

Abstract
Stereogenic-at-Mo monoalkoxide and monoaryloxide complexes promote enyne ring-closing metathesis (RCM) reactions, affording the corresponding endo products with high selectivity (typically >98:<2 endo:exo). All catalysts can be prepared and used in situ. Five-, six-, and seven-membered rings are obtained through reactions with enyne substrates that bear all-carbon tethers as well as those that contain heteroatom substituents. The newly developed catalytic protocols complement the related exo-selective Ru-catalyzed processes. In cases where Ru-based complexes deliver exo and endo products nondiscriminately, such as when tetrasubstituted cyclic alkenes are generated, Mo-catalyzed reactions afford the endo product exclusively. The efficiency of synthesis of N- and O-containing endo diene heterocycles can be improved significantly through structural modification of Mo catalysts. The modularity of Mo-based monopyrrolides is thus exploited in the identification of the most effective catalyst variants. Through alteration of O-based monodentate ligands, catalysts have been identified that promote enyne RCM with improved efficiency. The structural attributes of three Mo complexes are elucidated through X-ray crystallography. The first examples of catalytic enantioselective enyne RCM reactions are reported (up to 98:2 enantiomer ratio and >98% endo).
Introduction
Catalytic enyne ring-closing metathesis (RCM) is an effective method for preparation of cyclic dienes.1 Various mechanistic aspects of this class of transformations, nearly all reported cases of which are promoted by Ru-based carbenes, have been elucidated;2 the utility of this important branch of catalytic olefin metathesis has been demonstrated on several occasions in the context of natural product total synthesis.3 Nonetheless, solutions to a number of unresolved problems in reactivity and selectivity4 would substantially enhance the utility of metal-catalyzed enyne RCM.
Herein, we detail our studies regarding the development of Mo-catalyzed enyne RCM reactions,5 which in contrast to Ru-,1 W-,6 and Cr-catalyzed7 processes deliver cyclic endo diene isomers. None of the previously reported olefin metathesis-based methods allow selective access to endo products.8 Five-, six-, and seven-membered-ring endo dienes (vs exo dienes generated predominantly by Ru-based carbenes) bearing di-, tri-, and tetrasubstituted alkenes are obtained in 41–90% yield. Reactions generally proceed with exceptional endo selectivity (typically >98:<2 endo:exo). Ring-forming processes are promoted by readily prepared stereogenic-at-Mo complexes, which bear only monodentate ligands.9 Such attributes allow for modification of Mo complexes so that reactivity and selectivity of enyne RCM reactions are enhanced. We introduce several structurally modified stereogenic-at-Mo complexes that initiate enyne RCM with degrees of efficiency and selectivity not achievable through the use of previously reported catalysts. We conclude by presenting the first examples of efficient metal-catalyzed enantioselective enyne RCM reactions.10
The key challenge facing us in the course of investigations detailed herein originates from the availability of various pathways2 through which a catalyst might promote an enyne RCM reaction: a metal-based alkylidene may first either associate with the alkyne (pathways 1 and 2, Scheme 1) or the alkene (pathway 3, Scheme 1) unit of the substrate i. Transformation through complex ii gives rise to β-substituted metallacyclobutene iii, where the metal center forms a bond with the terminal carbon of the alkyne; conversion of iii to conjugated terminal alkylidene iv would then be followed by RCM to afford cyclic diene v (endo product). A metal alkylidene might alternatively coordinate to the alkyne in such a manner that, as depicted in pathway 2 in Scheme 1, generation of α-substituted metallacyclobutene vii is preferred; cycloreversion to disubstituted alkylidene viii culminates in the formation of the exo product ix. Pathway 3 (Scheme 1) commences by initial interaction of the catalyst with the alkene unit x, affording terminal carbene xi, which through an intramolecular cycloaddition is converted to α,β-metallacyclobutene xii. Subsequent transformation gives rise to cyclopentene-substituted terminal carbene xiii, which through a cross-metathesis process with another enyne substrate (i) affords exo product ix and regenerates terminal carbene xi.
Scheme 1.
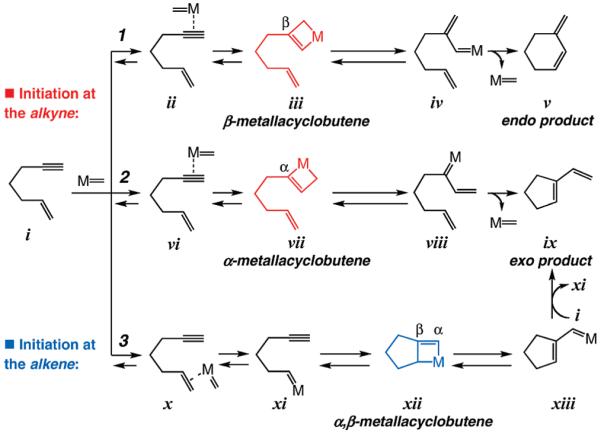
Various Modes of Reaction in Metal-Catalyzed Enyne RCM
Extensive previous research has established that when Ru-based carbene complexes are used to promote enyne RCM, the formation of exo products, represented by ix (Scheme 1), is strongly favored.1 Our interest in examining the ability of stereogenic-at-Mo complexes, recently developed in these laboratories to address a number of unresolved issues in selective catalytic olefin metathesis, arose from the principle that high oxidation state complexes should favor reaction through a pathway that commences with association with the alkyne site of an enyne substrate.11 The above considerations, together with the preference of the sterically demanding Mo center to form a bond with the terminal carbon of an alkyne, led us to surmise that β-metallacyclobutenes should prove to be favored intermediates, leading to selective formation of endo RCM products (pathway 1, Scheme 1). We were also aware, however, that the strong preference for formation of Mo-alkyne complex could lead to competitive oligomerization processes, involving reaction of intermediate alkylidenes (e.g., iv or viii, Scheme 1) with an alkyne of another substrate molecule. We will demonstrate that such complications can be addressed through modification of the catalyst structure so that intramolecular processes that lead to RCM products are preferred over intermolecular reactions that result in formation of oligomers.
Results and Discussion
1. Selective Synthesis of endo-1,3-Carbocyclic Dienes through Mo-Catalyzed Enyne RCM
1.1. Initial Evaluation of Mo- and Ru-Based Complexes
We began by examining the ability of representative Mo-based complexes to catalyze RCM of enyne 1 (Scheme 2). Use of Mo alkylidene 410b (5 mol %) does not lead to formation of either the endo or the exo cyclic diene isomer (2 and 3, respectively; 22 °C, 30 min). In contrast, under identical conditions, monopyrrolide–monoalkoxide 55 delivers endo RCM product 2 in 72% yield (<2% exo isomer 3, detected by 400 MHz 1H NMR spectroscopy). As has been described previously, stereogenic-at-Mo complexes, represented by 5, are prepared and often used in situ9a,b through reaction of a bispyrrolide (e.g., 6) with an alcohol.12 As also shown in Scheme 2, the latter precursor complex does not exhibit any catalytic activity in this case (<2% conversion). Ru-based carbene 713 initiates enyne RCM, albeit less effectively than 5 (2 h vs 30 min of reaction time). It is only the exo isomer 3 that is isolated in the Ru-catalyzed process (Scheme 2).14 When the RCM promoted by carbene 7 is performed under an atmosphere of ethylene,14c after 1 h there is only 24% conversion to 3, which is obtained along with a byproduct that is derived from homo-cross-metathesis of the product (3, 26%; ~50% recovered substrate).
Scheme 2.
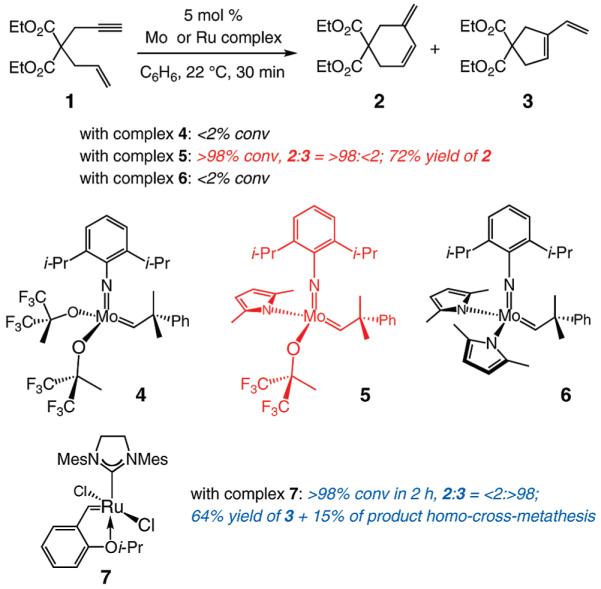
Mo and Ru Complexes as Enyne RCM Catalystsa
a Mes = 2,4,6-(Me)3C6H2.
