

SUMMARY
The somatic mutations in cytosolic isocitrate dehydrogenase 1 (IDH1) observed in gliomas can lead to the production of 2-hydroxyglutarate (2HG). Here, we report that tumor 2HG is elevated in a high percentage of patients with cytogenetically normal acute myeloid leukemia (AML). Surprisingly, less than half of cases with elevated 2HG possessed IDH1 mutations. The remaining cases with elevated 2HG had mutations in IDH2, the mitochondrial homolog of IDH1. These data demonstrate that a shared feature of all cancer-associated IDH mutations is production of the onco-metabolite 2HG. Furthermore, AML patients with IDH mutations display a significantly reduced number of other well characterized AML-associated mutations and/or associated chromosomal abnormalities, potentially implicating IDH mutation in a distinct mechanism of AML pathogenesis.
INTRODUCTION
Mutations in human cytosolic isocitrate dehydrogenase I (IDH1) occur somatically in >70% of grade II-III gliomas and secondary glioblastomas, and in 8.5% of acute myeloid leukemias (AML) (Mardis et al., 2009; Yan et al., 2009). Mutations have also been reported in cancers of the colon and prostate (Kang et al., 2009; Sjoblom et al., 2006). To date, all reported IDH1 mutations result in an amino acid substitution at a single arginine residue in the enzyme’s active site, R132. A subset of intermediate grade gliomas lacking mutations in IDH1 have been found to harbor mutations in IDH2, the mitochondrial homolog of IDH1. The IDH2 mutations which have been identified in gliomas occur at the analogous residue to IDH1 R132, IDH2 R172. Both IDH1 R132 and IDH2 R172 mutants lack the wild-type enzyme’s ability to convert isocitrate to α-ketoglutarate (Yan et al., 2009). To date, all reported IDH1 or IDH2 mutations are heterozygous, with the cancer cells retaining one wild-type copy of the relevant IDH1 or IDH2 allele. No patient has been reported with both an IDH1 and IDH2 mutation. These data argue against the IDH mutations resulting in a simple loss of function.
Normally both cytosolic IDH1 and mitochondrial IDH2 exist as homodimers within their respective cellular compartments, and the mutant proteins retain the ability to bind to their respective wild-type partner. Therefore, it has been proposed that mutant IDH1 can act as a dominant negative against wild-type IDH1 function, resulting in a decrease in cytosolic α-ketoglutarate levels and leading to an indirect activation of the HIF-1α pathway (Zhao et al., 2009). However, recent work has provided an alternative explanation. The R132H IDH1 mutation observed in gliomas was found to display a gain of function for the NADPH-dependent reduction of α-ketoglutarate to R(-)-2-hydroxyglutarate (2HG) (Dang et al., 2009). This in vitro activity was confirmed when 2HG was found to be elevated in IDH1-mutated gliomas. Whether this neomorphic activity is a common feature shared by IDH2 mutations was not determined.
IDH1 R132 mutations identical to those reported to produce 2HG in gliomas were recently reported in AML (Mardis et al., 2009). These IDH1 R132 mutations were observed in 8.5% of AML patients studied, and a significantly higher percentage of mutation was observed in the subset of patients whose tumors lacked cytogenetic abnormalities. IDH2 R172 mutations were not observed in this study. However, during efforts to confirm and extend these findings, we found an IDH2 R172K mutation in an AML sample obtained from a 77 year old female. This finding confirmed that both IDH1 and IDH2 mutations can occur in AML and prompted us to more comprehensively investigate the role of IDH2 in AML.
The present study was undertaken to see if IDH2 mutations might share the same neomorphic activity as recently reported for glioma-associated IDH1 R132 mutations. We also determined whether tumor-associated 2HG elevation could prospectively identify AML patients with mutations in IDH. To investigate the lack of reduction to homozygosity for either IDH1 or IDH2 mutations in tumor samples, the ability of wild-type IDH1 and/or IDH2 to contribute to cell proliferation was examined.
