

Abstract
Background
Identification of the Th17 T cell subset as important mediators of host defense and pathology, prompted us to determine their susceptibility to HIV infection.
Methods and Results
We found that a sizeable portion of Th17 cells express HIV co-receptor CCR5 and produce very low levels of CCR5 ligands MIP-1α and MIP-1β. Accordingly, CCR5+ Th17 cells were efficiently infected with CCR5-tropic HIV and were depleted during viral replication in vitro. Remarkably, HIV+ individuals under treatment showed significantly reduced Th17 cells compared to HIV− subjects, regardless of their viral loads or CD4 numbers, whereas treatment naïve subjects had normal levels. However, there was a preferential reduction in CCR5+ T cells that were also CCR6+, which is expressed on all Th17 cells, as compared to CCR6−CCR5+ cells, in both treated and untreated HIV+ subjects. This observation suggests preferential targeting of CCR6+CCR5+ Th17 cells by CCR5-tropic viruses in vivo. Th17 cell levels also inversely correlated with activated CD4+ T cells in HIV+ individuals under treatment.
Conclusion
Our findings suggest a complex perturbation of Th17 subsets during the course of HIV-disease potentially through both direct viral infection and virus indirect mechanisms such as immune activation.
Keywords: Th17, IL-17, IFNγ, MIP-1α, MIP-1β, CCR5, CCR6, CD38, HIV
Introduction
Th17 cells are a subset of helper T cells, which mediate inflammation [1] and development of autoimmunity [2–7]. Th17 cells are also important in protective immune responses to a variety of pathogens in animal models [8–13]. In humans, it was recently reported that patients with hyper immunoglobulin E syndrome, characterized by recurrent staphylococcal and candida infections, [14, 15] have impaired Th17 development [16, 17], further highlighting the importance of Th17 responses in normal host defense against these pathogens.
HIV infection perturbs T cell subsets [18], and the systemic depletion of CD4+ T cells leads to immunosuppression and development of opportunistic infections, such as Pneumocystis jirovecci and Candida [19]. Several groups have also reported the depletion of mucosa-associated memory CD4+CCR5+ T cells and linked this to HIV disease progression [20–22]. HIV-infected patients also have higher levels of chronic immune activation markers, which correlate with disease progression and CD4+ cell depletion [23, 24]. Th17 cells could play a role in host defense mechanisms against HIV-associated opportunistic infections [11, 13]. Th17 cells are also enriched in the lamina propria of the gastrointestinal tract (GI) [25, 26] and may play an important role in the defense against microbes particularly at mucosal surfaces [27]. Thus, perturbation of Th17 cells during HIV-infection could compromise mucosal defenses against resident and pathogenic microbes, which in turn could result in immune activation [28].
Little is known about the role of Th17 cells in HIV pathogenesis. Two recent studies on Th17 cells in HIV infection showed that HIV-infected children with detectable viremia had lower levels of IL-17 secreting cells compared to uninfected children [29] and in adults Th17 cells were lost in the GI tract of HIV-infected individuals [30]. However, another cross-sectional study suggested significantly higher levels of IL-17 in infected individuals as compared with HIV negative volunteers [31]. It is also not clear whether Th17 cells are directly infected and depleted by HIV or whether their numbers are perturbed due to generalized immune activation.
Here we sought to determine the susceptibility of Th17 subsets to CCR5-tropic (R5-tropic) HIV infection and the relative proportion of these effector cells in HIV-infected individuals. We found that a sizeable portion of Th17 cells expressed CCR5 and low levels of CCR5 ligands MIP-1α and MIP-1β and were highly susceptible to infection with R5-tropic viruses. Th17 cells were reduced in the blood of HIV-infected individuals under antiretroviral therapy (ART) but not in untreated subjects (naïve), compared to HIV negative subjects. Remarkably, reduction in the number of Th17 cells in HIV-infected individuals on ART with undetectable viral load was highly correlated with increased immune activation parameters, suggesting this may be a potential reason for perturbation of Th17 cells in this group of patients.
Materials and Methods
Subjects
Thirty-seven ART and 11 naive HIV-infected individuals were recruited in accordance with an IRB approved protocol and consent. The HIV viral loads (VL) were determined by HIV RNA PCR and reported as copies/ml. Clinical details for each subject are shown in Table 1. HIV+ individuals on ART had a median CD4 count of 336 cells/mm3. Twenty-six patients on ART had VL <50 copies/ml and the remaining had a median VL of 997 copies/ml. Treatment naïve HIV-infected individuals had a median VL of 23,300 copies/ml and CD4 count of 418 cells/mm3, which was not statistically different from CD4 cell counts of HIV+ individuals on ART. For HIV uninfected controls, thirty-three random blood samples were obtained from the blood bank.
Table 1. HIV+ subjects’ CD4 levels and HIV viral loads.