1.2. Enyne RCM Promoted by Monoalkoxide Mo Complex 5
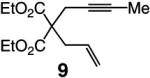
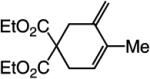
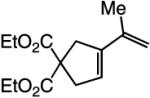
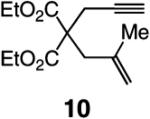
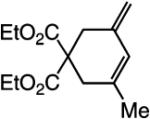
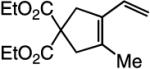
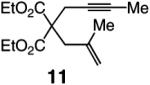
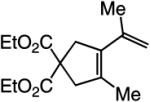
The findings summarized in Table 1 demonstrate that, through the use of Mo complex 5, a range of cyclic dienes can be accessed. Thus, five-, six-, and seven-membered ring products are isolated in 70–82% yield after purification; reactions proceed readily and selectively with terminal alkynes as well as 1,1-disubstituted alkenes (e.g., entry 3, Table 1). The RCM process in entry 4 of Table 1, involving enyne 11, which contains an internal alkyne and a disubstituted olefin, leads to the formation of a tetrasubstituted cyclic alkene; as a result, unlike cases where di- or trisubstituted olefins are generated (5 mol % 5), higher catalyst loading (20 mol %) is required for achieving complete substrate consumption. When 5 mol % 5 is used, there is only 23% conversion, and with 10 mol % Mo-alkylidene, 42% of the desired endo product is obtained after 30 min [70% conversion after 24 h; the remainder of the enyne remains unreacted (i.e., <2% oligomerization)]. Additional points regarding the RCM processes in Table 1 are worthy of note: Although most reactions described above are performed with 5 mol % 5, in at least some cases, efficient transformation can be achieved with lower catalyst loading. For example, RCM of 1 (Scheme 2) carried out in the presence of 1 mol % 5 proceeds to >98% conversion within 30 min at 22 °C to afford endocyclic diene 2 (Scheme 2) in 74% yield.
Table 1.
Selective Synthesis of endo-Carbocyclic Dienes by Mo-Catalyzed Enyne RCM (Diester Tether)a
| entry | substrate | endo product | exo product | endo:exob | yield (%)c |
|---|---|---|---|---|---|
| 1 |

|
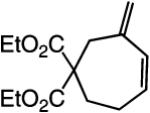
|
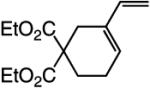
|
>98:<2 | 70 |
| 2 |
|
|
|
75:25 | 80d |
| 3 |
|
|
|
>98:<2 | 82 |
| 4e |
|

|
|
>98:<2 | 75 |
Reactions were performed under N2 atmosphere with 5 mol % 5 for 30 min in C6H6 at 22 °C.
Ratios were determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures prior to purification.
Yield of isolated products after purification.
Combined yields of the product mixture.
Reaction performed with 20 mol % 5.
High endo selectivity is observed in most instances (entries 1 and 3–4), except for the reaction of internal alkyne 9 (entry 2), where a mixture of endo and exo dienes is formed (75:25). It is plausible that the presence of the methyl substituent and the attendant steric repulsion that entails Mo alkylidene–alkyne association, represented by ii (Scheme 1, pathway 1), leads to partial reaction through pathway 3, involving initiation at the olefin site; some of the exo product could also result from reaction via vi and the corresponding α-metallacyclobutene vii (Scheme 1, pathway 2).15 The complete endo selectivity (<2% exo) observed in RCM of enyne 11 (entry 4, Table 1), which bears an internal alkyne in addition to a 1,1-disubstituted alkene (vs a terminal olefin in 9), is noteworthy; with the more substituted olefin, the Mo alkylidene likely initiates exclusively at the site of the internal alkyne. When Ru-based carbenes are used to promote RCM reactions of enynes bearing a terminal alkyne and a disubstituted alkene (e.g., 11), nearly equal mixtures of exo and endo isomers are generated.14g
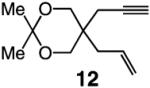
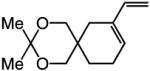
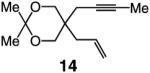
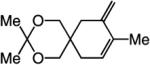
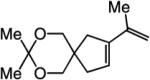
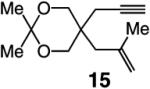
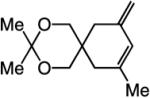
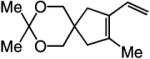
To probe further the effect of steric factors on endo vs exo selectivity, we prepared several acetonide-containing enynes (Table 2) and examined the corresponding RCM reactions (vs diesters in Table 1). Catalytic cyclizations of enynes 12 (entry 1, Table 2) and 15 (entry 4, Table 2) proceed to afford the endo products exclusively (>98:<2). Unlike the reaction of diester 8 (entry 1, Table 1), however, RCM of acetonide–enyne 13 (entry 2, Table 2) gives rise to a 75:25 mixture of endo and exo isomers. Similar to catalytic RCM of internal alkyne 9, illustrated in entry 3 of Table 1, acetonide 14 undergoes transformation readily to generate a mixture of endo and exo dienes (66:33 endo:exo). We suggest that, in the case of 13, exo isomer formation is competitive since generation of the α-metallacyclobutene intermediate (pathway 2, Scheme 1) is rendered more favorable as a result of reduced steric repulsion between the metal complex and the less sterically cumbersome and geometrically constrained acetonide moiety (vs the diester units of 8). The latter scenario does not lead to diminution of product selectivity in RCM of the structurally related acetonide–enyne 12 (entry 1, Table 2), likely because of the sterically accessible terminal alkyne and because the endo product is a readily generated six-membered-ring diene. In contrast, endo selectivity in RCM of acetonide–enyne 13 results in the generation of a less readily formed seven-membered-ring product (exo isomer is a six-membered ring). It merits mention that the validity of some of the scenarios mentioned depends on reversibility of some of the steps within the catalytic cycle (Scheme 1); a more sophisticated appreciation regarding any mechanistic preferences must await the outcome of more detailed studies.
Table 2.
| entry | substrate | endo product | exo product | endo:exob | yield (%)c |
|---|---|---|---|---|---|
| 1 |
|

|
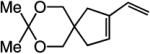
|
>98:<2 | 81 |
| 2 |
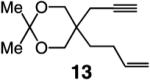
|
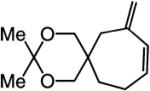
|
|
75:25 | 75d |
| 3 |
|
|
|
66:33 | 72d |
| 4 |
|
|
|
>98:<2 | 81 |
Reactions were performed under N2 atmosphere with 5 mol % 5 for 30 min in C6H6 at 22 °C.
Ratios were determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures prior to purification.
Yield of isolated products after purification.
Combined yields of the product mixture.
The above observations illustrate that the degree of endo:exo selectivity in Mo-catalyzed RCM of enynes can depend on whether the typically favored mode of reaction (via Mo-alkyne complex ii and β-metallacyclobutene iii in Scheme 1) engenders unfavorable steric repulsion between any of the substrate substituents and the relatively sizable ligands of the Mo center. Furthermore, diminished selectivity may arise if the endo product represents a ring size (e.g., a cycloheptadiene) that is typically formed more slowly than that which corresponds to the exo isomer (e.g., a cyclohexadiene).
2. Selective Synthesis of endo-1,3-Heterocyclic Dienes through Mo-Catalyzed Enyne RCM
A significant consequence of the ability of Mo-based monoaryloxide–monopyrrolides to promote selective formation of endo-1,3-cyclic dienes is the possibility of applying such protocols toward preparation of the corresponding heterocyclic structures.16 Accordingly, we investigated the effect of the presence of a heteroatom within the tether separating the alkyne and alkene units on the efficiency and selectivity of the catalytic RCM processes.