RESULTS
IDH2 is mutated in AML
A recent study employing a whole-genome sequencing strategy in an AML patient resulted in the identification of somatic IDH1 mutations in AML (Mardis et al., 2009). Based on the report that IDH2 mutations were also observed in the other major tumor type in which IDH1 mutations were implicated (Yan et al., 2009), we sequenced the IDH2 gene in a set of de-identified AML DNA samples. Several cases with IDH2 R172 mutations were identified. In the initial case, the IDH2 mutation found, R172K, was the same mutation reported in glioma samples. It has been recently reported that cancer-associated IDH1 R132 mutants display a loss-of-function for the use of isocitrate as substrate, with a concomitant gain-of-function for the reduction of α-ketoglutarate to 2HG (Dang et al., 2009). This prompted us to determine if the recurrent R172K mutation in IDH2 observed in both gliomas and leukemias might also display the same neomorphic activity. In IDH1, the role of R132 in determining IDH1 enzymatic activity is consistent with the stabilizing charge interaction of its guanidinium moiety with the β-carboxyl group of isocitrate (Figure 1A). This β-carboxyl is critical for IDH’s ability to catalyze the interconversion of isocitrate and α-ketoglutarate, with the overall reaction occurring in two steps through a β-carboxyl-containing intermediate (Ehrlich and Colman, 1976). Proceeding in the oxidative direction, this β-carboxyl remains on the substrate throughout the IDH reaction until the final decarboxylating step which produces α-ketoglutarate.
Figure 1. IDH1 R132 and IDH2 R172 are analogous residues that both interact with the β-carboxyl of isocitrate.
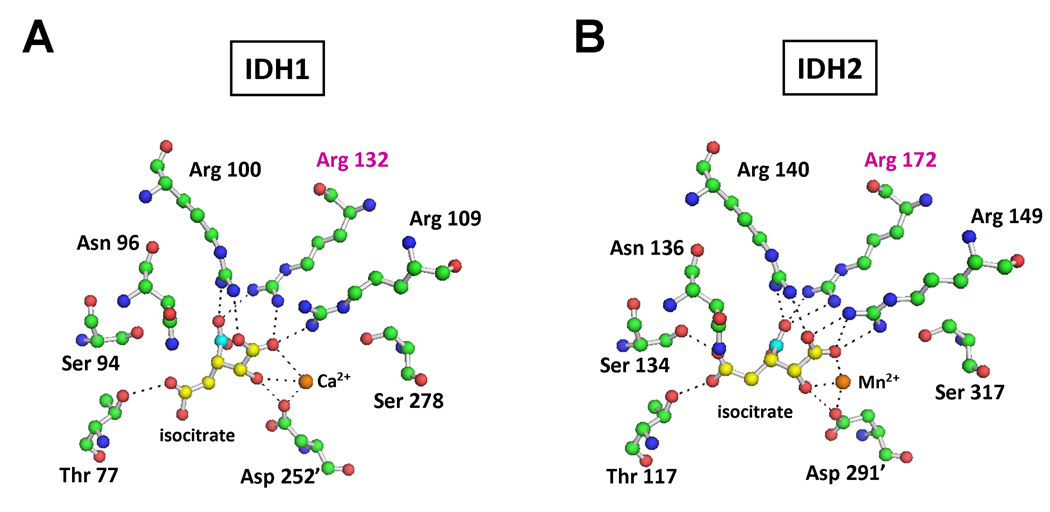
(A) Active site of crystallized human IDH1 with isocitrate. (B) Active site of human IDH2 with isocitrate, modeled based on the highly homologous and crystallized pig IDH2 structure. For (A) and (B), carbon 6 of isocitrate containing the β-carboxyl is highlighted in cyan, with remaining isocitrate carbons shown in yellow. Carbon atoms of amino acids (green), amines (blue), and oxygens (red) are also shown. Hydrogen atoms are omitted from the figure for clarity. Dashed lines depict interactions < 3.1 angstroms, corresponding to hydrogen and ionic bonds. Residues coming from the other monomer of the IDH dimer are denoted with a prime (') symbol.