Clinical details of HIV+ subjects in the study including CD4 cell counts and HIV viral load (VL) as measured by HIV RNA PCR in copies/ml.
| Subject | CD4+ (cells/mm3) | HIV VL (copies/ml) | Time on ART (months) |
|---|---|---|---|
| 1 | 23 | < 50 | 1 |
| 2 | 296 | 100,000 | 1 |
| 3 | 266 | 904 | 1 |
| 4 | 223 | < 50 | 2 |
| 5 | 600 | 3320 | 3 |
| 6 | 62 | < 50 | 4 |
| 7 | 176 | < 50 | 8 |
| 8 | 322 | < 50 | 9 |
| 9 | 344 | < 50 | 16 |
| 10 | 297 | < 50 | 16 |
| 11 | 375 | < 50 | 21 |
| 12 | 356 | < 50 | 24 |
| 13 | 525 | < 50 | 24 |
| 14 | 184 | < 50 | 24 |
| 15 | 111 | < 50 | 43 |
| 16 | 242 | 144 | 46 |
| 17 | 749 | < 50 | 54 |
| 18 | 205 | 158 | ≥60 |
| 19 | 517 | < 50 | ≥60 |
| 20 | 496 | < 50 | ≥60 |
| 21 | 484 | < 50 | ≥60 |
| 22 | 1083 | < 50 | ≥60 |
| 23 | 137 | < 50 | ≥60 |
| 24 | 953 | < 50 | ≥60 |
| 25 | 476 | < 50 | ≥60 |
| 26 | 200 | 600 | ≥60 |
| 27 | 25 | 13100 | ≥60 |
| 28 | 414 | < 50 | ≥60 |
| 29 | 419 | 5480 | ≥60 |
| 30 | 167 | < 50 | ≥60 |
| 31 | 1075 | 131 | ≥60 |
| 32 | 408 | 6670 | ≥60 |
| 33 | 651 | < 50 | ≥60 |
| 34 | 148 | 433 | ≥60 |
| 35 | 681 | < 50 | ≥60 |
| 36 | 472 | < 50 | ≥60 |
| 37 | 239 | 1090 | ≥60 |
| 38 | 686 | 4990 | naïve |
| 39 | 336 | 21900 | naïve |
| 40 | 332 | 100,000 | naïve |
| 41 | 418 | 24300 | naïve |
| 42 | 476 | 100,000 | naïve |
| 43 | 567 | 33000 | naïve |
| 44 | 104 | 70600 | naïve |
| 45 | 617 | 721 | naïve |
| 46 | 652 | 23300 | naïve |
| 47 | 322 | 10800 | naïve |
| 48 | 188 | 15700 | naïve |
Staining and FACS analysis
Cells were stained with corresponding antibodies as previously described [32]. For intracellular staining, fixation and permeabilization were performed using a kit (BD Biosciences) according to the manufacturer’s instructions. Analyses were performed using LSRII flow cytometer (BD Biosciences) and FlowJo software (Tree Star). The following anti-human antibodies were used for staining: CD3, CD25, CD38, CD45RO, CCR5, CCR6, MIP-1α, MIP-1β, (BD Biosciences), CD4, CD8, IFNγ and IL-17 (eBioscience). Intracellular HIV p24 staining was done using PE-conjugated p24 antibody (Coulter) as described above. Chemokine secretion was measured from T cells activated with plate-bound anti-CD3 and soluble anti-CD28 for 16 hours using a cytometric bead array (BD Biosciences).
HIV production
HIV pseudotyped with VSV-G envelope (VSV-G.HIV) was generated as previously described [33]. Viral supernatants from replication competent CCR5-tropic HIV were prepared by transfecting HEK293T cells with HIV that encoded R5-tropic(BaL) envelope. These viruses expressed either murine CD24 (mCD24) or green fluorescent protein (GFP) in place of the nef gene as marker genes, as previously described [32–34]. In some experiments R5-tropic viruses that also encode nef were used and the infections were monitored by intracellular p24 staining as described [32–34]. Viral titers were measured as described [32, 33] and ranged from 1 to 5 × 106 infectious units (IFU)/ml for replication-competent viruses and 10–30 × 106 IFU/ml for VSV-G.HIV.
T cell purifications
Peripheral Blood Mononuclear Cells (PBMCs) from blood of HIV-uninfected and HIV-infected individuals were prepared using Ficoll-paque plus (GE Health care). CD4+ T cells were isolated using Dynal CD4 Positive Isolation Kit (Invitrogen) and were >99% pure. CD4+ cells from healthy donors were further sorted by flow cytometry (FACSAria; BD Biosciences) based on expression of CD45RO and CCR6 or CCR5, and were used for the in vitro experiments. Sorted subsets were >99% pure and were kept at 37°C and 5% CO2 in RPMI 1640 medium with 10% fetal calf serum [32].
T cell activation and HIV infection
Sorted CD4+ T cell subsets were stimulated and maintained in IL-15 (10ng/ml, R&D Systems) during the course of infection experiments. Five days later, cells were infected with VSV-G.HIV, or R5.HIV at different multiplicities of infection (MOI). Alternatively, cells were activated with anti-CD3/CD28 conjugated beads. For intracellular cytokine or chemokine staining, cells were reactivated for 5 hours with Phorbol ester (PMA; 20ng/ml; Sigma), Ionomycin (500ng/ml; Sigma) and GolgiStop (Brefeldin A; BD Biosciences) to prevent protein secretion.
