

Abstract
IL-17R signaling is critical for pulmonary neutrophil recruitment and host defense against Gram-negative bacteria through the coordinated release of G-CSF and CXC chemokine elaboration. In this study, we show that IL-17R is localized to basal airway cells in human lung tissue, and functional IL-17R signaling occurs on the basolateral surface of human bronchial epithelial (HBE) cells. IL-17A and IL-17F were potent inducers of growth-related oncogene-α and G-CSF in HBE cells, and significant synergism was observed with TNF-α largely due to signaling via TNFRI. The activities of both IL-17A and IL-17F were blocked by a specific anti-IL-17R Ab, but only IL-17A was blocked with a soluble IL-17R, suggesting that cell membrane IL-17R is required for signaling by both IL-17A and IL-17F. Because IL-17A and IL-17F both regulate lung neutrophil recruitment, we measured these molecules as well as the proximal regulator IL-23p19 in the sputum of patients with cystic fibrosis (CF) undergoing pulmonary exacerbation. We found significantly elevated levels of these molecules in the sputum of patients with CF who were colonized with Pseudomonas aeruginosa at the time of pulmonary exacerbation, and the levels declined with therapy directed against P. aeruginosa. IL-23 and the downstream cytokines IL-17A and IL-17F are critical molecules for proinflammatory gene expression in HBE cells and are likely involved in the proinflammatory cytokine network involved with CF pathogenesis.
IL-17 is a proinflammatory cytokine that regulates both granulopoiesis and recruitment of neutrophils into sites of inflammation (1–5). This is due in part to the ability of IL-17A to induce the release of CXC chemokines (4, 6, 7) as well as regulate the expression of G-CSF (2, 7, 8), a critical granulopoietic growth factor. Mice with a homozygous deletion of the IL-17R have enhanced lethality, defective neutrophil recruitment, and granulopoiesis to experimental Gram-negative pneumonia (2), whereas they do not have an increased susceptibility to intracellular infections caused by Listeria monocytogenes or Mycobacteria tuberculosis (our unpublished observations). This defect in host defense is likely due in part to a >90% reduction in G-CSF in response to Gram-negative bacterial challenge in IL-17R-deficient mice compared with control mice as well as a significantly attenuated granulopoietic response to infection (2). Recently, five other members of the IL-17 family have been described (6, 9–13) with IL-17F (10, 14) having the closest sequence homology (58% at the protein level) as well as similar induction of CXC chemokines and neutrophil mobilization as IL-17 (12). IL-17A and IL-17F lie immediately downstream from each other on mouse chromosome 1 and human chromosome 6, and both cytokines are induced by T cells in response to IL-23 (15–17). Furthermore, IL-17A and IL-17F are induced in a similar time course in the lung, in experimental animal model of Gram-negative pneumonia (our unpublished observations). Due to similar biological activity, there has been speculation whether both IL-17A and IL-17F signal via the IL-17R, although IL-17F has at least an order of magnitude lower affinity for IL-17R than IL-17 (14).
Based on these data, we undertook studies to immunolocalize the IL-17R in human lung and investigate growth factor and chemokine induction by both IL-17 and IL-17F in polarized human bronchial epithelial (HBE)3 cells grown at an air-liquid interface (18). In human lung, the IL-17R is expressed in respiratory epithelial cells as well as in lung parenchymal cells. The greatest expression was observed on the basolateral surfaces of respiratory epithelial cells in lung tissue. Based on these data, studies designed to investigate apical vs basolateral signaling by IL-17A and IL-17F revealed that growth factor induction was significantly more potent with basolateral-supplied ligand. HBE cell supernatants were screened using Luminex cytokine beads, which assay IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13, IL-17, G-CSF, GM-CSF, IFN-γ, MCP-1, MIP-1b, and TNF-α, as well as growth-related oncogene (GRO)-α by ELISA. Among these cytokines/chemokines, we observed the greatest induction of GRO-α, G-CSF, IL-6, and IL-8 in HBE cells from at least seven donors. In each case, the response to both IL-17A and IL-17F was always greater with basolaterally applied ligand, and there was significant attenuation of cytokine/chemokine induction by blocking IL-17R with a neutralizing mAb. IL-17A and IL-17F had synergistic induction of GRO-α and G-CSF when combined with TNF-α. Both TNFRI and TNFRII were immunolocalized to the cell surface below apical tight junctions, and functional synergy occurred only with TNF-α applied basolaterally to HBE cells. Furthermore, this synergism was blocked by an anti-TNFRI Ab, demonstrating the critical role of this TNFR in IL-17A and IL-17F synergy. Moreover, the bioactivity of IL-17A and IL-17F were blocked with an anti-IL-17R mAb, whereas a soluble IL-17R only blocked IL-17A. These data suggest that cell surface IL-17R is critical for IL-17A and IL-17F bioactivity, but the ligand binding affinity of IL-17F for soluble IL-17R is not strong enough to permit effective neutralization. Finally, because IL-17A has been shown to be as critical for neutrophil recruitment in response to Gram-negative bacteria in the lung, we assayed IL-17A and IL-17F in the sputum of consecutive adult patients with cystic fibrosis (CF) undergoing a pulmonary exacerbation. We choose CF because these patients are most often colonized with Gram-negatives, and CF is characterized by chronic neutrophilic inflammation (19). IL-17A and IL-17F were detectable in all patients on day 1 of hospitalization and showed a significant decline with treatment of the pulmonary exacerbation. Taken together, these data demonstrate that IL-17R signaling occurs basolaterally in HBE cells, resulting in CXC chemokines and granulopoietic factors, which result in neutrophil recruitment and may be critical in endobronchial infection as observed in patients with CF. Moreover, use of a soluble IL-17R is specific to inhibit IL-17A bioactivity but not IL-17F, whereas targeting the cell surface receptor anti-IL-17R mAb would likely inhibit both IL-17A and IL-17F.