2.1. Mo-Catalyzed RCM of N-Containing Enynes
2.1.1. Catalytic RCM of Enynes Bearing Different N-Substituents
We began by investigating the RCM of a small set of enynes with various N-substituents. As the data summarized in Scheme 3 illustrate, in the presence of 5 mol % of Mo-based alkylidene 5, tosylamide 16a readily undergoes RCM to afford 17a in >98% endo selectivity and 92% yield after purification. Catalytic ring closure of 16b, carrying the more easily removable o-nosylamide, is equally efficient: the desired N-containing cyclic endo-1,3-diene 17b is isolated in 85% yield with only slightly diminished product selectivity (97:3 endo:exo). When Boc-amide 16c serves as the starting enyne (Scheme 3), oligomer formation is competitive with the desired RCM, which furnishes 17c in 48% yield and >98% endo selectivity. Adventitious oligomerization competes with formation of 17c, likely because the adjacent Boc unit renders the alkyne sterically more accessible (vs Ts or o-Ns), causing the intermolecular reaction to be favored. As the data below will demonstrate (see eq 5), it is less likely that the reduced conformational constraint imposed by the Boc-amide plays a significant role in the lower yield in which 17c is isolated.
Scheme 3.

Initial Examination of Mo-Catalyzed RCM of Various N-Containing Enynes
2.1.2. Selective Mo-Catalyzed RCM of Enynes Bearing an N-Tosylamide Tether
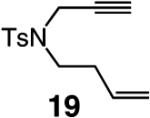
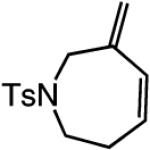
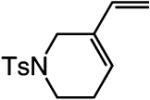
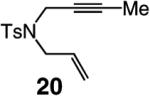
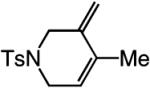
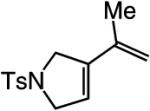
A range of N-containing enynes readily undergo RCM reactions to afford the derived cyclic diene products with high endo selectivity (Table 3). Similar to previous cases, even when a tetrasubstituted olefin is generated (entry 4, Table 3), the endo isomer is generated exclusively. As illustrated in entry 2 of Table 3, in only one case is a small amount of the exo product formed (endo:exo = 84:16). The arguments discussed above, regarding reactions of substrates bearing an internal alkyne and a terminal olefin, are likely valid here as well.
Table 3.
| entry | substrate | endo product | exo product | endo:exob | yield (%)c |
|---|---|---|---|---|---|
| 1 |
|
|
|
>98:<2 | 80 |
| 2 |
|
|
|
84:16 | 90d |
| 3 |
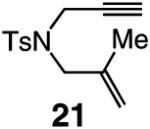
|

|
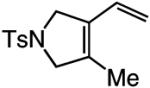
|
>98:<2 | 60 |
| 4 |
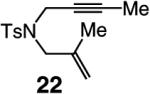
|
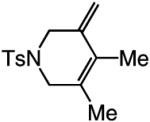
|
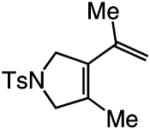
|
>98:<2 | 77 |
Reactions were performed under N2 atmosphere with 5 mol % 5 for 30 min in C6H6 at 22 °C.
Ratios were determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures prior to purification.
Yield of isolated products after purification.
Combined yields of the product mixture.
In cases where a medium ring is generated (entry 1, Table 3) or when the rate of ring closure is retarded due to increased substitution at the alkyne and/or the alkene site (entries 2–4, Table 3), the reaction must be performed under conditions that are designed to minimize oligomerization. That is, whereas the transformations in Schemes 2 and 3 and Tables 1 and 2 were performed in 0.16 M solutions, RCM processes in entries 1, 3, and 4 of Table 3 were carried out under more dilute conditions (1.3 × 10−3, 2 × 10−3, and 2 × 10−3 M, respectively).17 It is noteworthy, however, that even when substrate concentration is decreased significantly, the stereogenic-at-Mo complex remains highly effective as an olefin metathesis catalyst.
As the example illustrated in eq 1 demonstrates, Mo-catalyzed RCM reactions involving terminal alkynes and alkenes readily proceed to >98% conversion with only 1 mol % 5, delivering the corresponding endo products efficiently and with outstanding selectivity (>98:<2 endo:exo). In cases where the alkyne or the olefin unit is more sterically hindered, 5 mol % catalyst loading is required for complete conversion; for example, with 1,6-enyne 20 (entry 2, Table 3) as the substrate, RCM in the presence of 2.5 mol % 5 leads to 68% conversion after 90 min. It should also be noted that the Mo catalyst can be readily generated and used in situ without any diminution in reaction efficiency or product selectivity; the example illustrated in eq 2, regarding the preparation of endo diene 23, is representative.
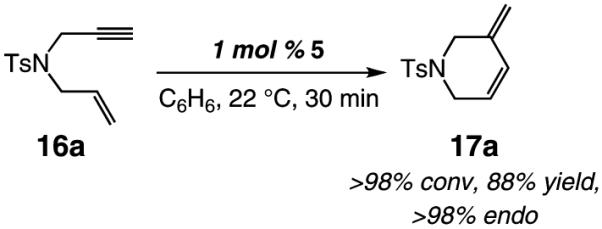 |
(1) |
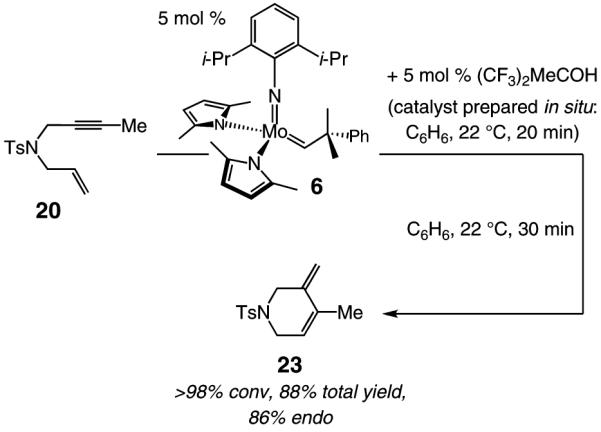 |
(2) |
The transformations illustrated in Scheme 4 involve RCM reactions of N-substituted 1,7-enyne substrates (vs 1,6-enynes examined above). We find that, with 5 mol % 5, aryl-substituted enyne 24 is converted within 30 min to seven-membered endo diene 25 in >98% selectivity (<2% exo product 26) and 68% yield. When Ru-based complex 7 is used, exo diene 26 is formed as the sole product (<2% endo product, 25) in 70% yield (Scheme 4). In contrast, when 1,7-enyne 27, bearing a less sterically congested alkyne unit (vs the benzylic alkyne in 24), is treated with the same conditions as mentioned above, only rapid oligomerization ensues.
Scheme 4.