Statistical analysis
All statistical analyses were performed with GraphPad Prism 5 software. The significance in in vitro studies was determined using a two-tailed Two-Sample Student’s t-test. Comparisons of HIV-infected and healthy individuals were analyzed by the Mann-Whitney U-test and considered significant if p<0.05. Correlation between IL-17, IFNγ, and T cell activation markers was calculated by Spearman’s rank test.
Results
Expression of HIV co-receptor CCR5 on Th17 cells
Human Th17 cells are part of the CD4+ memory subset and have been reported to express the chemokine receptor CCR6 [8, 35, 36]. Infection with R5- and X4-tropic HIV requires expression of CCR5 or CXCR4, respectively, in addition to CD4. We found that a sizeable and significantly higher proportion of CCR6+ T cells compared to CCR6− subset, expressed CCR5, which is in contrast to another recent study [37]. The expression of CXCR4 was high and similar in both subsets (Fig. 1a and Fig. 1b). CD45RO+ memory cells were then sorted into 4 subsets based on these chemokine receptor profiles shown in Figure 1c: 1) CCR6+CCR5−, 2) CCR6+CCR5+, 3) CCR6−CCR5+, 4) CCR6−CCR5−. The sorted cells were activated and the percent of IL-17 and IFNγ secreting cells within each subset was determined using intracellular staining. In accordance with previous publications [8, 35, 36], all IL-17-secreting T cells were CCR6+ (Fig. 1d). CCR6+CCR5+ T cells also included about 2-fold more IL-17-secreting T cells compared to the CCR6+CCR5− subset (Fig. 1e). Interestingly, the CCR6+CCR5+ subset contained the majority of Th17 cells that also produced IFNγ (IFNγ+IL-17+ cells).
Figure 1. CCR5 segregates Th17 cells into IFNγ−IL-17+ and IFNγ+IL-17+.

(a) Resting CD4+, CD45RO+, CCR6+ or CCR6− T cells were purified as described (methods) and stained with CCR5-PE or CXCR4-PerCP-cy5.5 antibodies (blue) or isotype-matched controls (red). (b) Percent of memory CCR5+ cells in CD4+ cells stained for CCR5 and CCR6. Each circle is representative of one adult healthy donor. (c) Resting memory CD4+ T cells were stained with CCR6-biotin and CCR5-PE (d) Stained cells in (c) were sorted into four memory (CD45RO+) subsets: CCR6+CCR5−, CCR6+CCR5+, CCR6−CCR5+, and CCR6−CCR5−. To detect IL-17 and IFNγ producing cells; we activated sorted subsets using PMA, Ionomycin and Golgi stop. Cells were fixed, permeabilized and stained with IFNγ-PE-cy7 and IL-17-FITC antibodies. An isotype-matched control did not show any background staining (data not shown). (e) Percent of IL-17+, IFNγ+ and IFNγ+IL-17+ in memory CCR6+CCR5−, CCR6+CCR5+, CCR6−CCR5+, and CCR6−CCR5− T cell subsets. Each symbol represents one adult healthy donor. P values were calculated using the Two-Sample Student’s t-test.
Expression of MIP-1α and MIP-1β in Th17 cells
The finding that Th17 cells expressed high levels of CCR5 prompted us to determine the percent of Th17 cells that secrete CCR5 ligands MIP-1α and MIP-1β, which can act as entry inhibitors of HIV infection [38]. For this experiment, memory CCR6+ and CCR6− T cells from healthy individuals were sorted and activated as described in the methods section. These cells were then stained for intracellular IL-17 and IFNγ in conjunction with MIP-1α and MIP-1β (Fig. 2a). We found that Th17 cells produced significantly less of either chemokine compared to IFNγ+ T cells both in CCR6+ and CCR6− subsets (Fig. 2b). This finding suggests that the Th17 T cell subset would be predicted to have less capability to suppress CCR5-mediated HIV entry through chemokine expression during T cell activation.
Figure 2. Th17 cells express lower levels of MIP-1α and MIP-1β.