Materials and Methods
Primary cell culture from human airway tissues
HBE cells were isolated from native lungs of transplant recipients, or unused sections of the donor lungs as described previously (20). Airways were dissected from surrounding adventitial tissue and placed in ice-cold HEPES-buffered MEM containing penicillin, streptomycin, and amphotericin B. After multiple washes with cold HBSS, cartilaginous airway segments were cut longitudinally and incubated overnight at 4°C in 0.1% protease XIV (Sigma-Aldrich). Airway epithelial cells were obtained by gently scraping the epithelium with the blunt end of forceps. Recovered cells were plated on type IV human placental collagen (Sigma-Aldrich)-coated tissue culture plates in 1:1 mixture of bronchial epithelial growth medium (Cambrex) and keratinocyte serum-free medium (Invitrogen Life Technologies). After 5–7 days under these conditions, cells were trypsinized, washed in HBSS, and seeded onto type IV human placental collagen-coated Corning/Costar Transwell filters at 100% confluence in bronchial epithelial growth medium/keratinocyte serum-free medium. After 24 h, cells were placed at air-liquid interface by removing apical media from the Transwell filter, and basolateral media were replaced with DMEM-F12 (Invitrogen Life Technologies) containing 2% Ultroser G (BioSepra) to promote differentiation. Under biphasic culture conditions, a mucociliary epithelium with the formation of cilia and mucus-secreting granules was observed. The cultures were deprived of serum 24 h before initiating cytokine treatment.
Cytokines and Ab treatment
IL-17A and IL-17F (R&D Systems) were dissolved in DMEM-F12 and added directly to both the apical and/or basal surfaces of primary HBE cultures at concentrations of 0, 1, 10, and 100 ng/ml. TNF-α (BioSource International) was used at a concentration of 1 ng/ml. For IL-17R neutralizing studies, a monoclonal anti-human IL-17R Ab or mouse IgG1 isotype control (R&D Systems) was added to the cultures at 2 μg/ml, which is 10-fold the ED50 to block IL-17-mediated IL-6 secretion in human dermal fibroblasts. A recombinant human IL-17R:Fc chimera (used at 1 μg/ml) was also purchased from R&D Systems. For TNFRs neutralization studies, we used anti-human TNF-RI (BioSource International) at a concentration of 10 μg/ml and/or recombinant human TNF-RII:Fc chimera (R&D Systems) at 0.5 μg/ml.
Bio-Plex and ELISA measurements
A Bio-Plex human cytokine assay (Bio-Rad) for simultaneous quantitation of IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13, IL-17, G-CSF, GM-CSF, IFN-γ, monocyte chemoattractant protein (MCP)-1, MIP-1b, and TNF-α in apical and basolateral media was run according to the recommended procedure. G-CSF and GRO-α were measured using separate ELISA kits (R&D Systems), following the manufacturer's instructions. Human IL-17F was measured using Abs provided by Wyeth.
Immunohistochemistry
Anti-human IL-17R Ab (Santa Cruz Biotechnology) was used to characterize the expression of IL-17R on respiratory epithelial cells from human lung tissue sections. The staining was conducted using Cy-3-conjugated rabbit anti-goat as secondary Ab (Sigma-Aldrich) and Fluoromount G as mounting medium. Rabbit serum was used for blocking prestaining. The staining photographs were captured by a camera attached to an Olympus Provis fluorescent microscope, and images were further analyzed with Magnafire software (Olympus).
To characterize the expression of TNFRs I and II on polarized HBE cells grown on air-liquid interface, we used mouse anti-human TNF-RI and TNF-RII mAbs (R&D Systems) and Alexa 488 goat anti-mouse as secondary Ab (Molecular Probes). Finally, we used ProLong Gold antifade with 4′,6′-diamidino-2-phenylindole as mounting medium (Molecular Probes). We captured the images by a camera attached to an Axioplan 2 universal imaging microscope (Intelligent Imaging Innovations) and further analyzed them with SlideBook 4.0 (Intelligent Imaging Innovations) and MetaMorph (Universal Imaging) software.
Human subjects
Adult patients with CF (mean age, 22 years) who were colonized with Pseudomonas aeruginosa and undergoing pulmonary exacerbation and requiring hospitalization were enrolled in a study to measure biomarkers of inflammation in sputum on days 1, 10, and 20 after initiation of antibiotics and intensified respiratory therapy. Sputum samples were processed using Sputolysin (Dade Behring). Briefly, 1 ml of 10% Sputolysin was added per 1 mg of sputum, and the sample was incubated for 5 min at 37°C with vigorous shaking and mixed vigorously with a transfer pipette. Samples were then centrifuged at 2000 × g rpm for 5 min at 4°C, and the supernatants were assayed by Bio-Plex and ELISA. All subjects gave written informed consent to procedures, and the study was approved by the local Institutional Review Board.
Western blot analysis
Western blot samples from processed sputum were separated (12.4 μg of protein per lane) on SDS-PAGE. Protein separated on gels were transferred onto Immobilon-P membranes (Millipore) at 140 mA for 1 h. The membranes were blocked overnight at 4°C with PBS containing 5% BSA. The blots were stained with anti-p19 Ab (rabbit anti-human) for 1 h at room temperature and developed by incubation with a secondary alkaline phosphatase-conjugated goat anti-rabbit IgG (Bio-Rad) and 5-bromo-4-chloro-3-indolyl phosphate/NBT reagent (Bio-Rad).
Statistical analysis
Data were analyzed using StatView statistical software (Brain Power). Comparisons between groups where data were normally distributed were made with Student's t test, and comparisons among multiple groups or nonparametric data were made with ANOVA. Scheffe's test was the post hoc test used. The Mann-Whitney U test or the Wilcoxon paired-sample test was used to make ordinal comparisons. Significance was accepted at a p value of <0.05.
