h. Thereafter, rFVIIa levels were decreased slightly. However, a significant amount of rFVIIa was present in the zone of calcified cartilage even after a week following the administration. Earlier studies suggested that the Ca2+ binding properties of the Gla domain mediate FVIIa accumulation in the bone and thus other vitamin K-dependent clotting factors, such as FIX, would also be accumulated in the bone [16]. Although our present data supports this hypothesis, we find significant differences in the retention of FVIIa and FIX in the mineralized bone. FIX sequestered in the bone disappeared within 24 h, whereas a significant amount of rFVIIa sequestered in the bone following the administration was retained for a week. In this context, it may be pertinent to point out that the region where FVIIa accumulated was stained positively for TF. Thus, it is possible that both FVIIa and FIX may initially sequester in the mineralized bone via the binding of the Gla residues to hydroxyapatite of the bone, and FVIIa, but not FIX, binding to TF in mineralized bone may slow down its dissociation from the bone.
In the liver, pharmacologically administered rFVIIa was localized initially to the portal vein and the sinusoidal capillaries. As both the portal vein and the sinusoidal capillaries were shown to express EPCR [21], the localization of rFVIIa in these regions clearly supports the concept that FVIIa binds to EPCR in vivo. Accumulation of rFVIIa, but not rFIX, in hepatocytes suggests that FVIIa binding and internalization in hepatocytes is a specific process. These data are consistent with earlier observations made during in vitro studies. Chang and Kisiel [22], based on the studies with human hepatoma cell line HuH7, suggested that hepatocytes contain a specific receptor for FVIIa and this receptor plays a role in FVIIa catabolism in the liver. At present, the identity of this receptor is unknown. Although our earlier studies [23] failed to provide unequivocal evidence for the presence of a specific high affinity receptor for FVIIa in hepatocytes, they revealed 5- to 10-fold more FVIIa, compared with FIX and FX, bound and internalized in primary hepatocytes. These data suggest that FVIIa binding to hepatocytes and the subsequent internalization represent a specific interaction of FVIIa with hepatocytes, rather than a general interaction of the Gla-domain with the cell surface phospholipids. Although rFVIIa levels in the liver were decreased after 6 h following the administration, traces of rFVIIa were found in the liver even 7 days after the administration of rFVIIa. The present data on distribution of rFVIIa to the liver are consistent with the earlier observations made with 125I-rFVIIa tissue distribution in rats, where liver tissue was found to consistently contain high concentrations of TCA precipitable radioactivity [20]. Overall, these findings coupled with the earlier data suggest that the liver could play an important role in rFVIIa metabolism.
As observed in the bone and liver, FVIIa was also retained for a longer time period in the kidney. In contrast to FVIIa, very little FIX was accumulated in the kidney, which was cleared within 3–6 h after the administration. Kidney tissue is rich in TF and the longer retention of FVIIa in the kidney may reflect FVIIa binding to TF in this organ. Alternatively, it is possible that secretion of FVIIa bound to the mineralized bone could have diffused slowly to the kidney for glomerular filtration.
A common trend of the present study is that pharmacologically administered rFVIIa initially binds to the endothelium and later enters the extravascular spaces, where it localizes to the regions that contain TF expressing cells. FVIIa binding TF appears to sequester FVIIa in extravascular tissues. For example, in the skin, the sebaceous gland and hair follicles that are rich in TF were shown to sequester FVIIa for a longer time period than other regions that are devoid of TF. In this context, it may be mentioned that one dose of rFVIIa in a wound healing model of hemophilia B mice resulted in 25% of the wounds being closed at 9 days compared with none after one dose of FIX [24].