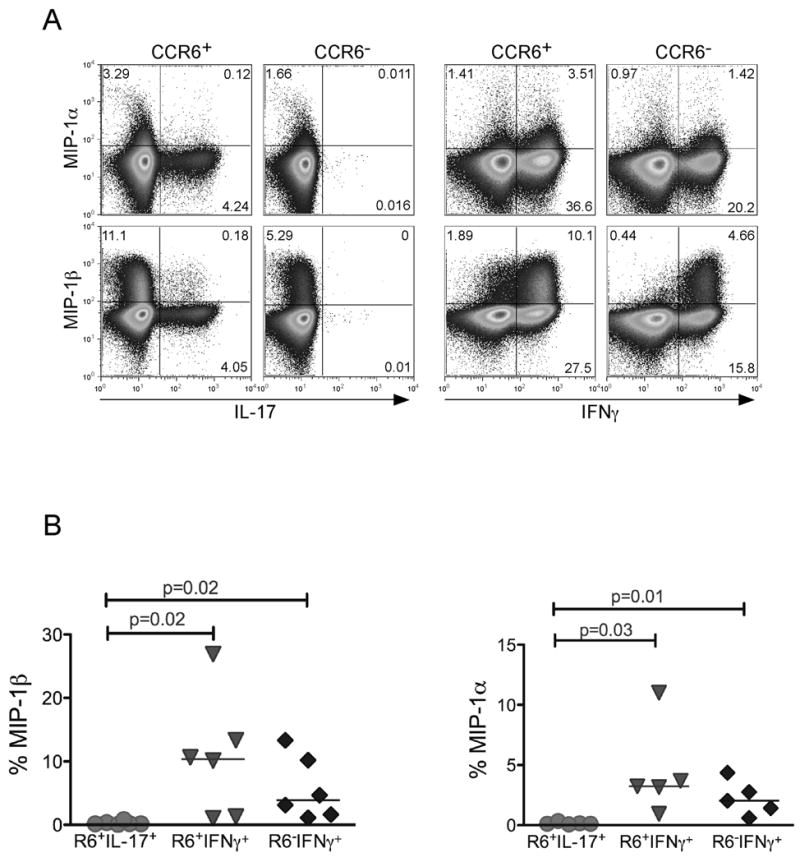
Resting CD4+ T cells from peripheral blood were further purified based on CD45RO and CCR6 markers into CD45RO+CCR6+ and CD45RO+CCR6−. Purified subsets were then stimulated with PMA and Ionomycin in the presence of Golgi Stop. (a) Cells were co-stained with anti-IL-17-FITC, IFNγ-pecy7, and either MIP-1α or MIP-1β-PE. (b) The percent MIP-1α and MIP-1β producing cells were determined in CCR6+IL-17+, CCR6+IFNγ+, and CCR6−IFNγ+ subsets in different healthy donors. P values were calculated using the Two-Sample Student’s t-test.
HIV infection of Th17 cells
Based on the above findings, we then determined the susceptibility of Th17 cells to HIV infection. T cells require activation either through the T cell receptor (TCR) or via cytokines to become susceptible to HIV [33]. For this purpose, we stimulated sorted memory CCR6+ and CCR6− cells either through the TCR or via IL-15 alone, which renders resting T cells susceptible to infection [33] and enhances the production of IL-17 [39]. Cells stimulated with IL-15 displayed three effector subsets, i.e. IL-17+, IFNγ+ or IFNγ+IL-17+ subsets after 6-day culture in vitro (Fig. 3a) and maintained this for up to 3 weeks in culture (data not shown). Th17 cells were not induced within the CCR6− subset with either stimulus (Fig. 3a).
Figure 3. HIV infects IL-15 stimulated Th17 cells.

(a) Resting CD4+ T cells were stained with CD45RO and CCR6 antibodies and sorted into CD45RO+CCR6+ and CD45RO+CCR6−. Purified cells were cultured in IL-15 for 5 days or were activated using anti-CD3/CD28 conjugated beads. Intracellular staining for IFNγ and IL-17 was performed at day 6 post activation as described in Figure 1. (b) IL-15 stimulated cells were infected with either CCR5-tropic HIV (R5.HIV), or VSV-G pseudotyped HIV (VSVG.HIV) having a GFP marker. They were then activated using PMA and Ionomycin and stained for IFNγ and IL-17 as above. GFP expression was determined in IFNγ+, IL-17+IFNγ+, and IL-17+ cell subsets. (c) T cells stimulated with IL-15 were infected with R5.HIV or VSVG.HIV having a GFP marker. At different time points post infection, cells were restimulated with PMA and Ionomycin and stained for IL-17 and IFNγ expression. Percent infected cells within each cytokine-secreting group were determined by GFP expression after gating on: IL-17+, IL-17+IFNγ+ and IFNγ+ cells following intracellular staining. (d) Percent infected IFNγ+ cells was determined at different time points post infection with R5.HIV or VSVG.HIV in CCR6+ and CCR6− cells. P values shown represent one time point post infection and were calculated using the Two-Sample Student’s t-test.
In subsequent experiments we focused on using IL-15 stimulation to determine susceptibility of Th17 subsets to HIV infection since we found that activation of T cells through the TCR, as opposed to IL-15, causes extensive proliferation of T cells, which skews IL-17-secreting effector populations in vitro from experiment to experiment. Further, the TCR activation-induced differentiation and proliferation also occasionally selected for cells that could have differential susceptibility to HIV, possibly due to changes in receptor expression or chemokine secretion, whereas IL-15 activation of resting T cells has minimal effect on T cell proliferation while still rendering them susceptible to HIV. We therefore challenged CCR6+ memory T cells cultured in the presence of IL-15 with either replication incompetent VSV-G-pseudotyped HIV (VSV-G.HIV), which bypasses a receptor requirement, or with replication competent R5-tropic HIV (R5.HIV). We found that IL-15 stimulation rendered Th17 cells susceptible to infection with both viruses (Fig. 3b).