Results
IL-17A and IL-17F up-regulate G-CSF, GRO-α, and MCP-1 in HBE cells: kinetic studies
Using Bio-Plex and ELISA, we screened both apical and basolateral media for cytokines/chemokines regulated by IL-17A and IL-17F in human primary bronchial epithelial cells grown at the air-liquid interface (see Material and Methods). In addition to IL-8 and IL-6, two factors already reported to be induced by IL-17A (data not shown), we detected a significant induction in G-CSF, GRO-α, and MCP-1 secretion at 24 h in primary HBE cells treated with IL-17A and IL-17F (Table I). Due to variability in the absolute amount of growth factor secreted from different airway donors, remaining data are graphed as fold induction. These effects were dose dependent (Fig. 1A, and Table I) with a maximal effect observed at a concentration of 100 ng/ml. IL-17A was more potent than IL-17F on a mass basis to induce G-CSF, GRO-α, and MCP-1 at 24 h. A time course performed with 10 ng/ml IL-17A and IL-17F showed that the effect of IL-17A and IL-17F on G-CSF, GRO-α, and MCP-1 were time dependent (Fig. 1B) with a maximum effect at 24 h. Based on these kinetic studies, we performed most of the next experiments using a concentration of IL-17A or IL-17F of 10 ng/ml and a incubation time of 24 h.
Table I. Concentration of G-CSF, GRO-α, and MCP-1 in basolateral media after 24 h of HBE stimulation with IL-17A and IL-17F.
| IL-17A | G-CSF | GRO-α | MCP-1 |
|---|---|---|---|
| IL-17A | |||
| 0 ng/ml | 401.2 ± 32.24 | 2,012.1 ± 102.34 | 22.01 ± 1.98 |
| 1 ng/ml | 829.56 ± 128.38 | 8,412.1 ± 503.02 | 82.8 ± 6.6 |
| 10 ng/ml | 2,029.3 ± 192.57 | 11,144.7 ± 643.87 | 98.83 ± 6.16 |
| 100 ng/ml | 3,546.24 ± 296.88 | 15,140.7 ± 1026.17 | 118.5 ± 8.8 |
| IL-17F | |||
| 0 ng/ml | 401.2 ± 32.24 | 2,012.1 ± 102.34 | 22.01 ± 1.98 |
| 1 ng/ml | 655.4 ± 44.13 | 5,798.1 ± 382.30 | 46.75 ± 2.64 |
| 10 ng/ml | 1,482 ± 112.33 | 9,729.2 ± 804.84 | 43.36 ± 4.13 |
| 100 ng/ml | 2,236 ± 164.49 | 14,175.4 ± 865.20 | 68.5 ± 6.61 |
FIGURE 1.
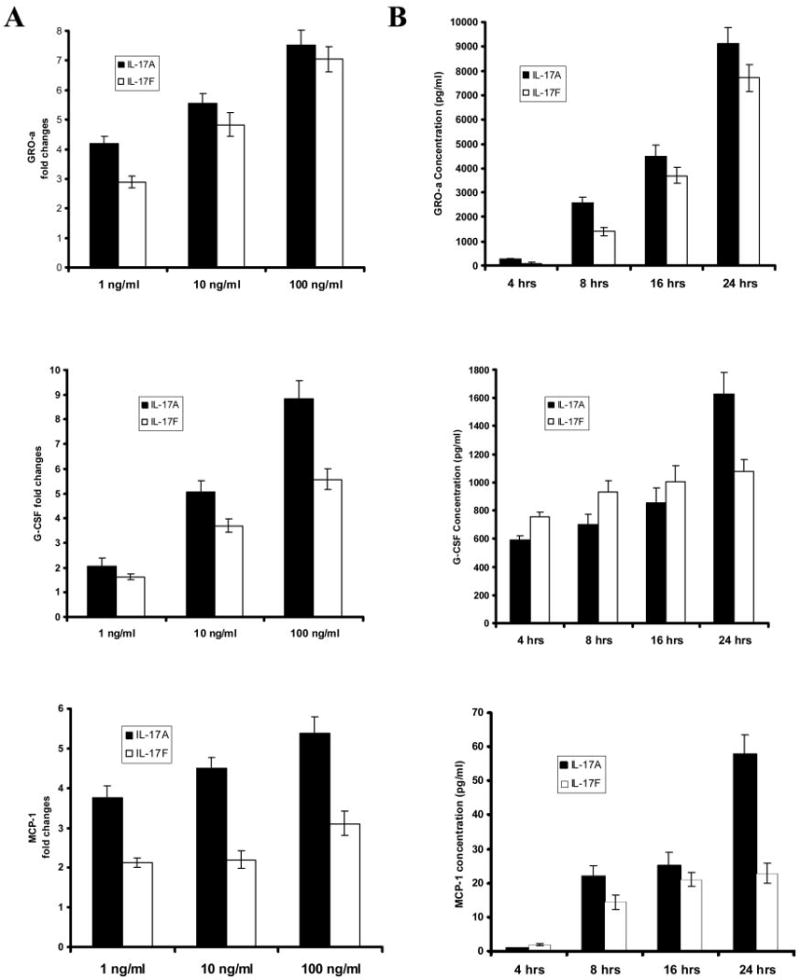
A, Dose-dependent elevation of GRO-α, G-CSF, and MCP-1 protein levels by recombinant human IL-17A and IL-17F. Primary HBE cells were treated with different doses of IL-17A and IL-17F, as indicated. Basolateral media were collected 24 h after the treatment, and cytokine concentrations were measured by ELISA. B, Time course study. HBE cells were stimulated with IL-17A and IL-17F (both at 10 ng/ml), and basolateral media were collected after 4, 8, 16, and 24 h for G-CSF, GRO-α, and MCP-1 measuring. Cytokine concentrations are shown in fold changes vs control. Results are expressed as the mean ± SEM of triplicate samples from one representative experiment.