To address the aforementioned complication, we turned to the modified stereogenic-at-Mo monopyrrolide complex 28,9a where the hexafluoro-tert-butoxide ligand of 5 has been replaced by a bulkier 2,6-di(isopropyl)phenoxide. We surmised that the presence of the more sizable aryloxide would discourage the competing intermolecular processes, leading to oligomeric products (vs the desired RCM). As illustrated in Scheme 4, in the presence of Mo monoaryloxide–monopyrrolide 28, the efficiency of the RCM is increased significantly: a mixture of endo-29 and exo-30 is obtained in 41% yield; the remainder of the substrate is consumed toward side reactions. The low endo:exo selectivity is likely due to favorable formation of the derived Mo-arylidene complex (derived from the styrenyl alkene) that gives rise to the competing mode of transformation (pathway 3, Scheme 1) involving initiation of the catalytic cycle at the styrenyl site of enyne 27. Ru-catalyzed RCM with 27,18 as indicated in Scheme 4, leads to formation of unidentified product mixtures when carried out under N2; when ethylene atmosphere is used at 22 °C, low efficiency is observed. It has, however, been previously reported that, with 5 mol % Ru carbene 7, RCM of the acetamide derivative of 27 proceeds readily at 80 °C (toluene, under N2 atm, reaction time not specified) to deliver the corresponding exo isomer in 65% yield.18d
2.2. Mo-Catalyzed RCM of O-Containing Enynes
2.2.1. Initial Studies
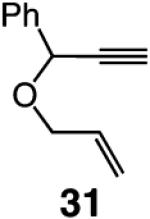
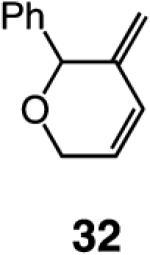
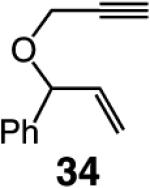
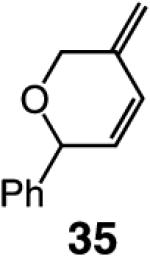
We began our studies by treatment of 31 with 5 mol % 5, which resulted in the formation of pyran 32 in 46% yield and >98% endo selectivity; a significant amount of oligomer generation accounts for complete consumption of the substrate. With the more sterically hindered monoaryloxide 28, there is little or no competing oligomerization, but there is only 20% conversion to the desired product (>98% endo), and 32 is obtained in 15% yield. When enyne 34 is used, a similar trend in selectivity is observed (Scheme 5). The lower yield of the endo diene 35 versus that of 32 is likely because the sterically less hindered alkyne unit of 34 is more prone to undergo the intermolecular cross-metathesis reactions that deliver oligomeric products. As indicated in Scheme 5, when performed under an atmosphere of ethylene, RCM with Ru complex 7 affords the exo products with high efficiency; otherwise, only a complex mixture of products is obtained.19
Scheme 5.
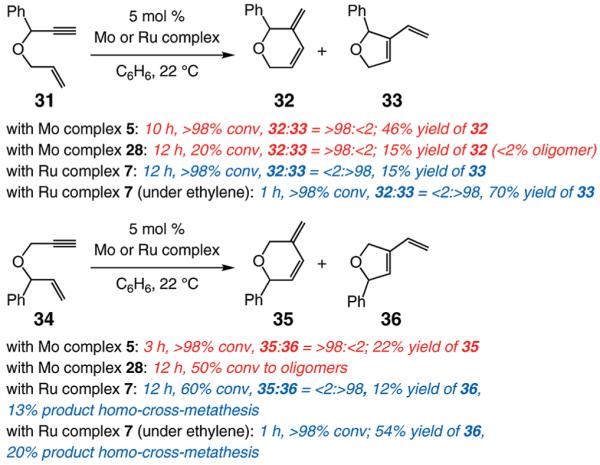
Initial Study of Mo-Catalyzed RCM of O-Containing Enynesa
a All reactions performed under N2 atmosphere, unless otherwise noted.
2.2.2. Improved Mo-Catalyzed RCM Efficiency and Minimization of Competitive Oligomerization through the Use of Coordinating Solvents
To enhance the efficiency of RCM reactions of O-containing enynes, we first turned our attention to investigating the effect of the reaction medium in diminishing the rate of competitive oligomerization. We reasoned that, in the presence of a coordinating solvent, which might associate with the metal center, the steric requirements of the substrate coordination site as well as the Lewis acidity of the Mo might be altered, such that oligomerization reactions, for reasons delineated above regarding the preparation and examination of monoaryloxide 28 (Scheme 4), would be rendered relatively sluggish. We have successfully used the above strategy in enhancing the enantioselectivity of RCM reactions of dienes promoted by chiral Mo diolates.20
Whereas reactions performed in Et2O proceed in a similar fashion as in benzene (Table 4, entries 1 and 5), when THF is used the desired RCM pathway becomes significantly more competitive: 32 and 35 constitute 76% and 46% of the mixture (entries 2 and 6, Table 4; vs 46% and 22%, respectively) and are isolated in 75% and 44% yield. The suggestion that the positive effect of THF is, at least partly, due to its coordinating ability is supported by the findings that, when 2-methyl-THF (entries 3 and 7, Table 4) and the more sizable 2,5-dimethyl-THF (entries 4 and 8) serve as solvent, reaction efficiency is improved.
Table 4.
| entry | endo product | solvent | time (h) | endo:exob | conv (%);b yield (%)c |
|---|---|---|---|---|---|
| 1 |
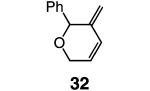
|
Et2O | 12 | >98:<2 | 40; nd |
| 2 | THF | 12 | >98:<2 | 76; 75 | |
| 3 | 2-MeTHF | 10 | >98:<2 | 55; nd | |
| 4 | 2,5-Me2THF | 10 | >98:<2 | 48; nd | |
| 5 |
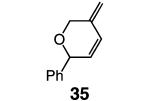
|
Et2O | 4 | >98:<2 | 21; nd |
| 6 | THF | 6 | >98:<2 | 46; 44 | |
| 7 | 2-MeTHF | 3 | >98:<2 | 31; nd | |
| 8 | 2,5-Me2THF | 3 | >98:<2 | 29; nd |
Reactions were performed under N2 atmosphere with 5 mol % 5 for 30 min in C6H6 at 22 °C.
Ratios were determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures prior to purification.
Yields of purified endo products; >98% substrate consumption in all cases (400 MHz 1H NMR analysis). nd = not determined.
2.2.3. Enhancement of RCM Efficiency and Minimization of Competitive Oligomerization through Catalyst Modification
We next turned our attention to altering the structure of the Mo catalyst. We reasoned that, although with alkylidene 28, bearing a 2,6-di(isopropyl)phenoxide, formation of unsaturated pyran 32 is less efficient than when complex 5 is used (see Scheme 5), other modified catalyst structures might furnish superior selectivity based on the aforementioned rationales (see Scheme 4 and related discussion). Several monoaryloxides were thus prepared in situ through treatment of bispyrrolide 6 with the appropriate phenol (Table 5), and their ability to promote RCM of enyne 31 was investigated. In two cases (entries 1 and 4, Table 5), a substantial amount of unreacted enyne is recovered; with the catalyst derived from 2,3,5,6-tetraphenylphenol (entry 4, Table 5), the desired heterocycle 32 constitutes a substantial portion of the product mixture (55% conversion of the substrate, 32% yield of 32). Furthermore, the Mo complex derived from 2,6-diphenylphenoxide (entry 3, Table 5) delivers the desired monomeric product 32 in 62% yield after purification (>98% endo; complete consumption of enyne 31).
Table 5.
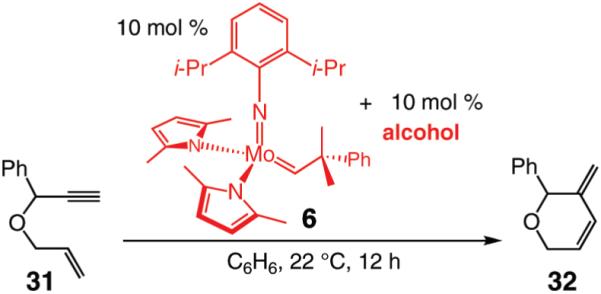
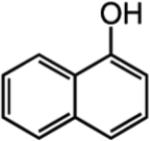
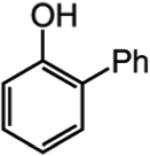
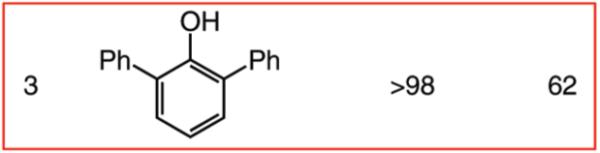
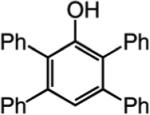
Reactions were performed under N2 atmosphere with 5 mol % 5 for 30 min in C6H6 at 22 °C.
Ratios were determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures prior to purification.
Yield of isolated products after purification.
In light of the promising activity and selectivity levels furnished by the two modified stereogenic-at-Mo alkylidenes, we isolated and characterized complexes 38a,b which, as illustrated in eq 3, are generated in 80% and 76% yield, respectively. The ability of 38a,b, prepared and used in situ, to promote RCM reactions of a select number of O-containing enynes was then probed.