IDH1 R132 and IDH2 R172 are analogous residues that both interact with the β-carboxyl of isocitrate
To understand how R172 mutations in IDH2 might relate to the R132 mutations in IDH1 characterized for gliomas, we modeled human IDH2 based on the pig IDH2 structure containing bound isocitrate (Ceccarelli et al., 2002). Human and pig IDH2 protein share over 97% identity and all active site residues are identical. The active site of human IDH2 was structurally aligned with human IDH1 (Figure 1). Similar to IDH1, in the active site of IDH2 the isocitrate substrate is stabilized by multiple charge interactions throughout the binding pocket. Moreover, like R132 in IDH1, the analogous R172 in IDH2 is predicted to interact strongly with the β-carboxyl of isocitrate. This raised the possibility that cancer-associated IDH2 mutations at R172 might affect enzymatic interconversion of isocitrate and α-ketoglutarate similarly to IDH1 mutations at R132.
Mutation of IDH2 R172K enhances α-ketoglutarate-dependent NADPH consumption
To test whether cancer-associated IDH2 R172K mutations shared the gain of function in α-ketoglutarate reduction observed for IDH1 R132 mutations (Dang et al., 2009), we overexpressed wild-type or R172K mutant IDH2 in cells with endogenous wild-type IDH2 expression, and then assessed isocitrate-dependent NADPH production and α-ketoglutarate-dependent NADPH consumption in cell lysates. As reported previously (Yan et al., 2009), extracts from cells expressing the R172K mutant IDH2 did not display isocitrate-dependent NADPH production above the levels observed in extracts from vector-transfected cells. In contrast, extracts from cells expressing a comparable amount of wild-type IDH2 markedly increased isocitrate-dependent NADPH production (Figure 2A). However, when these same extracts were tested for NADPH consumption in the presence of α-ketoglutarate, R172K mutant IDH2 expression was found to correlate with a significant enhancement to α-ketoglutarate-dependent NADPH consumption. Vector-transfected cell lysates did not demonstrate this activity (Figure 2B). Although not nearly to the same degree as with the mutant enzyme, wild-type IDH2 overexpression also reproducibly enhanced α-ketoglutarate-dependent NADPH consumption under these conditions.
Figure 2. Expression of R172K mutant IDH2 results in enhanced α-ketoglutarate-dependent consumption of NADPH.

(A) 293T cells transfected with wild-type or R172K mutant IDH2, or empty vector, were lysed and subsequently assayed for their ability to generate NADPH from NADP+ in the presence of 0.1 mM isocitrate. (B) The same cell lysates described in (A) were assayed for their consumption of NADPH in the presence of 0.5 mM α-ketoglutarate. Data for (A) and (B) are each representative of 3 independent experiments. Data are presented as the mean ± SEM from 3 independent measurements at the indicated time points. (C) Expression of wild-type and R172K mutant IDH2 was confirmed by Western blotting of the lysates assayed in (A) and (B). Re-probing of the same blot with IDH1 antibody as a control is also shown.