IL-17F is synergistic with TNF-α for G-CSF and GRO-α secretion
Because synergy of IL-17A with TNF-α has been reported, we determined the effect of combining IL-17F (10 ng/ml) and TNF-α (1 ng/ml) to up-regulate G-CSF and GRO-α secretion by primary HBE cells. Optimal concentration of cytokines had been determined in previous experiments (data not shown). HBE cells showed a synergistic effect in GRO-α and G-CSF secretion when IL-17F was combined with TNF-α for 24 h (Fig. 2, A and B). This synergistic effect was neutralized by preincubating the stimulating cytokine mixture with an anti-IL-17R mAb, but not with a soluble IL-17R:Fc chimera recombinant protein or an isotype-matched control Ab (isotype data not shown). However, both anti-IL-17R mAb and soluble IL-17R:Fc proteins were effective in inhibiting IL-17A-induced increases in G-CSF (Fig. 2C). These data strongly suggest that membrane IL-17R is critical for both IL-17A and IL-17F-induced G-CSF responses.
FIGURE 2.

GRO-α (A) and G-CSF (B) secreted after stimulation with IL-17F and/or TNF-α. primary HBE cells were treated with IL-17F (10 ng/ml) and TNF-α (1 ng/ml). The cytokine mixture (IL-17F + TNF-α) was also preincubated with anti-IL-17 mAb (2 μg/ml), recombinant human IL-17R:Fc (1 μg/ml), or isotype-matched controls. Basolateral media were collected 24 h after the treatment, and cytokine levels were determined by ELISA. Results are expressed as the mean ± SEM of triplicate samples from one representative experiment (*, p < 0.05). C, G-CSF secreted after stimulation with IL-17A (10 ng/ml) and/or TNF-α. Cells were treated as outlined above. Results are expressed as the mean ± SEM of triplicate samples from one representative experiment (*, p < 0.05).
GRO-α and G-CSF secretion induced by IL-17A and IL-17F is decreased by anti-IL-17R Ab
To determine polarization of GRO-α and G-CSF secretion in response to IL-17A and IL-17F, primary HBE cells were stimulated with IL-17A and IL-17F for 24 h, and GRO-α and G-CSF were assayed in apical or basolateral fluid. Both GRO-α and G-CSF were secreted both apically and basolaterally, with GRO-α showing a greater induction in basolateral secretion compared with G-CSF (Fig. 3). Preincubation with anti-IL-17R Ab significantly abrogated GRO-α and G-CSF secretion induction mediated by both IL-17A and IL-17F in apical and basolateral media (Fig. 3). These results support the notion that the IL-17R is required for both IL-17A and IL-17F activity on HBE cells to induce G-CSF and GRO-α production.
FIGURE 3.
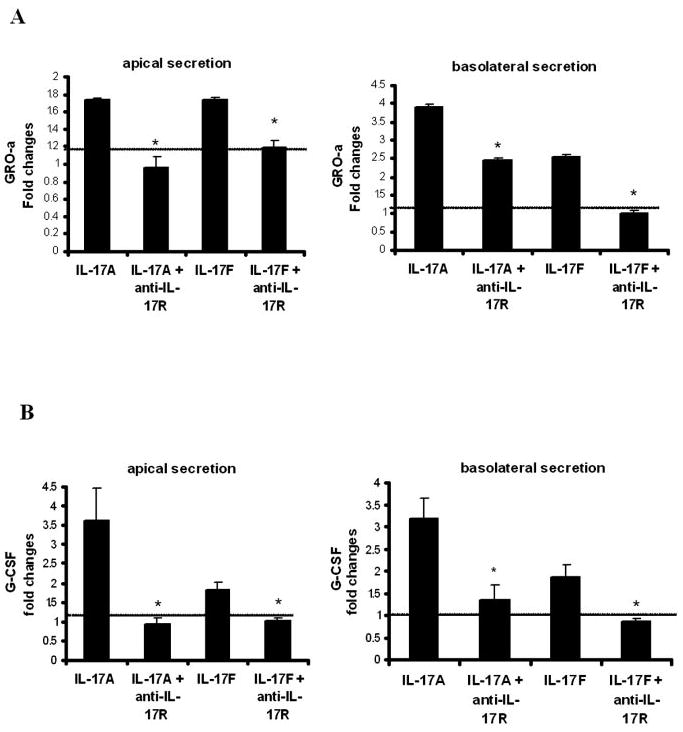
Effects of blocking IL-17R on GRO-α (A) and G-CSF (B) production by IL-17A and IL-17F. Primary HBE cells were pretreated with IL-17R Ab (2 μg/ml) 30 min before IL-17A and IL-17F treatment (both at 10 ng/ml). Apical and basolateral medium was collected 24 h later. Results are expressed as the mean ± SEM of three separate experiments (*, p < 0.05).
IL-17R is functionally expressed on the basolateral surface of respiratory epithelial cells
Immunohistochemical staining for IL-17R was performed on frozen sections of human lung specimens. The IL-17R was found to be expressed in respiratory epithelial cells as well as in lung parenchymal cells. Moreover, it was localized mainly to the basolateral surface of respiratory epithelial cells (Fig. 4A, left panel). As a negative control, a section was stained only with secondary Ab, and it did not show unspecific staining (Fig. 4A, right panel). To confirm the immunohistochemical findings, we designed an experiment in which HBE cells were incubated with IL-17A or IL-17F in basolateral or apical media for 24 h. We assayed conditioned basolateral media for G-CSF and GRO-α and found that both growth factors were up-regulated when IL-17A and IL-17F were applied in basolateral media, but no induction of GRO-α or G-CSF was observed when IL-17A or IL-17F were applied apically (Fig. 4B). Taken together, these data strongly suggest that IL-17R signaling occurs basolaterally in HBE cells.
FIGURE 4.