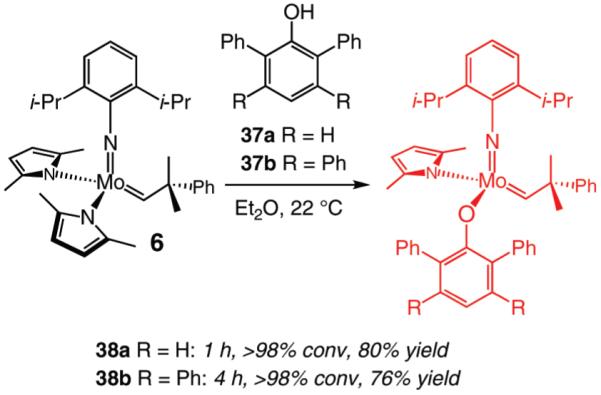 |
(3) |
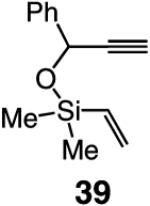
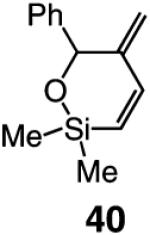
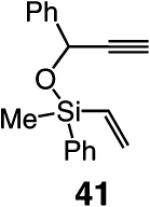
As the data in Table 6 indicate, the two modified monoaryloxides allow access to the desired O-substituted cyclic endo dienes in useful levels of efficiency; none of the corresponding exo product isomers are detected (as judged by analysis of 400 MHz 1H NMR spectra). It is noteworthy that Mo-catalyzed RCM reactions of sterically congested vinylsilanes 39 and 41 (entries 5–8) proceed readily to afford the desired cyclic unsaturated siloxanes in up to 85% yield. A notable feature of the findings in Table 6 is the difficulty of gauging a priori whether the diphenylphenoxide 38a (e.g., in the case of 31 and 41 in entries 1–2 and 7–8) or the more sterically hindered tetraphenylphenoxide 38b promotes a more efficient reaction (e.g., with enynes 34 and 39 in entries 3–6). A significantly improved ability to predict the outcome of RCM reactions requires a more detailed understanding of the mechanistic nuances of the catalytic process, including the relationship between the structure of a Mo complex and the level of endo:exo selectivity.
Table 6.
| entry | substrate | endo product | Mo complex; mol (%) |
conv (%);b time (h) |
endo:exob | yield (%)c |
|---|---|---|---|---|---|---|
| 1 |
|
|
38a; 10 | >98; 12 | >98:<2 | 62 |
| 2 | 38b; 10 | 55; 12 | >98:<2 | 32 | ||
| 3 |
|
|
38a; 5 | >98; 3 | >98:<2 | 43 |
| 4 | 38b; 5 | >98; 3 | >98:<2 | 67 | ||
| 5 |
|
|
38a; 5 | >98; 1 | >98:<2 | 75 |
| 6 | 38b; 5 | >98; 1 | >98:<2 | 85 | ||
| 7 |
|
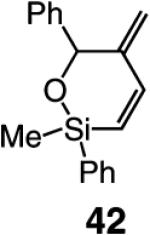
|
38a; 5 | >98; 3 | >98:<2 | 67d |
| 8 | 38b; 5 | >98; 6 | >98:<2 | 61d |
Reactions were performed under N2 atmosphere with 5 mol % 5 for 30 min in C6H6 at 22 °C.
Ratios were determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures prior to purification.
Yield of isolated products after purification.
Mixture of diastereomers (~60:40; stereogenic at Si).
Several additional points regarding the data in Table 6 merit mention: (a) The reaction performed in the presence of 38a (entry 1, Table 6), although superior to that involving Mo complex 5 (62% vs 46% yield of 32), remains less efficient than when 5 is utilized in THF (see entry 2, Table 4: 75% yield of 32). In the RCM involving enyne 34, however, it is the tetraphenylphenoxide 38b that delivers 35 with the highest degree of efficiency (entry 4, Table 6: 67% yield vs 22% and 44% yield with alkylidene 5 in C6H6 and THF, respectively). (b) The amount of catalyst required depends on the structure of the substrate. Thus, 5 mol % loading is sufficient for achieving >98% conversion with enyne 34, which bears the less sterically congested alkyne (vs 10 mol % for 31). The same amount of 38a,b engenders complete conversion in reactions of vinylsilanes 39 and 41 (entries 5–8, Table 6). The higher degree of efficiency observed for RCM of 39 (vs 31) is likely due to the favorable entropic (Thorpe–Ingold) effects due to the tetrasubstituted Si-based tether within such substrates. Finally, it should be noted that use of complexes 38a,b in THF (see Table 4) does not give rise to any additional increase in RCM efficiency. It is plausible that association of the Lewis basic solvent with the metal center is less favorable with the more sterically demanding aryloxide ligands.
3. Structural Attributes of Stereogenic-at-Mo Monopyrrolide Complexes
Several stereogenic-at-Mo complexes, bearing different O-based ligands, have played a critical role in the development of enyne RCM reactions described above. A brief analysis of some of the structural features of these alkylidenes is thus warranted. The X-ray structures of monopyrrolide monoalkoxide complex 5 as well as monoaryloxides 38a,b are illustrated in Scheme 6. The structural details of complex 28 (Scheme 4) have been previously disclosed.8a
Scheme 6.

X-ray Structures and Select Physical Properties of Mo-Based Monopyrrolidesa
a See the Supporting Information for details regarding the X-ray structures of complexes 5, 38a, and 38b.
Selected bond lengths and angles are depicted in Scheme 6. Noteworthy is the relatively long Mo–O bond in 2,6-diphenylphenoxide 38a and 2,3,5,6-tetraphenyphenoxide 38b, a characteristic that is likely the result of minimization of unfavorable steric interactions between the sizable aryloxides and the other ligands. Similar considerations can be used to offer a rationale for the unusually large C–Mo–O angle in 38b (115.03° vs ~109° for 5 and 28). The above attributes can give rise to a more spacious binding pocket, allowing for facile RCM reactions of sterically demanding enyne substrates. Two noteworthy examples within this context are the reactions of vinylsilanes 39 and 41, shown in entries 5–8 of Table 6. In contrast to RCM promoted by monopyrrolides 38a and 38b (>98% conversion, 61–85% yields), <2% consumption of the substrates is observed with Mo-based complexes 5 and 28 (as well as the Ru-based carbene 7).
Somewhat surprisingly, Mo complex 38a (138.51°) has the smallest Mo–O–C and C–Mo–O angles of the four complexes. This unusual attribute may be due to π-stacking associations21 between the phenyl unit of the neophylidene substituent and the aryloxide group, as suggested by the distance between these two aromatic planes (see X-ray of 38a, Scheme 6). Such an interaction would be less favored with the sterically more demanding tetraphenylphenoxide of 38b. It is important to note that, upon initiation and loss of the neophylidene group, the Mo–O–C angle of the complexes derived from 38a would likely resemble that of 38b. Thus, in general, whereas the bond length values shown in Scheme 6 may prove to be relevant to the properties of the catalytically active complex in solution, the corresponding bond angles might be largely influenced by crystal packing forces and steric interactions that arise from the presence of the neophylidene unit. One set of observations supports the validity of such a caveat: The alkylidene proton in 38b resides only ~28 Å above the π surface of an aryl unit of the aryloxide ligand, as observed in the X-ray structure of this complex (Scheme 6), and would thus be expected to appear more upfield as a result of well-established anisotropic effects. The 1H NMR spectrum of 38b (in C6D6), however, exhibits a signal at δ 11.67 ppm, which is closely related to the chemical shifts detected for the alkylidene proton in 38a (δ 11.70 ppm).
4. Enantioselective Mo-Catalyzed Enyne RCM Reactions
Varying degrees of progress have been achieved in connection with enantioselective versions of several types of olefin metathesis reactions.22-24 One subset of transformations that has not yet yielded to enantioselective catalysis is the RCM reactions of enynes. Based on the outcome of the investigations summarized above, and encouraged by the recent discoveries regarding the exceptional ability of stereogenic-at-Mo complexes to promote enantioselective RCM9a-b and ring-opening/cross-metathesis reactions of olefinic substrates,9c we set out to establish whether the same class of chiral alkylidenes can promote the formation of enantiomerically enriched cyclic endo dienes.