Mutation of IDH2 R172K results in elevated 2HG levels
R172K mutant IDH2 lacks the guanidinium moiety in residue 172 that normally stabilizes β-carboxyl addition in the interconversion of α-ketoglutarate and isocitrate. Yet R172K mutant IDH2 exhibited enhanced α-ketoglutarate-dependent NADPH consumption in cell lysates (Figure 2B). A similar enhancement of α-ketoglutarate-dependent NADPH consumption has been reported for R132 mutations in IDH1, resulting in conversion of α-ketoglutarate to 2HG (Dang et al., 2009). To determine if cells expressing IDH2 R172K shared this property, we expressed IDH2 wild-type or IDH2 R172K in cells. The accumulation of organic acids, including 2HG, both within cells and in culture medium of the transfectants was then assessed by gas chromatography-mass spectrometry (GC-MS) after MTBSTFA derivatization of the organic acid pool. We observed a metabolite peak eluting at 32.5 min on GC-MS which was of minimal intensity in the culture medium of IDH2 wild-type expressing cells, but which in the medium of IDH2 R172K expressing cells had a markedly higher intensity approximating that of the glutamate signal (Figures 3A–3B). Mass spectra of this metabolite peak fit that predicted for MTBSTFA-derivatized 2HG, and the peak’s identity as 2HG was additionally confirmed by matching its mass spectra with that obtained by derivatization of commercial 2HG standards (Figure 3C). Similar results were obtained when the intracellular organic acid pool was analyzed. IDH2 R172K expressing cells were found to have an approximately 100-fold increase in the intracellular levels of 2HG compared with the levels detected in vector-transfected and IDH2 wild-type overexpressing cells (Figure 3D). Consistent with previous work, IDH1 R132H expressing cells analyzed in the same experiment had comparable accumulation of 2HG in both cells and in culture medium. 2HG accumulation was not observed in cells overexpressing IDH1 wild-type (data not shown).
Figure 3. Expression of R172K mutant IDH2 elevates 2HG levels within cells and in culture medium.

293T cells transfected with IDH2 wild-type (A) or IDH2 R172K (B) were provided fresh culture medium the day following transfection. 24 hours later, the medium was collected, from which organic acids were extracted, purified, and derivatized with MTBSTFA. Shown are representative gas chromatographs for the derivatized organic acids eluting between 30 to 34 minutes, including aspartate (Asp) and glutamate (Glu). The arrows indicate the expected elution time of 32.5 minutes for MTBSTFA-derivatized 2HG, based on similar derivatization of a commercial R(-)-2HG standard. Metabolite abundance refers to GC-MS signal intensity. (C) Mass spectrum of the metabolite peak eluting at 32.5 minutes in (B), confirming its identity as MTBSTFA-derivatized 2HG. The structure of this derivative is shown in the inset, with the tert-butyl dimethylsilyl groups added during derivatization highlighted in green. m/e− indicates the mass (in atomic mass units) to charge ratio for fragments generated by electron impact ionization (D) Cells were transfected as in (A) and (B), and after 48 hours intracellular metabolites were extracted, purified, MTBSTFA-derivatized, and analyzed by GC-MS. Shown is the quantitation of 2HG signal intensity relative to glutamate for a representative experiment. See also Figure S1.
Mutant IDH2 produces the (R) enantiomer of 2HG
Cancer-associated mutants of IDH1 produce the (R) enantiomer of 2HG (Dang et al., 2009). To determine the chirality of the 2HG produced by mutant IDH2 and to compare it with that produced by R132H mutant IDH1, we used a two step derivatization method to distinguish the stereoisomers of 2HG by GC-MS: an esterification step with R-(-)-2-butanolic HCl, followed by acetylation of the 2-hydroxyl with acetic anhydride (Kamerling et al., 1981). Test of this method on commercial S(+)-2HG and R(-)-2HG standards demonstrated clear separation of the (S) and (R) enantiomers, and mass spectra of the metabolite peaks confirmed their identity as the O-acetylated di-(-)-2-butyl esters of 2HG (Figure S1A and S1B). By this method, we confirmed the chirality of the 2HG found in cells expressing either R132H mutant IDH1 or R172K mutant IDH2 corresponded exclusively to the (R) enantiomer (Figure S1C and S1D).
Leukemic cells bearing heterozygous R172K IDH2 mutations accumulate 2HG
To determine whether 2HG also accumulates in leukemic cells with either IDH1 R132 or IDH2 R172 mutations, we next analyzed the 2HG levels of selected leukemic samples. To date, every IDH1 R132H or IDH2 R172K mutant AML sample has exhibited evidence of 2HG accumulation, demonstrating a 2HG signal intensity by GC-MS analysis that is ≥ 30% of the intrasample glutamate signal. In contrast, control extracts have either had undetectable 2HG or a 2HG signal ≤ 1% of that for glutamate (n=13, p<0.001). Thus leukemic cells bearing either IDH1 R132 or IDH2 R172 mutation share the ability to accumulate 2HG in vivo with glioma cells exhibiting IDH1 R132 mutations.