The IL-15-stimulated and HIV-infected T cells were then harvested every three days and evaluated for GFP expression as a marker for infected T cells and stained for IL-17 and IFNγ to monitor the changes of effector cells in the cultures. We found that while IFNγ+ and IL-17+ subsets were similarly infected with R5.HIV or VSV-G.HIV (Fig. 3c), the IFNγ+IL-17+ T cells were relatively more susceptible with R5.HIV and VSV-G.HIV compared to the other two subsets of T cells (Fig. 3c). Of interest, CCR6− T cells that were IFNγ+ were significantly less susceptible to HIV infection both with R5.HIV as well as VSVG.HIV compared to CCR6+ IFNγ+ T cells (Fig. 3d). Based on this result we hypothesized that T cell subsets infected with replicating HIV would also be preferentially depleted during in vitro culture. We therefore determined the proportion of IFNγ+, IFNγ+IL-17+ and IL-17+ producing cells in culture at each indicated time point post-infection. We found that cells that were infected with VSV-G.HIV persisted stably at similar percentages during the period of culture, while R5-tropic replication competent viruses profoundly depleted IFNγ or IL-17 secreting T cells within these cultures over time (Fig. 4a and 4b). Within these cytokine producing populations infected with R5.HIV, IFNγ+IL-17+ cells were depleted faster and more drastically compared to IFNγ+ and IL-17+ cells. Consistent with infection results, CCR6− IFNγ+ persisted longer in cultures infected with R5.HIV compared to CCR6+IFNγ+(Fig. 4b). Similar results were obtained from different donors at different time points after infection and using wild-type viruses that also contain the nef gene (data not shown).
Figure 4. Th17 cells are depleted post infection with R5-tropic HIV.

(a) FACS plot of CD4+ CD45RO+CCR6+ and CD45RO+CCR6− cells cultured in IL-15 and stained with IL-17 and IFNγ at 3 and 16 days post infection with VSVG.HIV or R5.HIV. (b) Relative proportion of IFNγ+ in CCR6+ and CCR6− T subsets and that of IL-17+ and IFNγ+IL-17+ cells in CCR6+ cultures at different time points post infection with VSV-G. HIV or R5. HIV. P values were calculated for the entire data set using the paired Two-Sample Student’s t-test.
Analysis of the Th17 cell subset in peripheral blood of HIV-infected individuals
To complement in vitro HIV infection studies, we determined levels of IL-17− and IFNγ-producing cells in the peripheral blood of HIV-infected and non-infected individuals. Because Th17 cells are found within the memory cell population, and most HIV-infected individuals have a predominance of memory over naïve cells, we report the percent of IL-17 or IFNγ producing cells in the memory population gated on CD45RO+ cells. We divided HIV-infected individuals by CD4+ cell counts of less than or greater than 350. HIV-infected individuals with CD4 cell counts of less than 350 had a marked decrease in IL-17+ and IFNγ+IL-17+ cells (Fig. 5a). Those with CD4 cell counts greater than 350 also had lower IL-17+ and IFNγ+IL-17+ cells, but of less statistical significance (Fig. 5a). Similarly IFNγ–secreting cells were also significantly lower in HIV-infected groups regardless of the CD4+ cell numbers (Fig. 5a). There was no correlation between total CD4 levels and either IL-17+ or IFNγ+ T cells (data not shown). We next compared the HIV-infected population based on antiretroviral treatment and viral loads. All treatment naïve subjects had HIV copy numbers greater than 50 copies/ml by HIV RNA PCR. The subjects on ART were further divided by HIV viral load less than or greater than 50 copies/ml. Remarkably, the portion of IL-17+ and IFNγ+IL-17+ T cells in treatment naïve subjects was not significantly different from HIV negative subjects (Fig. 5b). This observation was confirmed even when we calculated the absolute cell numbers of each cytokine producing subset where we did not find significant difference between absolute cell counts of IL-17+ and IFNγ+IL-17+ T cells in naïve subjects as compared to HIV negative individuals (Fig. 5c). In contrast, subjects on ART with or without detectable HIV viral loads had significantly lower IL-17+ and IFNγ+IL-17+ T-cells both in percentage (Fig. 5b) and more significantly in absolute numbers (Fig 5c). All HIV infected individuals had significantly fewer IFNγ+ producing cells regardless of ART therapy and HIV viral load (Fig. 5b).
Figure 5. Th17 cells are depleted in the peripheral blood of HIV infected individuals on ART but not in treatment naive.

Purified CD4+ cells were stimulated with PMA and Ionomycin in the presence of Golgi stop. They were then stained for IL-17-FITC and IFNγ-APC and analyzed by LSR-II. IFNγ+, IL-17+, and IFNγ+IL-17+ subsets are shown in HIV-infected versus uninfected individuals gated on memory CD4+ cells. The memory population was determined by gating on CD3+CD4+CD45RO+ cells. (a) IFNγ+, IL-17+, and IFNγ+Th17 cell subsets are shown in healthy individuals compared to HIV+ individuals with CD4+ cell counts of less than or greater than 350 cells/mm3. (b) IFNγ+, IL-17+, and IFNγ+Th17 cell subset presented as percentages or (c) as absolute cell counts, are shown in healthy individuals compared with HIV+ individuals either ART naïve or on ART with HIV RNA PCR <50 copies/ml or >50 copies/ml.