Treatment of dienyne 43 with 10 mol % of 44a, prepared and used in situ, leads to complete consumption of the substrate (Scheme 7). The product from enantioselective RCM affords the endo cycloadduct 45 in 79:21 enantiomer ratio (er) and 30% yield after purification; a significant portion of the product mixture consists of oligomeric materials. We then surmised that if the RCM process is performed under an atmosphere of ethylene (vs N2), the efficiency with which the monomeric heterocycle is generated might be improved. This strategy, previously utilized in relation to Ru-catalyzed enyne metathesis reactions,14c is based on the principle that initial cross-metathesis of the alkyne unit with ethylene might deliver tetraene 46 (Scheme 7), which would undergo RCM to generate endocyclic product 45. Increased RCM efficiency would arise from the more expeditious removal of the terminal alkyne, largely responsible for the formation of oligomeric side products. When RCM of 43 is carried out under an atmosphere of ethylene (Scheme 7), unsaturated tosylpiperidine 45 is isolated in 60% yield (vs 30% under N2) in the same degree of enantiomeric purity (79:21 er). Enantioselectivity is improved with di-iodo complex 44b: the endo product is obtained in 85:15 er. Although unreacted substrate is recovered when 44b is used (69% conversion), there is minimal oligomerization: endo product 45 is formed in 63% yield after purification (Scheme 7). It should be noted that, despite repeated attempts, we did not detect the purported diene intermediate 46 (see below for an additional related observation).
Scheme 7.
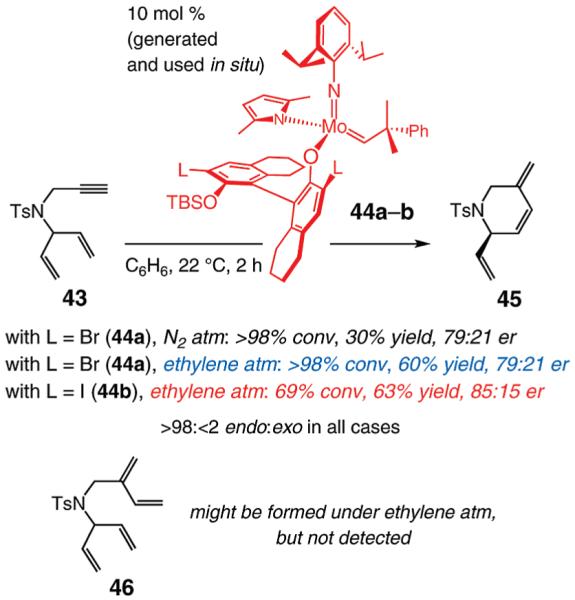
Mo-Catalyzed Enantioselective RCM of a Dienyne
Next, we evaluated the corresponding transformation of dienyne 47, which bears two 1,1-disubstituted alkenes (vs terminal olefins in 43, Scheme 7). As shown in Scheme 8, treatment of 47 with 10 mol % 44a under an atmosphere of ethylene gives rise to >98% conversion within 2 h at 22 °C. Tetraene 48 is isolated in 70% yield, and none of the expected RCM product is detected (49, as judged by analysis of the 400 MHz 1H NMR spectrum of the unpurified mixture). Resubjection of 48 to 5 mol % 44a, this time under an atmosphere of N2, gives rise to formation of 49 in >98% endo selectivity, 92% yield, and 98:2 er. Minimal oligomerization is observed in the transformations depicted in Scheme 8. It should be noted that there is <10% conversion when 47 is subjected to 10 mol % 44a at 22 °C under an atmosphere of N2; longer reaction times do not lead to additional transformation. The origin of the aforementioned lack of reactivity is not clear at the present time; it is feasible, however, that initial oligomerization leads to formation of an unreactive Mo alkylidene, resulting in sequestration of all catalytically competent complexes. The two-step procedure shown in Scheme 8 can be carried out in a single vessel: treatment of 47 with 10 mol % 44a in benzene under ethylene atmosphere for 10 min, followed by removing the balloon and allowing the mixture to stir for 12 h, delivers 49 in 62% yield and 95:5 er.
Scheme 8.
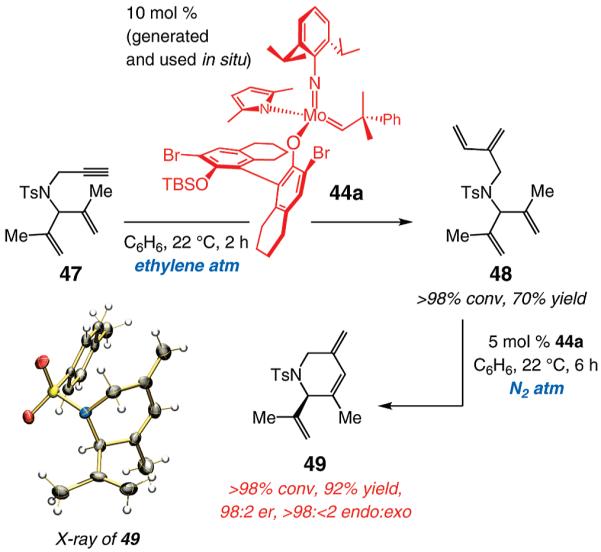
Effect of Ethylene on Catalytic Cross-Metathesis/Enantioselective RCM of a Dienyne
Initial investigations indicate that use of an ethylene atmosphere as the means to improve the efficiency of an enyne RCM process does not apply to all substrate classes. As an example, when O-containing enyne 34 (Table 6) is subjected to 10 mol % Mo-based monopyrrolide complex 38b (prepared and used in situ, as with 44a,b) under an ethylene atmosphere, after 2 h at 22 °C, there is >98% substrate consumption but endo product 35 is obtained in 42% yield (vs 67% yield under N2; see entry 4, Table 6). It is plausible that sterically less congested alkynes (e.g., those bearing an allylic ether vs an NTs unit) undergo oligomerization at a rate with which ethylene cross-metathesis cannot effectively compete.
Attempts to promote Mo-catalyzed enantioselective RCM reactions of the related enyne substrates bearing all-carbon or O-containing tethers proved significantly less selective (<65:35 er). Clearly, development of chiral catalyst variants that exhibit a wider range of substrate generality is required; studies along these lines are in progress.
Conclusions
We report the first endo-selective set of protocols for catalytic RCM of enynes, including those that bear an N- or an O-based tether. Cyclization reactions typically proceed with high endo:exo selectivity and in yields that render the method of utility, particularly since, in most instances, the Ru-catalyzed reactions deliver the corresponding exo products. The endo cyclic dienes, as illustrated by the highly site-selective oxidation25 in eq 4, can be functionalized to allow access to a range of useful heterocyclic small organic molecules (e.g., through alkylations or substitution reactions of the allylic epoxide).26
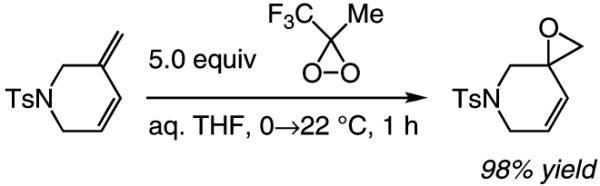 |
(4) |
Reactions are promoted by stereogenic-at-metal monoalkoxide– or monoaryloxide–monopyrrolide Mo complexes, prepared from the corresponding bispyrrolide (6 in Scheme 2) and the appropriate alkyl or aryl alcohols. Isolation and purification of the metal complexes is not required, as they can be synthesized and used in situ. The above characteristic, together with the fact that the Mo-based catalysts bear only monodentate ligands, lends itself to facile modification of the catalyst structure. The latter attribute has been used in this study to alter the catalyst structure in order to maximize the efficiency of certain enyne RCM reactions (e.g., Tables 5and 6). The present investigations have therefore led to identification of several new catalyst variants that enhance the utility of this class of transformations. An additional example is provided in eq 5. Catalytic RCM of enyne 50 with Mo complex 5 affords diene 51 in only 45% yield as a result of a significant oligomerization, likely due to the absence of the quaternary carbon, which facilitates intramolecular cyclization (compare to RCM of 1 in Scheme 2). The catalysts bearing substituted phenoxide ligands, however, promote formation of 51 with substantially higher efficiency; complex 38a furnishes the desired product in 80% yield with a similarly exceptional degree of endo selectivity.
 |
(5) |
The first examples of enantioselective enyne RCM reactions are presented as well. Products are obtained in up to 98:2 er and >98:<2 endo:exo selectivity through desymmetrization reactions of N-containing dienynes in transformations promoted by enantiomerically pure stereogenic-at-Mo complexes, which have been shown to be exceptionally effective for RCM of various diene substrates. Many shortcomings remain to be addressed, including identification of catalysts that promote enantioselective reactions of enynes with O-containing tethers, a class of substrates particularly prone toward oligimerization. It is important to note that not only will discovery and development of new chiral catalysts be beneficial to enantioselective variants of endo-selective enyne RCM reactions, but such chiral complexes may prove to be superior catalysts in general and highly effective in cases where the desired product is achiral. Every Mo complex used in the present investigation is chiral; this class of chiral catalysts, used in the racemic form, promotes efficient endo-selective reactions. As has been described previously, the stereogenic Mo center and the attendant stereoelectronic attributes of such chiral catalysts render the complexes examined here effective RCM catalysts.9b,c These investigations provide a clear example of the principle that chiral catalysts should not be considered as potentially viable solutions only in cases where control of enantioselectivity is desired.