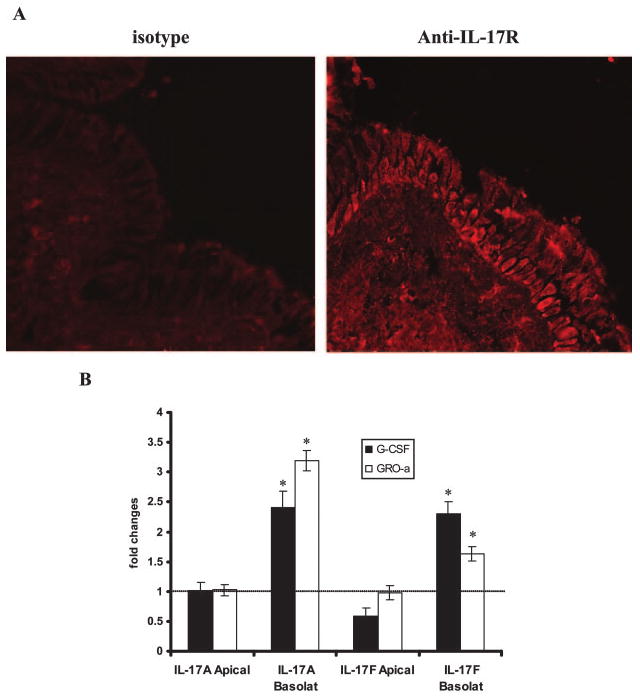
Detection and localization of the IL-17R. A, Representative immunohistochemical staining for IL-17R in a human lung section with a specific detecting monoclonal anti-IL-17R Ab, showing basolateral localization of the IL-17R. B, G-CSF and GRO-α secretion by primary HBE cells after addition of IL-17A and IL-17F (both at 10 ng/ml) to basolateral or apical surface. Basolateral media were collected after 24 h, and cytokine levels were measured by ELISA. Results are expressed as the mean ± SEM of triplicate samples from one representative experiment (*, p < 0.05).
TNFRs I and II are structurally and functionally expressed on the basolateral surface of respiratory epithelial cells
TNFRs I and II were immunohistochemically stained on polarized primary HBE cells grown on Transwell membranes using anti-human TNF-RI and anti-human TNF-RII mAbs. Both receptors were found to be expressed in HBE cells (Fig. 5A, left and middle upper panels). As a negative control, a filter was stained only with secondary Ab, and it did not show unspecific staining (Fig. 5A, upper right panel). Furthermore, x–z axis reconstruction showed that TNF-RI and TNF-RII localized to the lateral membranes of HBE cells, below tigh junctions (Fig. 5A, lower panels). To confirm the immunohistochemical findings, we designed an experiment in which HBE cells were incubated with IL-17F and/or TNF-α in basolateral or apical media for 24 h. We assayed conditioned basolateral media for G-CSF and found that it was up-regulated when IL-17F and/or TNF-α were applied in basolateral media, but no induction of G-CSF was observed when IL-17F and/or TNF-α were applied apically (Fig. 5B). Taken together, these data suggest that the signaling that leads to synergism between IL-17F and TNF-α occurs basolaterally in HBE cells.
FIGURE 5.
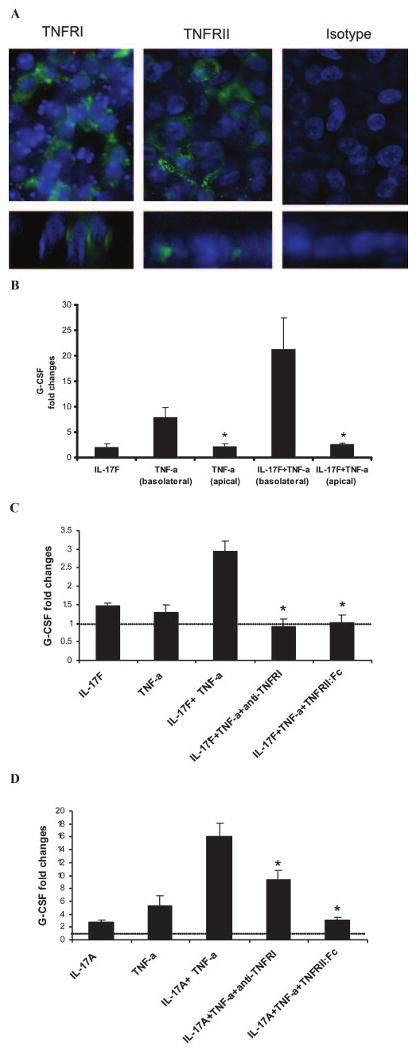
Detection and localization of TNFRs, TNF-RI, and TNF-RII. A, Immunohistochemical staining of TNF-RI, TNF-RII, and isotype-matched control. Conventional xy images are shown on upper panels, and x–z axis reconstructions are shown in lower panels. B, G-CSF secretion by primary HBE cells after addition of IL-17F and/or TNF-α (both at 10 ng/ml) to basolateral or apical surface. Basolateral media were collected after 24 h, and G-CSF levels were measured by ELISA. Results are expressed as the mean ± SEM of triplicate samples from one representative experiment (*, p < 0.05). C, Primary HBE cells were pretreated with anti-human TNF-RI and/or recombinant TNF-RII:Fc chimera (0.5 μg/ml) 2 h before IL-17F and/or TNF-α treatment (both at 10 ng/ml). Basolateral media were collected after 24 h, and G-CSF levels were measured by ELISA. Results are expressed as the mean ± SEM of triplicate samples from one representative experiment (*, p < 0.05). D, Primary HBE cells were pretreated with anti-human TNF-RI and/or recombinant TNF-RII:Fc chimera (0.5 μg/ml) 2 h before IL-17A and/or TNF-α treatment (both at 10 ng/ml). Basolateral media were collected after 24 h, and G-CSF levels were measured by ELISA. Results are expressed as the mean ± SEM of triplicate samples from one representative experiment (*, p < 0.05).
To address the importance of the TNFRs I and II on the signaling required for synergism between IL-17F and TNF-α, we preincubated HBE cells with anti-human TNF-RI, recombinant human TNF-RII:Fc chimera, and with both neutralizers for 2 h before the addition of the cytokines. We observed that the synergistic effect on G-CSF secretion after combining IL-17F and TNF-α was blocked by anti-human TNF-RI and by recombinant TNF-RII:Fc chimera. Unexpectedly, the levels of G-CSF secreted by HBE cells in response to the combination of IL-17F and TNF-α in the presence of either of the TNFR neutralizers were lower than the levels of G-CSF secreted by HBE cells in response to IL-17F stimulation, suggesting that even when IL-17F is applied alone to HBE cultures, it has a synergistic effect by interacting with TNF-α that is tonically secreted by these cells.