IDH2 is critical for proliferating cells and contributes to the conversion of α-ketoglutarate into citrate in the mitochondria
A peculiar feature of the IDH-mutated cancers described to date is their lack of reduction to homozygosity. All tumors with IDH mutations retain one IDH wild-type allele. To address this issue we examined whether wild-type IDH1 and/or IDH2 might play a role in either cell survival or proliferation. Consistent with this possibility, we found that siRNA knockdown of either IDH1 or IDH2 can significantly reduce the proliferative capacity of a cancer cell line expressing both wild-type IDH1 and IDH2 (Figure 4A).
Figure 4. Both IDH1 and IDH2 are critical for cell proliferation.

(A) SF188 cells were treated with either of two unique siRNA oligonucleotides against IDH1 (siIDH1-A and siIDH1-B), either of two unique siRNA oligonucleotides against IDH2 (siIDH2-A and siIDH2-B), or control siRNA (siCTRL), and total viable cells were counted five days later. Data are the mean ± SEM of four independent experiments. In each case, both pairs of siIDH nucleotides gave comparable results. A representative Western blot from one of the experiments, probed with antibody specific for either IDH1 or IDH2 as indicated, is shown on the right-hand side. (B) Model depicting the pathways for citrate +4 (blue) and citrate +5 (red) formation in proliferating cells from [13C-U]-L-glutamine (glutamine +5). (C) Cells were treated with two unique siRNA oligonucleotides against IDH2 or control siRNA, labeled with [13C-U]-L-glutamine, and then assessed for isotopic enrichment in citrate by LC-MS. Citrate +5 and Citrate +4 refer to citrate with five or four 13C-enriched atoms, respectively. Reduced expression of IDH2 from the two unique oligonucleotides was confirmed by Western blot. Blotting with actin antibody is shown as a loading control. (D) Cells were treated with two unique siRNA oligonucleotides against IDH3 (siIDH3-A and siIDH3-B) or control siRNA, and then labeled and assessed for isotopic citrate enrichment by GC-MS. Shown are representative data from three independent experiments. Reduced expression of IDH3 from the two unique oligonucleotides was confirmed by Western blot. In (C) and (D), data are presented as mean ± SD of 3 replicates per experimental group.
IDH1 is one of only three cytosolic enzymes that contribute to the NADPH production required for nucleotide and lipid biosynthesis during cell growth (DeBerardinis et al., 2007). In addition, IDH1 contributes to the maintenance of cytosolic redox state (Yan et al., 2009). Therefore, there are several reasons why IDH1 might be important for cell proliferation. However, unlike IDH1, IDH2 resides in the mitochondrial matrix. Mitochondria normally contain a high level of NADPH and readily interconvert NADH and NADPH (Rydstrom, 2006). Furthermore, it is IDH3, the NAD+-dependent isocitrate dehydrogenase, that is believed to be responsible for isocitrate conversion into α-ketoglutarate in the mitochondrial citric acid cycle (McCormack and Denton, 1979). This raised the possibility that wild-type IDH2 might be contributing to the ability of cancer cells to produce citrate from glutamine, as illustrated in Figure 4B. Such an enzymatic activity, though not ascribed to a particular IDH isoform, was characterized in early metabolic studies (Ochoa, 1948; Siebert et al., 1957). As shown in Figure 2B, when wild-type IDH2 was over-expressed in cells, it was found to consume NADPH in an α-ketoglutarate-dependent manner.