Future studies will be directed toward the design of more efficient catalysts for enyne RCM reactions, further development of the enantioselective variants, and applications to the synthesis of biologically active molecules.
Supplementary Material
Acknowledgment
The NIH (GM-59426) provided financial support. We are grateful to Dr. Bo Li (Boston College) for obtaining the X-ray crystal structures. We thank S. J. Malcolmson and S. J. Meek (Boston College) for valuable discussions. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
References
- (1).For reviews on metal-catalyzed enyne metathesis, see: Poulsen CS, Madsen R. Synthesis. 2003:1–18. Mori M. In: Handbook of Metathesis. Grubbs RH, editor. Vol. 2. Wiley-VCH; Weinheim: 2003. pp. 176–204. Diver ST, Giessert AJ. Chem. Rev. 2004;104:1317–1382. doi: 10.1021/cr020009e. Mori M. J. Mol. Catal. A. 2004;213:73–79. Villar H, Frings M, Bolm C. Chem. Soc. Rev. 2007;36:55–66. doi: 10.1039/b508899m. Maifield SV, Lee D. Chem. Eur. J. 2005;11:6118–6126. doi: 10.1002/chem.200500407.
- (2).For studies on mechanistic aspects of Ru-catalyzed enyne RCM reactions, see: Hansen EC, Lee D. J. Am. Chem. Soc. 2004;126:15074–15080. doi: 10.1021/ja045422d. Lippstreu JJ, Straub BF. J. Am. Chem. Soc. 2005;127:7444–7457. doi: 10.1021/ja042622g. Lloyd-Jones GC, Margue RG, de Vries JG. Angew. Chem., Int. Ed. 2005;44:7442–7447. doi: 10.1002/anie.200502243. Hansen EC, Lee D. Acc. Chem. Res. 2006;39:509–519. doi: 10.1021/ar050024g. Dieltiens N, Moonen K, Stevens CV. Chem. Eur. J. 2007;13:203–214. doi: 10.1002/chem.200600789.
- (3).For examples of application of exo-selective catalytic enyne RCM to natural product synthesis, see: Kinoshita A, Mori M. J. Org. Chem. 1996;61:8356–8357. Boyer F-D, Hanna I, Ricard L. Org. Lett. 2004;6:1817–1820. doi: 10.1021/ol049452x. Aggarwal VK, Astle CJ, Rogers-Evans M. Org. Lett. 2004;6:1469–1471. doi: 10.1021/ol049665m. Kitamura T, Sato Y, Mori M. Tetrahedron. 2004;60:9649–9657. Cesati RR, III, de Armas J, Hoveyda AH. J. Am. Chem. Soc. 2004;126:96–101. doi: 10.1021/ja0305407. Brenneman JB, Martin SF. Org. Lett. 2004;6:1329–1331. doi: 10.1021/ol049631e. Orain D, Mattes H. Synlett. 2005:3008–3010. Movassaghi M, Piizzi G, Siegel DS, Piersanti G. Angew. Chem., Int. Ed. 2006;45:5859–5863. doi: 10.1002/anie.200602011. Satcharoen V, McLean NJ, Kemp SC, Camp NP, Brown RCD. Org. Lett. 2007;9:1867–1869. doi: 10.1021/ol070255i. Hu F, Zhang Y-H, Yao Z-J. Tetrahedron Lett. 2007;48:3511–3515. Boyer FD, Hanna I. Org. Lett. 2007;9:715–718. doi: 10.1021/ol0630548. Paquette LA, Lai KW. Org. Lett. 2008;10:2111–2113. doi: 10.1021/ol800418m. Yang X, Zhai H, Li Z. Org. Lett. 2008;10:2457–2460. doi: 10.1021/ol800737d. Ramharter J, Mulzer J. Org. Lett. 2009;11:1151–1153. doi: 10.1021/ol9000137. Li J, Park S, Miller RL, Lee D. Org. Lett. 2009;11:571–574. doi: 10.1021/ol802675j. For examples of application of endo-selective catalytic enyne RCM to natural product synthesis, see: Layton ME, Morales CA, Shair MD. J. Am. Chem. Soc. 2002;124:773–775. doi: 10.1021/ja016585u.
- (4).Hoveyda AH, Zhugralin AR. Nature. 2007;450:243–251. doi: 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]
- (5).For a preliminary account of this work, see: Singh R, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2007;129:12654–12655. doi: 10.1021/ja075569f.
- (6).(a) Katz TJ, Sivavec TM. J. Am. Chem. Soc. 1985;107:737–738. [Google Scholar]; (b) Katz TJ. In: Handbook of Metathesis. Grubbs RH, editor. Vol. 1. Wiley-VCH; Weinheim: 2003. pp. 47–60. [Google Scholar]
- (7).Watanuki S, Ochifuji N, Mori M. Organometallics. 1994;13:4129–4130. [Google Scholar]
- (8).It is noteworthy that other, non-metathesis-based catalytic protocols, such as various cycloisomerization processes that are catalyzed by different metal-based complexes and involve enyne substrates, predominantly generate the corresponding exo products. For selected examples, see the following: (a) Pd-catalyzed: Trost BM, Tanoury GJ. J. Am. Chem. Soc. 1988;110:11863–11869. (b) Ru-catalyzed: Chatani N, Morimoto T, Muto T, Murai S. J. Am. Chem. Soc. 1994;116:6049–6050. (c) Pt-catalyzed: Fürstner A, Szillat H, Stelzer F. J. Am. Chem. Soc. 2001;123:6785–6786. doi: 10.1021/ja0109343. (d) Ir-catalyzed: Chatani N, Inoue H, Morimoto T, Muto T, Murai S. J. Org. Chem. 2001;66:4433–4436. doi: 10.1021/jo010091y. (e) Ag-catalyzed: Porcel S, Echavarren AM. Angew. Chem., Int. Ed. 2007;46:2672–2676. doi: 10.1002/anie.200605041. (f) Au-catalyzed: Cabello N, Jiménez-Núñez E, Bunuel E, Cardenas DJ, Echavarren AM. Eur. J. Org. Chem. 2007:4217–4223. (g) In-catalyzed: Miyanohana Y, Chatani N. Org. Lett. 2006;8:2155–2158. doi: 10.1021/ol060606d. (h) Rh-catalyzed: Ota K, Chatani N. Chem. Commun. 2008:2906–2907. doi: 10.1039/b805100c.
- (9).For previous reports regarding stereogenic-at-Mo complexes, see: Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933–937. doi: 10.1038/nature07594. Sattely ES, Meek SJ, Malcolmson SJ, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:943–953. doi: 10.1021/ja8084934. Ibrahem I, Yu M, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3844–3845. doi: 10.1021/ja900097n.
- (10).For reviews on metal-catalyzed enantioselective olefin metathesis reactions, see: Hoveyda AH. In: Handbook of Metathesis. Grubbs RH, editor. Vol. 2. Wiley-VCH; Weinheim: 2003. pp. 128–150. Schrock RR, Hoveyda AH. Angew. Chem., Int. Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576.