IL-23, IL-17A, and IL-17F are increased in CF patients undergoing pulmonary exacerbation
CF is a lung disease characterized by persistent endobronchial infection, neutrophilic lung inflammation (21), and high sputum CXCL8 levels (22, 23). Because we have shown previously that IL-17R signaling is critical for CXCL2 expression in murine lung in response to Gram-negative infection, we hypothesized that IL-17A and IL-17F would be up-regulated in the sputum of CF patients undergoing pulmonary exacerbation. In support of this, preliminary studies demonstrated higher IL-17A levels in patients with CF undergoing bronchoscopy for ongoing pulmonary exacerbation compared with controls with chronic cough due to asthma or gastroesophageal reflux disease (data not shown). Because these samples could be subject to selection bias due to the decision to clinically perform bronchoscopy, we elected to investigate IL-17A, IL-17F, and the proximal mediator IL-23 (p19) in sputum samples from eight adult CF patients (mean age, 22 years) undergoing pulmonary exacerbation requiring hospitalization and i.v. antibiotics. On day 1 of hospitalization, IL-17A and IL-17F were readily detectable when compared with sputum samples collected from four non-CF patients (mean ± SEM, 59.58 ± 5.22 vs 4.17 ± 2.13 pg/ml for IL-17A and 84.67 ± 10.87 vs 20.1 ± 3.25 pg/ml for IL-17F). Sputum was collected and analyzed serially during the antibiotic treatment. IL-17A and IL-17F concentration dramatically decreased by day 20 (Fig. 6A), reaching levels similar to non-CF patients. We also measured a panel of 18 other cytokines in the sputum of these patients using Luminex cytokine beads and found that that IL-8, G-CSF, IL-6, GRO-α, MCP-1, MIP-1b, TNF-α, GM-CSF, and IL-1b were also increased at day 1 of hospitalization and impressively reduced by day 20 (Fig. 6B), showing a pattern similar to IL-17A and IL-17F. Similar expression patterns were seen whether cytokine/chemokine concentrations were corrected for total protein content or not. Finally, because IL-23, a product largely of macrophages and dendritic cells, is a proximal regulator of IL-17A and IL-17F, we assayed for the presence of IL-23 p19 protein by Western blot. We observed detectable IL-23 in all of the patients undergoing CF exacerbation, which was higher at day 0 of hospitalization and declined by day 20 (Fig. 6C).
FIGURE 6.
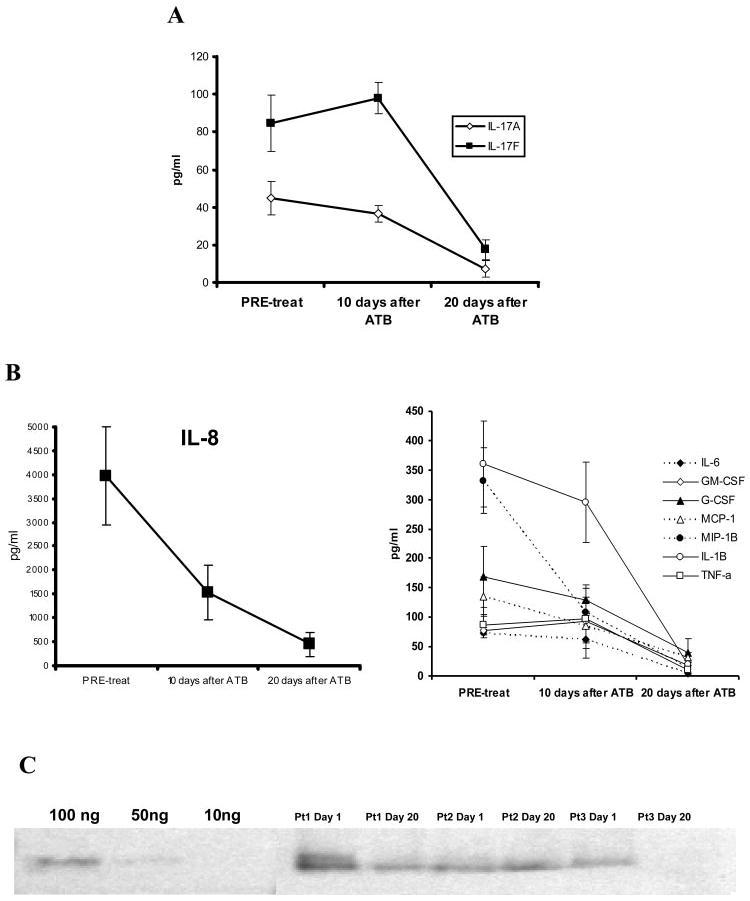
Cytokines levels in sputum from CF patients undergoing pulmonary exacerbation. Panel A: IL-17A and IL-17F concentration in sputum from CF patients at different time points, as indicated, were measured by ELISA. Panel B: IL-17- induced cytokines IL-8 (upper figure) G-CSF, GRO-a, MCP-1, MIP-1b, GM-CSF and IL-1b (lower figure) levels were measured in sputum samples at the same time points, using Luminex cytokine beads and ELISA. Panel C: Western blot of IL-23p19 in the sputum of patients undergoing pulmonary exacerbation of CF.
Discussion
IL-17A and IL-17F are products of activated T cells (6) in response to both infectious (8) and antigenic stimuli (24). Gram-negative bacteria and specifically LPS appear to induce IL-17A and IL-17F through TLR4-dependent and IL-23-dependent pathways (17, 25, 26). Overexpression of IL-17A or IL-17F in the lung results in the induction CXC chemokines and neutrophil recruitment (8, 12). Deficiency of IL-17R signaling through gene targeting results in an enhanced susceptibility to Gram-negative bacterial pulmonary infection with defects both in granulopoiesis and pulmonary neutrophil recruitment (2). Neutralization of IL-17A also has been reported to diminish LPS-induced lung neutrophil recruitment (4) (27). The defect in granulopoiesis in IL-17R KO mice is associated with a >90% reduction in G-CSF release (2). Moreover, systemic overexpression of IL-17A results in a marked induction in granulopoiesis, which is in part G-CSF dependent (28, 29).