Since one would predict that R5-tropic viruses would preferentially deplete CCR5+ T cells, we hypothesized that there would be fewer CCR5+ Th17 cells in HIV infected individuals. Because it was difficult to measure CCR5 expression in conjunction with IL-17 staining due to transient down regulation of CCR5 after activation and lower sensitivity during intracellular staining (data not shown), we determined the percent of memory CCR5+ T cells as well as the percent of CCR5+ cells within the CCR6+ subset, since all Th17 cells express the latter marker. HIV-infected subjects overall had lower CCR5+ memory cells both in naive and ART HIV+ subjects (Fig. 6a). Interestingly, the total proportion of CCR6+ cells was significantly increased in both naïve and ART groups compared to HIV-subjects (Fig. 6a). In contrast, CCR5+ within CCR6+ T cells were greatly reduced in both HIV+ groups, (Fig. 6b), while the proportion of CCR5+ within CCR6− T cells was not different compared to healthy donors (Fig. 6b). Taken together, these findings demonstrate a preferential reduction in the proportion of CCR5+CCR6+ memory T cells in vivo in HIV-infected individuals, which may be a consequence of direct infection and depletion with R5-tropic HIV due to high susceptibility of these cells to infection.
Figure 6. Changes in CCR6+ and CCR5+ T cells and correlation of T cell activation with Th17 cells during HIV infection.

PBMCs were stained for CD45RO-FITC, CCR5-PE and CCR6-biotin. Cells were gated first on CD45RO. (a) Total CCR6+ and CCR5+ T cells within the memory subset are shown for both HIV+ and HIV− donors. (b) The percent of memory CCR5+ cells within CCR6+ and CCR6− memory T cells was determined in HIV− and HIV+ subjects either treatment naïve or on ART. (c) PBMCs from HIV− and HIV+ individuals were stained for CD3-PerCP-cy5.5, CD4-Alexa 750, and activation marker CD38-PE as described in Methods, then gated first on CD3+CD4+ T cells and then for CD38+ populations. A representative FACS data plot comparing the percent of CD4+CD38+ cells in one HIV− and one HIV+ subject is shown. (d) CD4+CD38+ cells are shown in HIV− and HIV+ individuals. (e) IL-17 or IFNγ producing cells were identified in memory CD4 T cells and the percent of IL-17+ or IFNγ+ cells was plotted against the percent of CD38+ cells within CD4+ T cells in HIV− and HIV+ individuals on ART with HIV <50 copies/ml. Statistical correlations were determined by Spearman’s rank test.
Despite T cell depletion, it is well known that HIV-infected individuals demonstrate evidence of chronic immune activation. Consistent with prior studies, HIV-infected individuals had significantly higher expression of immune activation marker CD38 within CD4+ T cells, (Fig. 6d), an independent predictor of disease progression [40, 41]. Remarkably, in the group of HIV-infected individuals on ART and with undetectable HIV RNA, the reduction in IL-17 correlated positively with the percentage of CD4+CD38+ cells (Fig. 6e). A similar correlation was seen between CD4+CD38+ cells and IFNγ-secreting cells. There was no statistical correlation between CD4+CD38+ T cells and IL-17 or IFNγ-secreting cells both in HIV negative individuals (Fig 6e) and in HIV+ individuals who were treatment naïve or on ART with HIV RNA greater than 50 copies/ml (data not shown).
Discussion
Our findings that a portion of Th17 cells express high levels of CCR5 and lower levels of CCR5 ligands MIP-1α and MIP-1β, correlated with a high infection rate of this subset with R5-tropic HIV. This finding suggests that CCR5+ Th17 cells are conceivably infected with HIV in vivo and provide an explanation for their preferential depletion in the gut of HIV+ patients [30]. Interestingly, we found that infection and depletion of IFNγ+IL-17+ cells were more pronounced compared to IFNγ− IL-17+ T cells. This finding may be partly due to the level of CCR5 expression, which was consistently higher on IFNγ+IL-17+ cells. In addition, we found that the IFNγ+IL-17+ cells were also more susceptible to VSV-G-pseudotyped HIV, suggesting post-entry intrinsic host factors that modulate HIV infection of this subset compared to other effector T cell subsets. More detailed characterization of this subset including their phenotypic profile and effector functions might give us clues as to their origin and function in normal immune responses as well as their role in HIV pathogenesis.
Variations in MIP-1α gene dose and CCR5 genotypes were shown to act as host factors that affect HIV transmission, viral load, disease progression and immune recovery during antiretroviral therapy. In fact, the gene encoding MIP-1α encompasses many segmental duplications and people with a copy number of the MIP-1α gene lower than the population average have markedly enhanced HIV/AIDS susceptibility [42, 43]. It will be interesting in the future to determine the level of Th17 cells in these individuals with genetic differences in their CCR5 ligand levels or expression.