To test the possibility that wild-type IDH2 contributes to the conversion of α-ketoglutarate into citrate in the mitochondria of proliferating cells, we labeled cells with [13C-U]-L-glutamine (glutamine +5), and subsequently measured isotopic enrichment in citrate by GC-MS. Enrichment of citrate with five 13C atoms (citrate +5) can arise from the reductive carboxylation of α-ketoglutarate +5 to isocitrate +5, while citrate enriched with four 13C atoms (citrate +4) can arise from oxidative metabolism of α-ketoglutarate through the traditional citric acid cycle (Figure 4B). Both forms of citrate production were reproducibly observed. To examine the role of IDH2 in citrate production, cells were treated with one of two independent IDH2 siRNAs. IDH2 siRNA treated cells displayed a significant reduction in levels of citrate +5, supporting a role for mitochondrial IDH2 in reductive carboxylation. Of note, no change in citrate +4 levels was observed, indicating that the effect of IDH2 knockdown was specific and not affecting other fundamental processes regulating the citric acid cycle (Figure 4C). Similar labeling patterns were observed in aconitate, an intermediate in the isomerization of isocitrate to citrate, with aconitate +5 also reduced following IDH2 knockdown, and aconitate +4 levels remaining unchanged (data not shown). In contrast, when cells were treated with one of two independent siRNAs against IDH3, there was a reproducible increase in citrate +5, while citrate +4 was unchanged (Figure 4D). These data support a role for IDH2 and not IDH3 in the conversion of α-ketoglutarate into citrate by reductive carboxylation in the mitochondria, and provide a possible explanation for the lack of reduction to homozygosity for IDH2 mutations in cancer.
2HG can be used as a screening test for neomorphic IDH mutants
The above data suggest that the common feature of the IDH1 R132 and IDH2 R172 mutants is an ability to produce 2HG that can be measured directly in tumor samples. This suggested that screening for the presence of 2HG in tumor samples could be used as an assay to detect IDH mutations. To test this hypothesis, we obtained frozen samples of AML cells from 18 patients aged 50 years or older who presented with normal karyotype AML. All 18 samples were first screened for the intensity of their 2HG signal on GC-MS as a percentage of the intrasample glutamate signal (Table 1 and Figure 5). GC-MS analysis demonstrated that 9 of 18 samples had elevated 2HG. This fraction of samples was much higher than expected from the reported incidence of IDH1 mutation (Mardis et al., 2009). To correlate the results with mutation status, IDH1 and IDH2 were sequenced for all 18 samples in a blinded fashion. All samples displaying a signal ratio for 2HG/glutamate > 1% had either an IDH1 or IDH2 mutation. In contrast, none of the samples with a 2HG/glutamate signal ratio ≤ 1% had an IDH mutation (Table 1). Thus in this sample set, 2HG measurement was predictive of IDH mutation status.
Table 1.
Patient characteristics, metabolite analysis, and IDH status.
| Age (yr.) | Sex | 2HG / glutamate | IDH mutation |
|---|---|---|---|
| 50 | M | 0.01 | none |
| 54 | M | n.d. | none |
| 55 | M | n.d. | none |
| 57 | F | 1.94 | IDH1 R132G |
| 59 | F | n.d. | none |
| 60 | M | 0.01 | none |
| 61 | M | 0.11 | IDH2 R140Q |
| 62 | M | 0.47 | IDH2 R140Q |
| 65 | M | n.d. | none |
| 66 | M | 2.98 | IDH1 R132C |
| 68 | M | 0.61 | IDH1 R132G |
| 69 | M | n.d. | none |
| 69 | F | 1.38 | IDH1 R132C |
| 69 | M | 0.46 | IDH2 R140Q |
| 70 | F | 0.66 | IDH2 R140Q |
| 71 | F | n.d. | none |
| 72 | F | 0.32 | IDH2 R140Q |
| 78 | M | 0.01 | none |
Figure 5. Primary human AML samples with IDH1 or IDH2 mutations display marked elevations of 2HG.

(A) and (B) AML patient peripheral blood, bone marrow, or pheresis samples were extracted for analysis of intracellular metabolites. Organic acids were purified, derivatized with MTBSTFA, and then analyzed by GC-MS as in Figure 4. Shown are representative gas chromatograms from samples subsequently determined to lack IDH1 or IDH2 mutations (A) or to have a R140Q mutation in IDH2 (B). (C) 2HG signal intensity relative to the intrasample glutamate signal was quantified in a total of 27 serial samples where adequate tumor tissue was available, and then segregated by IDH mutation status. Horizontal bars depict the group mean.