To better define IL-17A and IL-17F's regulation of G-CSF and the CXC chemokine GRO-α in the lung, we examined IL-17R expression in lung tissue and found significant expression in basal respiratory epithelial cells. Incubation of polarized HBE cells with both IL-17A and IL-17F resulted in similar profiles of cytokine responses as measured by Bio-Plex with the induction of IL-8, IL-6 (data not shown), in addition to G-CSF and GRO-α. We also demonstrated that IL-17F synergizes with TNF-α to further induce G-CSF and GRO-α by bronchial epithelial cells isolated from the human lung. In contrast to our findings, Numasaki et al. (30) reported that IL-17F has an inhibitory effect on TNF-α-induced secretion of G-CSF. However, this study was performed in lung microvascular endothelial cells, which may differ in this response.
Both IL-17A and IL-17F appear to require the cell surface IL-17R for induction of GRO-α and G-CSF secretion because a mAb specific for the IL-17R significantly attenuated the release of these cytokines to IL-17A and IL-17F. However, IL-17F has a low ligand binding efficiency with this receptor (14), and IL-17F has recently been shown in vitro to bind to IL-17RC (31). In support of these data, a soluble IL-17R was efficient in inhibiting IL-17A bioactivity but not IL-17F in HBE cells. These data suggest that binding affinity of IL-17F is different for the cell membrane receptor or that a coreceptor complex involving IL-17R is required (15) for IL-17F responses. One other possibility, which we cannot exclude at this time, is cross-reactivity of the mAb to IL-17RC; however, this is unlikely because homology of IL-17RC to IL-17R is only 15% (32). Moreover, the bioactivity of both IL-17A and IL-17F and TNF-α was greatest when the ligands were applied basolaterally, suggesting that functional IL-17A and IL-17F and TNF-α signaling likely occurs through the basolateral surface of airway epithelial cells. This receptor localization teleologically makes sense because a prominent potential source of IL-17A and IL-17F are activated T cells, which can reside in the submucosal space (15). In fact, Langrish et al. (40) have recently defined a population of ThIL-17 cells, which coexpress IL-17A and IL-17F as well as TNF-α. Thus, ThIL-17 cells may represent a critical population of cells that interact with HBE that mediate inflammatory responses. Using soluble TNF-α, we demonstrate that TNFRI is critical for synergy with IL-17A and IL-17F. However, because HBE cells also express TNFRII, these cells may also respond to cell surface TNF expressed on ThIL-17 cells, which signals preferentially via TNFRII (33). Notably, the concentrations used to elicit G-CSF and GRO-α responses in HBE cells is ∼ 10–100 times higher than that detected in sputum (Fig. 6). This likely reflects the fact that local tissue concentration in the lung may be higher than that in sputum, which is rich in proteases, or the fact that IL-17A and IL-17F may require synergistic cytokines such as TNF-α to signal at picograms/milliliter concentrations (32). The mechanism of synergy of TNF-α and IL-17A and IL-17F has not been elucidated completely, but one mechanism may be synergistic induction of transcription factors such as C/EBPδ that drive subsequent gene transcription (34).
IL-17A has been reported to be up-regulated in many inflammatory autoimmune diseases including rheumatoid arthritis (35), multiple sclerosis (36), and in inflammatory bowel disease (37). It has been shown recently that T cell-derived IL-17A and IL-17F are regulated by TLR4 on macrophages and dendritic cells and subsequent IL-23 production by these cells (38–40). Moreover, IL-17A and IL-17F have similar chromosomal location and likely arose from a gene duplication event. Based on their ability to mediate lung neutrophilia (41), and the fact that chronic inflammation in CF is neutrophil predominant, we hypothesized that IL-17A and IL-17F likely play a role in airway inflammation in the setting of chronic Gram-negative bacterial infections such as bronchiectasis or CF.
Toward this end, we found that both IL-17A and IL-17F were elevated in the sputum of adult CF patients undergoing a pulmonary exacerbation. Moreover, IL-17A and IL-17F elevations were associated with previously identified inflammatory mediators such as IL-8 (42) and G-CSF (43), suggesting that these IL-17 family members may play a role in ongoing neutrophil recruitment into the airway of these patients. Furthermore, we postulate that IL-17A and IL-17F may regulate CXC chemokine and G-CSF release in patients with CF. We also found delectable IL-23p19 by Western blot in concentrated sputum that may approach levels of 100 ng/ml, which is well within the range for human T cell production of IL-17 (44).
These data are the first to measure IL-17F in clinical samples. Because chronic inflammation is thought to be critical to loss of lung function in the setting of CF, our data suggest that IL-17A and IL-17F are two IL-17 family members that represent excellent therapeutic targets to antagonize neutrophil-mediated inflammation. Moreover, a strategy that antagonizes cell surface IL-17R signaling may likely block both the action of IL-17A and IL-17F, whereas a strategy using soluble IL-17R will predominately block IL-17A.
Acknowledgments
We thank Victor VanCleave at Wyeth Research for development of the human IL-17F ELISA.
Footnotes
This work was supported by Public Health Service Grants HL061271 and HL062052 (to J.K.K.).