We showed here that HIV targets Th17 cells in in vitro experiments, which predicted that they would be depleted preferentially in subjects with higher viral loads. However, we found that Th17 subsets, along with IFNγ+ T cells, were greatly reduced in HIV+ subjects who were under treatment and even with undetectable viremia. It was also puzzling that in treatment naïve HIV+ individuals there was no decrease in IL-17+ T cells. How then can our results showing in vitro HIV-susceptibility of Th17 cells be reconciled with these perplexing ex vivo findings? We think during early stages of infection, Th17 cells are indeed targeted by HIV in vivo, but one would expect only the CCR5+ Th17 subset to be susceptible to infection with R5-tropic viruses. Indeed we found a dramatic decrease in the CCR5+ portion of CCR6+ T cells both in naïve and ART subjects (Fig. 6b). This finding is also consistent with a recent report describing preferential and rapid depletion of Th17 cells in the gut mucosa of HIV-infected individuals, which would be expected to be mostly CCR5 positive [30]. We postulate that perhaps the relative decrease in CCR5+CCR6+ T cells, which contain a substantial portion of the Th17 subset, causes a perturbation that results in a compensatory increase in CCR5 negative Th17 cells in naïve HIV+ subjects. In addition, while naïve subjects in our study may not have reached their CD4 nadir counts, some of the ART subjects could have started with a lower CD4 nadir prior to initiation of ART. It is conceivable that as CD4+ T cells progressively decline during the course of HIV infection, Th17 cells are depleted, and ART treatment may fail to restore their levels once lost to pre-infection levels, perhaps because of a compromise in Th17 cell differentiation or survival. However, because our study was not longitudinal it is difficult to predict when during the course of HIV infection this decline occurs and whether antiretroviral therapy restores Th17 cells. An alternative possibility is that, IL-17 as a marker underestimates the presence of lineage committed Th17 cells. It is conceivable that constant immune activation or immune-exhaustion suppresses Th17 or Th1 cells from displaying their effector functions such as secretion of IL-17 and IFNγ. In support of this notion of immune-exhaustion, Nixon and colleagues recently showed that a molecule called Tim-3 is upregulated in chronically activated T cells in HIV+ subjects and that these cells did not secrete IFNγ unless the signals from Tim-3 were blocked by neutralizing its interaction with its ligand [44]. It will be interesting to determine in future studies whether secretion of IL-17 can also be rescued by suppressing Tim-3 or other negative signaling molecules such as PD-1 on CCR6+ T cells.
How could reduced levels of IL-17-secreting cells impact HIV pathogenesis? A potential scenario can be envisaged, where the loss of Th17 cells may compromise the mucosal immune system during HIV infection. Th17 cells assist in vivo in mucosal host defense against many pathogens in the gut, lungs and skin through regulating antimicrobial peptide production by epithelial cells and keratinocytes [27, 45]. The loss of Th17 cells during HIV infection might also contribute to microbial translocation of bacterial products from mucosal tissues to the circulation resulting in immune activation [28, 46]. Hence, the loss of Th17 cells during HIV infection could also potentially be a biomarker for disease progression. More studies will be needed to understand the impact of Th17 depletion in HIV pathogenesis and its relationship with other T cell subsets such as regulatory T cells. Future prospective studies will also be important to determine if Th17 cells decline immediately post-treatment of naïve subjects and whether Th17 cells are reconstituted when immune activation is subdued.
In conclusion, we have shown that Th17 cells represent ideal targets for HIV by virtue of high expression of CCR5 and lower secretion of CCR5 ligands MIP-1α and MIP-1β. The high susceptibility of Th17 cells to HIV in vitro was reflected by their in vivo depletion in the peripheral blood of HIV+ individuals. Association of this preferential depletion with immune activation was particularly striking and highlights the potential significance of Th17 cell depletion during HIV immunopathogenesis.
Acknowledgments
We thank Dr. Ned Landau, Frances Mercer and Qi Wan for critical reading and valuable suggestions on the manuscript. This study was supported by CFAR NIH developmental grant P30AI027742 (A.K., L.K. and D.U.); NIH grant P01AI057127 (F.V.); NIH grant R01AI033856 (D.R.L.); NIH grant T32 AI07647-09 (A.E); R01 AI065303 (D.U), the European Molecular Biology Organization (N.M.) and the Irvington Institute Fellowship Program of the Cancer Research Institute (N.M.).
Abbreviations
- VSV-G
Vesicular Stomatitis Virus Glycoprotein
- CD
Cluster of differentiation
Footnotes
The authors report no potential conflicts on interest.
A partial data from this study was presented at the Keystone Symposia: HIV Immunobiology: From infection to Immune Control. March 22–27, 2009 Keystone Resort. Keystone, Colorado, USA. (Oral presentation and poster presentation #203).