The genetic analysis of these tumor samples revealed two neomorphic IDH mutations that produce 2HG. Among the IDH1 mutations, tumors with IDH1 R132C or IDH1 R132G accumulated 2HG. This result is not unexpected, as a number of mutations of R132 to other residues have also been shown to accumulate 2HG in glioma samples (Dang et al., 2009).
The other neomorphic allele was unexpected. All five of the IDH2 mutations producing 2HG in this sample set contained the same mutation, R140Q. As shown in Figure 1, both R140 in IDH2 and R100 in IDH1 are predicted to interact with the β-carboxyl of isocitrate. Additional modeling revealed that despite the reduced ability to bind isocitrate, the R140Q mutant IDH2 is predicted to maintain its ability to bind and orient α-ketoglutarate in the active site (Figure 6). This potentially explains the ability of cells with this neomorph to accumulate 2HG in vivo. As shown in Figure 5, samples containing IDH2 R140Q mutations were found to have accumulated 2HG to levels 10–100 fold greater than the highest levels detected in IDH wild-type samples.
Figure 6. Structural modeling of R140Q mutant IDH2.
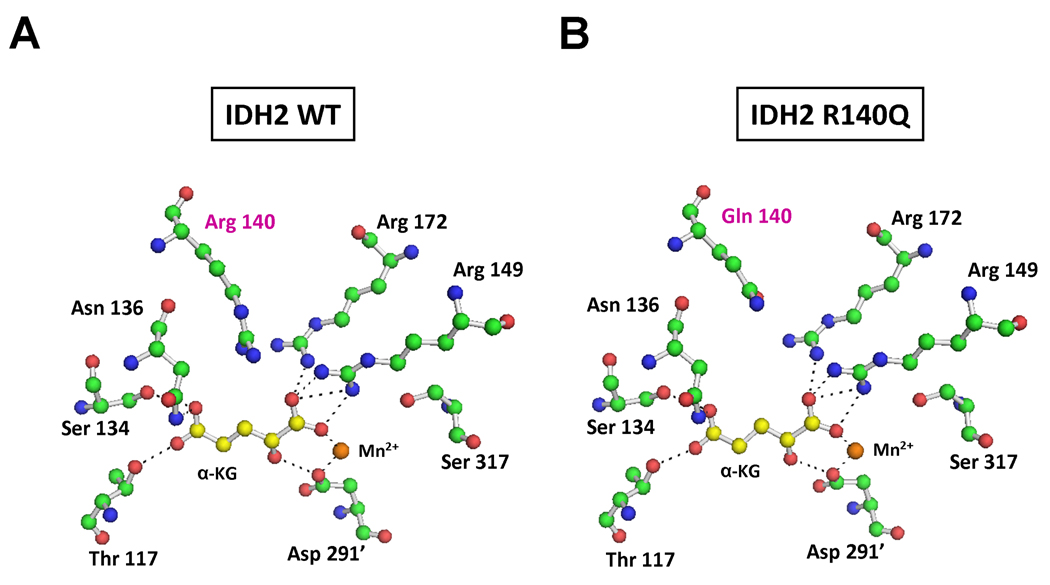
(A) Active site of human wild-type IDH2 with isocitrate replaced by α-ketoglutarate (α-KG). R140 is well positioned to interact with the β-carboxyl group that is added as a branch off carbon 3 when α-ketoglutarate is reductively carboxylated to isocitrate. (B) Active site of R140Q mutant IDH2 complexed with α-ketoglutarate, demonstrating the loss of proximity to the substrate in the R140Q mutant. This eliminates the charge interaction from residue 140 that stabilizes the addition of the β-carboxyl required to convert α-ketoglutarate to isocitrate.