rFVIIa association and accumulation in various cell types and tissues appears to be specific for rFVIIa metabolism and not a common pathway involving vitamin K-dependent clotting protein catabolism, as we found significant differences between rFVIIa and rFIX distribution following their administration to mice. Factor VIIa association with the endothelium of large blood vessels may reflect the binding of FVIIa to EPCR, as the endothelial cells of large blood vessels were shown to express EPCR [25] and FVIIa was shown to bind to EPCR on endothelial cells [26]. In contrast to rFVIIa, rFIX failed to associate with endothelial cells. Within 1 h, rFVIIa bound to endothelial cells was transferred to extravascular tissue surrounding the blood vessels and thereafter diffused throughout the tissue. Our recent studies of cell model systems demonstrated that FVIIa bound to EPCR at the cell surface was internalized, but some of the internalized FVIIa was transcytosed to the basolateral side as an intact protein [27]. Such a mechanism may be responsible for the transport of rFVIIa from the blood stream to the extravascular space. Our earlier in vitro studies also showed that only a fraction of the internalized FVIIa would be degraded, whereas the rest of it would be recycled back to the cell surface as an intact molecule [27,28]. Thus, FVIIa-TF complexes formed extravascularly would be capable of generating thrombin, which could facilitate the platelet plug in order to stop leakage of blood from small blood vessels. Interestingly, extravascular distribution of FVIIa appears to be limited to tissues where generation of thrombin would be helpful to achieve hemostasis and not lead to serious thrombotic risk. For example, FVIIa failed to enter the lung and brain tissue, which were very rich in TF and where thrombin generation could have devastating thrombotic effects. It is unclear what prevents FVIIa from binding to endothelium in these tissues. Lack of EPCR expression on endothelial cells in these tissues could not be a reason as EPCR expression is clearly documented in the large blood vessels of the lung [21]. Low circulatory shear stress in the lung or other differences in the lung vasculature could be responsible for this anomaly.
A number of caveats exist in the present study. First, we have used AF488 conjugated FVIIa to investigate the bio-distribution of pharmacologically administered rFVIIa. We chose to monitor FVIIa distribution by injecting AF488 tagged rFVIIa and detecting the AF488 by immunostaining as it offered unique advantages. They include (i) circumventing the necessity of having antibodies specific to mouse FVII, (ii) differentiating pharmacologically administered rFVIIa from that of endogenous FVII, and (iii) allowing accurate comparison of rFVIIa distribution with that of other vitamin K-dependent clotting proteins. Second, a positive immunostaining does not distinguish whether the intact AF488-FVIIa conjugate or the degraded product of AF488-FVIIa was stained. However, this limitation would exist even if the antibodies specific to FVIIa were used for the immunohistochemistry. It is unlikely that the observed immunostaining reflects the staining of free AF488 as we observed no immunostaining in tissue sections of mice that received free AF488 (data not shown). Third, the present study is designed to evaluate the bio-distribution of a single dose of pharmacologically administered rFVIIa, which may differ from rFVIIa distribution under prophylactic conditions. The limited availability of mouse rFVIIa and other practical/technical limitations prevented us from extending the present studies to a prophylactic mouse model system. In this context, it may be pertinent to note that long-term expression of canine rFVIIa in hemophilia dogs (A and B) prevented any spontaneous bleeding episodes, whereas untreated hemophilia A or B dogs experienced multiple spontaneous bleeding episodes [29]. Remarkably, one of these dogs, a hemophilia B dog, which had no significant increase in circulating levels of FVIIa, still did not develop any spontaneous bleeding during the 34 months of observation. These findings support the hypothesis that rFVIIa deposited perivascularly would prevent joint bleeding in hemophilia by stopping early bleeding in the microvasculature induced by normal moving pattern.
Overall, the present study suggests that pharmacologically administered rFVIIa readily associates with the endothelium and enters extravascular spaces where it can bind to TF and remain there for longer time periods than the circulatory half-life. This may be responsible for prolonged effects of the drug in prophylaxis. However, alternative explanations such as rFVIIa-mediated vascular healing cannot be ruled out as a probable reason for the prolonged effect of rFVIIa. Further studies are needed to evaluate these possibilities objectively.
Supplementary Material
Acknowledgments
The authors are thankful to M. Ezban and L. Peterson, Novo Nordisk, Denmark, for providing mouse recombinant FVIIa and antibodies against mouse tissue factor, and to C. Esmon, Oklahoma Medical Research Foundation, USA, for providing antibodies specific to murine EPCR. This work was supported partly by National Institutes of Health grants HL58869 and HL65500.
Addendum
R. Gopalakrishan performed the majority of the experiments described in the manuscript, analyzed the data, arranged the images and prepared the preliminary draft of the manuscript. S. Ghosh performed some of the experiments and contributed to preparation of the preliminary draft of the manuscript. R. Nayak prepared AF488-FVIIa and AF488-FIX conjugates, and helped in exsanguinations of mice and immunohistochemistry. T. Allen contributed to interpretation of immunohistochemistry. U. R. Pendurthi participated in the study design and analysis of the data and contributed to the preparation of the manuscript. U. Hedner conceived the research, participated in the research design, and contributed to data analysis and preparation of the manuscript. L. V. M. Rao designed and reviewed the research, analyzed the data and wrote the manuscript.
Footnotes
Disclosure of Conflict of Interests: The study was supported primarily by a research grant from Novo Nordisk, Denmark. During the course of this investigation, U. Hedner was an employee of Novo Nordisk, Denmark, and currently is a consultant to Novo Nordisk A/S, Zurich, Switzerland.