References
- 1.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 2.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 4.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 6.Murphy CA, Langrish CL, Chen Y, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–7. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 8.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 9.Cooper AM, Kipnis A, Turner J, Magram J, Ferrante J, Orme IM. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J Immunol. 2002;168:1322–7. doi: 10.4049/jimmunol.168.3.1322. [DOI] [PubMed] [Google Scholar]
- 10.Happel KI, Dubin PJ, Zheng M, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–9. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–31. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 12.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–15. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 13.Rudner XL, Happel KI, Young EA, Shellito JE. Interleukin-23 (IL-23)-IL-17 cytokine axis in murine Pneumocystis carinii infection. Infect Immun. 2007;75:3055–61. doi: 10.1128/IAI.01329-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 15.Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 16.Milner JD, Brenchley JM, Laurence A, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Beaucoudrey L, Puel A, Filipe-Santos O, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543–50. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hazenberg MD, Hamann D, Schuitemaker H, Miedema F. T cell depletion in HIV-1 infection: how CD4+ T cells go out of stock. Nat Immunol. 2000;1:285–9. doi: 10.1038/79724. [DOI] [PubMed] [Google Scholar]
- 19.Hage CA, Goldman M, Wheat LJ. Mucosal and invasive fungal infections in HIV/AIDS. Eur J Med Res. 2002;7:236–41. [PubMed] [Google Scholar]
- 20.Brenchley JM, Schacker TW, Ruff LE, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–59. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehandru S, Poles MA, Tenner-Racz K, et al. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. 2004;200:761–70. doi: 10.1084/jem.20041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paiardini M, Frank I, Pandrea I, Apetrei C, Silvestri G. Mucosal immune dysfunction in AIDS pathogenesis. AIDS Rev. 2008;10:36–46. [PubMed] [Google Scholar]
- 23.Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol. 2008;214:231–41. doi: 10.1002/path.2276. [DOI] [PubMed] [Google Scholar]
- 24.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat Med. 2006;12:289–95. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- 25.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 26.Ivanov II, Frutos Rde L, Manel N, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–49. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense. Semin Immunol. 2007;19:377–82. doi: 10.1016/j.smim.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 29.Ndhlovu LC, Chapman JM, Jha AR, et al. Suppression of HIV-1 plasma viral load below detection preserves IL-17 producing T cells in HIV-1 infection. Aids. 2008;22:990–2. doi: 10.1097/QAD.0b013e3282ff884e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brenchley JM, Paiardini M, Knox KS, et al. Differential Th17 CD4 T cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008 doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maek ANW, Buranapraditkun S, Klaewsongkram J, Ruxrungtham K. Increased interleukin-17 production both in helper T cell subset Th17 and CD4-negative T cells in human immunodeficiency virus infection. Viral Immunol. 2007;20:66–75. doi: 10.1089/vim.2006.0063. [DOI] [PubMed] [Google Scholar]
- 32.Oswald-Richter K, Grill SM, Leelawong M, et al. Identification of a CCR5-expressing T cell subset that is resistant to R5-tropic HIV infection. PLoS Pathog. 2007;3:e58. doi: 10.1371/journal.ppat.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J Exp Med. 1999;189:1735–46. doi: 10.1084/jem.189.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oswald-Richter K, Grill SM, Shariat N, et al. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLoS Biol. 2004;2:E198. doi: 10.1371/journal.pbio.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM. Human T Cells That Are Able to Produce IL-17 Express the Chemokine Receptor CCR6. J Immunol. 2008;180:214–21. doi: 10.4049/jimmunol.180.1.214. [DOI] [PubMed] [Google Scholar]
- 36.Liu H, Rohowsky-Kochan C. Regulation of IL-17 in human CCR6+ effector memory T cells. J Immunol. 2008;180:7948–57. doi: 10.4049/jimmunol.180.12.7948. [DOI] [PubMed] [Google Scholar]
- 37.Sato W, Aranami T, Yamamura T. Cutting edge: Human Th17 cells are identified as bearing CCR2+CCR5− phenotype. J Immunol. 2007;178:7525–9. doi: 10.4049/jimmunol.178.12.7525. [DOI] [PubMed] [Google Scholar]
- 38.Lederman MM, Penn-Nicholson A, Cho M, Mosier D. Biology of CCR5 and its role in HIV infection and treatment. Jama. 2006;296:815–26. doi: 10.1001/jama.296.7.815. [DOI] [PubMed] [Google Scholar]
- 39.Hoeve MA, Savage ND, de Boer T, et al. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol. 2006;36:661–70. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 40.Giorgi JV, Liu Z, Hultin LE, Cumberland WG, Hennessey K, Detels R. Elevated levels of CD38+ CD8+ T cells in HIV infection add to the prognostic value of low CD4+ T cell levels: results of 6 years of follow-up. The Los Angeles Center, Multicenter AIDS Cohort Study. J Acquir Immune Defic Syndr. 1993;6:904–12. [PubMed] [Google Scholar]
- 41.Liu Z, Cumberland WG, Hultin LE, Prince HE, Detels R, Giorgi JV. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16:83–92. doi: 10.1097/00042560-199710010-00003. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–40. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 43.Dolan MJ, Kulkarni H, Camargo JF, et al. CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat Immunol. 2007;8:1324–36. doi: 10.1038/ni1521. [DOI] [PubMed] [Google Scholar]
- 44.Jones RB, Ndhlovu LC, Barbour JD, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–79. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchetti G, Bellistri GM, Borghi E, et al. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. Aids. 2008;22:2035–8. doi: 10.1097/QAD.0b013e3283112d29. [DOI] [PubMed] [Google Scholar]