Abbreviations used in this paper: HBE, human bronchial epithelial; GRO, growth-related oncogene; CF, cystic fibrosis; MCP, monocyte chemoattractant protein; ATB, antibiotics.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- 2.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–528. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolls JK, Kanaly ST, Ramsay AJ. Interleukin-17: an emerging role in lung inflammation. Am J Respir Cell Mol Biol. 2003;28:9–11. doi: 10.1165/rcmb.2002-0255PS. [DOI] [PubMed] [Google Scholar]
- 4.Laan M, Cui ZH, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, Skoogh BE, Lindén A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 5.Lindén A, Hoshino H, Laan M. Airway neutrophils and interleukin-17. Eur Respir J. 2000;15:973–977. doi: 10.1034/j.1399-3003.2000.15e28.x. [DOI] [PubMed] [Google Scholar]
- 6.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 7.Jones CE, Chan K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-α, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am J Respir Cell Mol Biol. 2002;26:748–753. doi: 10.1165/ajrcmb.26.6.4757. [DOI] [PubMed] [Google Scholar]
- 8.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335–340. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, Dowd P, Gurney AL, Wood WI. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc Natl Acad Sci USA. 2000;97:773–778. doi: 10.1073/pnas.97.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, Hromas R. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol. 2001;167:4137–4140. doi: 10.4049/jimmunol.167.8.4137. [DOI] [PubMed] [Google Scholar]
- 11.Starnes T, Broxmeyer HE, Robertson MJ, Hromas R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol. 2002;169:642–646. doi: 10.4049/jimmunol.169.2.642. [DOI] [PubMed] [Google Scholar]
- 12.Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, Menon S, Seymour B, Jackson C, Kung TT, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol. 2002;169:443–453. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukocyte Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- 14.Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20:5332–5341. doi: 10.1093/emboj/20.19.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chmiel JF, Berger M, Konstan MW. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol. 2002;23:5–27. doi: 10.1385/CRIAI:23:1:005. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal S, Ghilardi N, Xie MH, De Sauvage FJ, Gurney AL. Interleukin 23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin 17. J Biol Chem. 2002 doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 17.Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S, Kolls JK. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitcutt MJ, Adler KB, Wu R. A biphasic chamber system for maintaining polarity of differentiation of cultured respiratory tract epithelial cells. In Vitro Cell Dev Biol. 1988;24:420–428. doi: 10.1007/BF02628493. [DOI] [PubMed] [Google Scholar]
- 19.Chmiel JF, Berger M, Konstan MW. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol. 2002;23:5–27. doi: 10.1385/CRIAI:23:1:005. [DOI] [PubMed] [Google Scholar]
- 20.Devor DC, Bridges RJ, Pilewski JM. Pharmacological modulation of ion transport across wild-type and ΔF508 CFTR-expressing human bronchial epithelia. Am J Physiol Cell Physiol. 2000;279:C461–C479. doi: 10.1152/ajpcell.2000.279.2.C461. [DOI] [PubMed] [Google Scholar]
- 21.Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, Yang R, Uddin J, Guggino WB, Atabani SF, et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat Immunol. 2004;5:388–392. doi: 10.1038/ni1056. [DOI] [PubMed] [Google Scholar]
- 22.Chmiel JF, Konstan MW, Saadane A, Krenicky JE, Lester KH, Berger M. Prolonged inflammatory response to acute Pseudomonas challenge in interleukin-10 knockout mice. Am J Respir Crit Care Med. 2002;165:1176–1181. doi: 10.1164/ajrccm.165.8.2107051. [DOI] [PubMed] [Google Scholar]
- 23.Chmiel JF, Konstan MW, Berger M. Murine models of CF airway infection and inflammation. Methods Mol Med. 2002;70:495–515. doi: 10.1385/1-59259-187-6:495. [DOI] [PubMed] [Google Scholar]
- 24.Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 25.Aggarwal S, Ghilardi N, Xie MH, De Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 26.Lindén A, Adachi M. Neutrophilic airway inflammation and IL-17. Allergy. 2002;57:769–775. doi: 10.1034/j.1398-9995.2002.02164.x. [DOI] [PubMed] [Google Scholar]
- 27.Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–2112. doi: 10.4049/jimmunol.170.4.2106. [DOI] [PubMed] [Google Scholar]
- 28.Schwarzenberger P, La Russa V, Miller A, Ye P, Huang W, Zieske A, Nelson S, Bagby GJ, Stoltz D, Mynatt RL, et al. IL-17 stimulates granulopoiesis in mice: use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J Immunol. 1998;161:6383–6389. [PubMed] [Google Scholar]
- 29.Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z, Bagby G, Nelson S, Kolls JK. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. 2000;164:4783–4789. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 30.Numasaki M, Takahashi H, Tomioka Y, Sasaki H. Regulatory roles of IL-17 and IL-17F in G-CSF production by lung microvascular endothelial cells stimulated with IL-1β and/or TNF-α. Immunol Lett. 2004;95:97–104. doi: 10.1016/j.imlet.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 31.Kuestner R, Gao Z, Taft D, Brandt C, Ostrander C, Bort S, Bilsborough J, Lewis P, Rixon M, Chan C, et al. Keystone Symposia: Cytokines, Disease, and Therapuetic Intervention, February 12, 2005. Keystone Symposia Publishing; Silverthorne, Co: 2005. Human and mouse IL-17A and IL-17F differentially bind IL-17RA and IL-17RC; p. 49. [Google Scholar]
- 32.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 33.Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 34.Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-α-induced genes in bone cells. J Leukocyte Biol. 2005;77:388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 35.Lubberts E. The role of IL-17 and family members in the pathogenesis of arthritis. Curr Opin Investig Drugs. 2003;4:572–577. [PubMed] [Google Scholar]
- 36.Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 37.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S, Kolls JK. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukocyte Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- 40.Langrish CL, McKenzie BS, Wilson NJ, de Waal MR, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 41.Laan M, Cui ZH, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, Skoogh BE, Lindén A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 42.Sagel SD, Kapsner R, Osberg I, Sontag MK, Accurso FJ. Airway inflammation in children with cystic fibrosis and healthy children assessed by sputum induction. Am J Respir Crit Care Med. 2001;164:1425–1431. doi: 10.1164/ajrccm.164.8.2104075. [DOI] [PubMed] [Google Scholar]
- 43.Schuster A, Haarmann A, Wahn V. Cytokines in neutrophil-dominated airway inflammation in patients with cystic fibrosis. Eur Arch Otorhinolaryngol. 1995;1(Suppl 252):S59–S60. doi: 10.1007/BF02484436. [DOI] [PubMed] [Google Scholar]
- 44.Eijnden SV, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur J Immunol. 2005;35:469–475. doi: 10.1002/eji.200425677. [DOI] [PubMed] [Google Scholar]