References
- 1.Hedner U, Ezban M. Tissue factor and factor VIIa as therapeutic targets in disorders of hemostasis. Annu Rev Med. 2008;59:29–41. doi: 10.1146/annurev.med.59.061606.095605. [DOI] [PubMed] [Google Scholar]
- 2.Saxon BR, Shanks D, Jory CB, Williams V. Effective prophylaxis with daily recombinant factor VIIa (rFVIIa-Novoseven) in a child with high titer inhibitors and a target joint. Thromb Haemost. 2001;86:1126–7. [PubMed] [Google Scholar]
- 3.Young G, McDaniel M, Nugent DJ. Prophylactic recombinant factor VIIa in haemophilia patients with inhibitors. Haemophilia. 2005;11:203–7. doi: 10.1111/j.1365-2516.2005.01096.x. [DOI] [PubMed] [Google Scholar]
- 4.Blatny J, Kohlerova S, Zapletal O, Fiamoli V, Penka M, Smith O. Prophylaxis with recombinant factor VIIa for the management of bleeding episodes during immune tolerance treatment in a boy with severe haemophilia A and high-response inhibitors. Haemophilia. 2008;14:1140–2. doi: 10.1111/j.1365-2516.2008.01767.x. [DOI] [PubMed] [Google Scholar]
- 5.Morfini M, Auerswald G, Kobelt RA, Rivolta GF, Rodriguez-Martorell J, Scaraggi FA, Altisent C, Blatny J, Borel-Derlon A, Rossi V. Prophylactic treatment of haemophilia patients with inhibitors: clinical experience with recombinant factor VIIa in European Haemophilia Centres. Haemophilia. 2007;13:502–7. doi: 10.1111/j.1365-2516.2007.01455.x. [DOI] [PubMed] [Google Scholar]
- 6.Tcheng WY, Donkin J, Konzal S, Wong WY. Recombinant factor VIIa prophylaxis in a patient with severe congenital factor VII deficiency. Haemophilia. 2004;10:295–8. doi: 10.1111/j.1365-2516.2004.00885.x. [DOI] [PubMed] [Google Scholar]
- 7.Mathijssen NC, Masereeuw R, Verbeek K, Lavergne JM, Costa JM, van Heerde WL, Novakova IR. Prophylactic effect of recombinant factor VIIa in factor VII deficient patients. Br J Haematol. 2004;125:494–9. doi: 10.1111/j.1365-2141.2004.04942.x. [DOI] [PubMed] [Google Scholar]
- 8.Konkle BA, Ebbesen LS, Erhardtsen E, Bianco RP, Lissitchkov T, Rusen L, Serban MA. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5:1904–13. doi: 10.1111/j.1538-7836.2007.02663.x. [DOI] [PubMed] [Google Scholar]
- 9.Le DT, Borgs P, Toneff TW, Witte MH, Rapaport SI. Hemostatic factors in rabbit limb lymph: relationship to mechanisms regulating extravascular coagulation. Am J Physiol Heart Circ Physiol. 1998;274:H769–76. doi: 10.1152/ajpheart.1998.274.3.H769. [DOI] [PubMed] [Google Scholar]
- 10.Miller GJ, Howarth DJ, Attfield JC, Cooke CJ, Nanjee MN, Olszewski WL, Morrissey JH, Miller NE. Haemostatic factors in human peripheral afferent lymph. Thromb Haemost. 2000;83:427–32. [PubMed] [Google Scholar]
- 11.Almus FE, Rao LVM, Fleck RA, Rapaport SI. Properties of factor VIIa/tissue factor complexes in an umbilical vein model. Blood. 1990;76:354–60. [PubMed] [Google Scholar]
- 12.Almus FE, Rao LVM, Rapaport SI. Regulation of factor VIIa/tissue factor functional activity in an umbilical vein model. Arterioscler Thromb. 1993;13:105–11. doi: 10.1161/01.atv.13.1.105. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman M, Colina CM, McDonald AG, Arepally GM, Pedersen L, Monroe DM. Tissue factor around dermal vessels has bound factor VII in the absence of injury. J Thromb Haemost. 2007;5:1403–8. doi: 10.1111/j.1538-7836.2007.02576.x. [DOI] [PubMed] [Google Scholar]
- 14.Lindley CM, Sawyer WT, Macik BG, Lusher J, Harrison JF, Baird-Cox K, Birch K, Glazer S, Roberts HR. Pharmacokinetics and pharmacodynamics of recombinant factor VIIa. Clin Pharmacol Ther. 1994;55:638–48. doi: 10.1038/clpt.1994.80. [DOI] [PubMed] [Google Scholar]