- (11).Based on previous theoretical studies (e.g., ref 2b), it is plausible that Ru-based complexes as well as high-oxidation-state Mo-based alkylidenes prefer initial association with an alkyne site of an enyne, unless the olefin presents a significantly less hindered alternative. It is, however, the more reactive Mo complexes that can readily proceed through the higher energy metallacycobutenes (vs metallacyclobutanes). That is, in Ru-catalyzed processes, it is the rate of metallacyclobutane or metallacyclobutene formation that is critical (vs the site of initial metal carbene binding). See also: Sohn J-H, Kim KH, Lee H-Y, No ZS, Ihee H. J. Am. Chem. Soc. 2008;130:16506–16507. doi: 10.1021/ja807717s.
- (12).(a) Hock AS, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2006;128:16373–16375. doi: 10.1021/ja0665904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Singh R, Czekelius C, Schrock RR, Müller P, Hoveyda AH. Organometallics. 2007;26:2528–2539. doi: 10.1021/om061134+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179. For an overview of Ru-based carbenes bearing bidentate styrene ether ligands, see: Hoveyda AH, Gillingham DG, Van Veldhuizen JJ, Kataoka O, Garber SB, Kingsbury JS, Harrity JPA. Org. Biomol. Chem. 2004;2:8–23. doi: 10.1039/b311496c.
- (14).For Ru-catalyzed RCM reactions of enynes with all-carbon tethers, see: Kim S-H, Bowden N, Grubbs RH. J. Am. Chem. Soc. 1994;116:10801–10802. Kim S-H, Zuercher WJ, Bowden NB, Grubbs RH. J. Org. Chem. 1996;61:1073–1081. Mori M, Sakakibara N, Kinoshita A. J. Org. Chem. 1998;63:6082–6083. doi: 10.1021/jo980896e. Renaud J, Graf C-D, Oberer L. Angew. Chem., Int. Ed. 2000;39:3101–3104. doi: 10.1002/1521-3773(20000901)39:17<3101::aid-anie3101>3.0.co;2-i. Ackermann L, Bruneau C, Dixneuf PH. Synlett. 2001:397–399. Schramm MP, Srinivasa Reddy D, Kozmin SA. Angew. Chem., Int. Ed. 2001;40:4274–4277. doi: 10.1002/1521-3773(20011119)40:22<4274::AID-ANIE4274>3.0.CO;2-#. Kitamura T, Sato Y, Mori M. Chem. Commun. 2001:1258–1259. Kitamura T, Sato Y, Mori M. Adv. Synth. Catal. 2002;344:678–693. Sashuk V, Grela K. J. Mol. Cat. A. 2006;257:59–66. See ref 3 for additional examples and applications to natural product synthesis.
- (15).For a catalytic RCM reaction involving allenynes as substrates, promoted by Mo-based complex 4, that delivers exo products selectively, see: Murakami M, Kadowaki S, Matsuda T. Org. Lett. 2005;7:3953–3956. doi: 10.1021/ol0514348.
- (16).For recent reviews regarding the use of catalytic olefin metathesis for preparation of heterocyclic structures in the context of natural product syntheses, see: Deiters A, Martin SF. Chem. Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872. Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem., Int. Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. Mori M. Adv. Synth Catal. 2007;349:121–135.
- (17).See the Supporting Information for more details.
- (18).For Ru-catalyzed RCM reactions of enyne substrates with N-containing tethers, see: (a) Reference 3c. (b) References 14c-i. Castarlenas R, Eckert M, Dixneuf PH. Angew. Chem., Int. Ed. 2005;44:2576–2579. doi: 10.1002/anie.200462865. Rosillo M, Domínguez G, Cassarubios L, Amador U, Pérez-Castells J. J. Org. Chem. 2004;69:2084–2093. doi: 10.1021/jo0356311. See ref 3 for additional examples and applications to natural product synthesis.
- (19).For Ru-catalyzed RCM reactions of enyne substrates withO-containing tethers, see: (a) Reference 14. Picquet M, Bruneau C, Dixneuf PH. Chem. Commun. 1998:2249–2250. Hoye TR, Donaldson SM, Vos TJ. Org. Lett. 1999;1:277–280. doi: 10.1021/ol9905912. Clark JS, Elustondo F, Trevitt GP, Boyall D, Robertson J, Blake AJ, Wilson C, Stammen B. Tetrahedron. 2002;58:1973–1982. See ref 3 for additional examples and applications to natural product synthesis.
- (20).Teng X, Cefalo DR, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2002;124:10779–10784. doi: 10.1021/ja020671s. [DOI] [PubMed] [Google Scholar]
- (21).Suezawa H, Yoshida T, Umezawa Y, Tsuboyama S, Nishio M. Eur. J. Inorg. Chem. 2002:3148–3155. [Google Scholar]
- (22).For Mo-catalyzed enantioselective RCM processes that deliver N-containing heterocycles, see: Dolman SJ, Sattely ES, Hoveyda AH, Schrock RR. J. Am. Chem. Soc. 2002;124:6991–6997. doi: 10.1021/ja012534l. Dolman SJ, Schrock RR, Hoveyda AH. Org. Lett. 2003;5:4899–4902. doi: 10.1021/ol036026n. Sattely ES, Cortez GA, Moebius DC, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2005;127:8526–8533. doi: 10.1021/ja051330s. For Mo-catalyzed enantioselective RCM reactions that afford other types of ring structures, see: Alexander JB, La DS, Cefalo DR, Hoveyda AH, Schrock RR. J. Am. Chem. Soc. 1998;120:4041–4042. La DS, Alexander JB, Cefalo DR, Graf DD, Hoveyda AH, Schrock RR. J. Am. Chem. Soc. 1998;120:9720–9721. Zhu SS, Cefalo DR, La DS, Jamieson JY, Davis WM, Hoveyda AH, Schrock RR. J. Am. Chem. Soc. 1999;121:8251–8259. Cefalo DR, Kiely AF, Wuchrer M, Jamieson JY, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2001;123:3139–3140. Kiely AF, Jernelius JA, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2002;124:2868–2869. doi: 10.1021/ja012679s. Jernelius JA, Schrock RR, Hoveyda AH. Tetrahedron. 2004;60:7345–7351. Lee A-L, Malcolmson SJ, Puglisi A, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2006;128:5153–5157. doi: 10.1021/ja058428r. (k) For applications of catalytic enantioselective RCM to natural product total synthesis, see refs 8b and c.
- (23).For Mo-catalyzed enantioselective ring-opening/cross-metathesis and/or ring-opening/ring-closing reactions, see: Weatherhead GS, Ford JG, Alexanian EJ, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2000;122:1828–1829. La DS, Sattely ES, Ford JG, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2001;123:7767–7778. doi: 10.1021/ja010684q. Cortez GA, Schrock RR, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:4534–4538. doi: 10.1002/anie.200605130. (d) Reference 8d. For Ru-catalyzed enantioselective ring-opening/cross-metathesis reactions, see: Gillingham DG, Kataoka O, Garber SB, Hoveyda AH. J. Am. Chem. Soc. 2004;126:12288–12290. doi: 10.1021/ja0458672. For a comparison of Mo- and Ru-based enantioselective ring-opening/cross-metathesis processes, see: Cortez GA, Baxter CA, Schrock RR, Hoveyda AH. Org. Lett. 2007;9:2871–2874. doi: 10.1021/ol071008h. For applications of catalytic enantioselective ring-opening reactions to natural product total synthesis, see: Weatherhead GS, Cortez GA, Schrock RR, Hoveyda AH. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5805–5809. doi: 10.1073/pnas.0307589101. Gillingham DG, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:3860–3864. doi: 10.1002/anie.200700501.
- (24).For Ru-catalyzed enantioselective ring-opening/cross-metathesis and cross-metathesis reactions developed in other laboratories, see: Berlin JM, Goldberg SD, Grubbs RH. Angew. Chem., Int. Ed. 2006;45:7591–7595. doi: 10.1002/anie.200602469.
- (25).For site-selective epoxidation reactions of endocyclic dienes, see: Evarts JB, Jr., Fuchs PL. Tetrahedron Lett. 2001;42:3673–3675. Yang D, Wong M-K, Yip YC. J. Org. Chem. 1995;60:3887–3889.
- (26).For representative procedures regarding functionalization of allylic epoxides, see: Cahiez C, Alexakis A, Normant JF. Synthesis. 1978:528–530. Yang D, Xu M. Org. Lett. 2001;3:1785–1788. doi: 10.1021/ol015722p.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.