- 15.Beeby TL, Chasseaud LF, Taylor T, Thomsen MK. Distribution of the recombinant coagulation factor 125I-rFVIIa in rats. Thromb Haemost. 1993;70:465–8. [PubMed] [Google Scholar]
- 16.Thomsen MK, Wildgoose P, Nilsson P, Hedner U. Accumulation of the recombinant factor VIIa in rat bone: importance of the Gla-domain and relevance to factor IX, another vitamin K-dependent clotting factor. Pharmacol Toxicol. 1993;73:127–32. doi: 10.1111/j.1600-0773.1993.tb01549.x. [DOI] [PubMed] [Google Scholar]
- 17.Petersen LC, Elm T, Ezban M, Krogh TN, Karpf DM, Steino A, Olsen EH, Sorensen BB. Plasma elimination kinetics for factor VII are independent of its activation to factor VIIa and complex formation with plasma inhibitors. Thromb Haemost. 2009;101:818–26. [PubMed] [Google Scholar]
- 18.Erhardtsen E. Pharmacokinetics of recombinant activated factor VII (rFVIIa) Semin Thromb Hemost. 2000;26:385–91. doi: 10.1055/s-2000-8457. [DOI] [PubMed] [Google Scholar]
- 19.Erhardtsen E, Nilsson P, Johannessen M, Thomsen MS. Pharmacokinetics and safety of FFR-rFVIIa after single doses in healthy subjects. J Clin Pharmacol. 2001;41:880–5. doi: 10.1177/00912700122010780. [DOI] [PubMed] [Google Scholar]
- 20.Thomsen MK, Diness V, Nilsson P, Rasmussen SN, Taylor T, Hedner U. Pharmacokinetics of recombinant factor VIIa in the rat–a comparison of bio-, immuno- and isotope assays. Thromb Haemost. 1993;70:458–64. [PubMed] [Google Scholar]
- 21.Li W, Zheng X, Gu J, Hunter J, Ferrell GL, Lupu F, Esmon NL, Esmon CT. Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost. 2005;3:1351–9. doi: 10.1111/j.1538-7836.2005.01385.x. [DOI] [PubMed] [Google Scholar]
- 22.Chang GTG, Kisiel W. Internalization and degradation of recombinant human coagulation factor VIIa in the human hepatoma cell line HuH7. Thromb Haemost. 1995;73:231–8. [PubMed] [Google Scholar]
- 23.Hjortoe G, Sorensen BB, Petersen LC, Rao LV. Factor VIIa binding and internalization in hepatocytes. J Thromb Haemost. 2005;3:2264–73. doi: 10.1111/j.1538-7836.2005.01542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald A, Hoffman M, Hedner U, Roberts HR, Monroe DM. Restoring hemostatic thrombin generation at the time of cutaneous wounding does not normalize healing in hemophilia B. J Thromb Haemost. 2007;5:1577–83. doi: 10.1111/j.1538-7836.2007.02647.x. [DOI] [PubMed] [Google Scholar]
- 25.Laszik Z, Mitro A, Taylor FB, Jr, Ferrell G, Esmon CT. Human protein C receptor is present primarily on endothelium of large blood vessels: implications for the control of the protein C pathway. Circulation. 1997;96:3633–40. doi: 10.1161/01.cir.96.10.3633. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LV. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007;282:11849–57. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nayak RC, Sen P, Ghosh S, Gopalakrishan R, Esmon CT, Pendurthi UR, Rao LVM. Endothelial cell protein C receptor cellular localization and trafficking. Blood. 2009;114:1974–86. doi: 10.1182/blood-2009-03-208900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iakhiaev A, Pendurthi UR, Voigt J, Ezban M, Rao LVM. Catabolism of factor VIIa bound to tissue factor in fibroblasts in the presence and absence of tissue factor pathway inhibitor. J Biol Chem. 1999;274:36995–7003. doi: 10.1074/jbc.274.52.36995. [DOI] [PubMed] [Google Scholar]
- 29.Margaritis P, Roy E, Aljamali MN, Downey HD, Giger U, Zhou S, Merricks E, Dillow A, Ezban M, Nichols TC, High KA. Successful treatment of canine hemophilia by continuous expression of canine FVIIa. Blood. 2009;113:3682–9. doi: 10.1182/blood-2008-07-168377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.