IDH2 mutations are more common than IDH1 mutations in AML
In the above sample set, just over half of the AML samples with 2HG accumulation had IDH2 mutations. This is in contrast to the data reported for gliomas in which less than 5% of IDH mutations were in IDH2 (Hartmann et al., 2009; Yan et al., 2009). Initial studies of IDH genes in leukemia had failed to detect IDH2 mutations in R172 (Mardis et al., 2009). In the 18 cases we examined initially, all of the IDH2 mutations were IDH2 R140Q. However, this sample may have been biased by either the sample size and/or patient selection. We therefore analyzed 78 serial AML samples collected from a single center to determine the frequency of IDH1/2 mutation and investigated the co-association of IDH1/2 mutations with other genes known to contribute to AML pathogenesis (Table 2). While IDH1 mutations were found in 7.7% of samples in this analysis (6/78), twice as many mutations were detected in IDH2, at a frequency of 15.4% (12/78, p<0.05). Both the R140Q and R172K neomorphic alleles of IDH2 were identified. Of note, the IDH2 R140Q mutation was observed in more patients (7/78) than the R172K allele of IDH2 or R132 mutations in IDH1. None of the IDH2-mutated samples were found to also have somatic mutations in Flt-3, NPM1, or ASXL1. Furthermore, IDH2 and IDH1 mutations were only observed in normal karyotype AML patients, whereas known cytogenetic abnormalities were observed in 7/51 samples without IDH1/2 mutations (13.7%; all IDH mutants vs. IDH wild-type, p<0.05). We also examined whether IDH1 and/or IDH2 mutation status had any effect on the overall survival of this set of patients. As was previously reported (Mardis et al., 2009), mutations in IDH1 did not appear to affect overall survival (p=0.37, Figure S2A). However, we noted a trend towards improved survival in patients who presented with IDH2 mutations (p=0.08, Figure S2B).
Table 2.
Clinical and genetic parameters of IDH1/2 wild-type and mutant AML samples.
| IDH status | Average age at diagnosis (range) |
Average months Survival (range) |
# Complex cytogeneticsA (%) |
# MLL rearrangement (%) |
# Flt-3 mutant (%) |
# NPM1 mutant (%) |
# ASXL1 mutant (%)B |
|---|---|---|---|---|---|---|---|
| Wild-type (n=60) |
58.3 (6–86) |
19.7 (1–70) |
7/51 (13.7%) |
4/60 (6.7%) |
11/60 (18.3%) |
4/60 (6.7%) |
8/56 (14.3%) |
|
IDH1 mutant (n=6) |
69.5 (51–91) |
12.5 (2–20) |
0/6 (0%) |
0/6 (0%) |
1/6 (16.7%) |
1/6 (16.7%) |
0/6 (0%) |
|
IDH2 mutant (R140Q n=7; R172K n=5) |
71.6 (48–85) |
45.8 (4–107) |
0/12 (0%) |
2/12 (16.7%) |
0/11 (0%) |
0/9 (0%) |
0/10 (0%) |
| All IDH mutants (n=18) |
70.9 (48–91) |
34.7 (2–107) |
0/18 (0%) |
2/18 (11.1%) |
1/17 (5.9%) |
1/15 (6.7%) |
0/16 (0%) |
Complex cytogenetics defined as ≥ 3 cytogenetic abnormalities.
Samples with alterations which could not be confirmed to be somatic where excluded from analysis.
See also Figure S2.
DISCUSSION
Neomorphic enzymatic activity to produce 2HG is the shared feature of IDH1 and IDH2 mutations
Mutations in the cytosolic enzyme IDH1 have been reported in a high percentage of gliomas and a significant subset of AML patients (Mardis et al., 2009; Yan et al., 2009). Recently, IDH1 mutations of R132 were associated with the accumulation of the metabolite 2HG in glial tumor samples (Dang et al., 2009). The discovery of an elderly AML patient with an IDH2 R172 mutation, the analogous residue to IDH1 R132, prompted us to test whether this IDH2 mutation also resulted in a gain-of-function ability to catalyze the conversion of α-ketoglutarate to 2HG. In this study of IDH1 and IDH2 mutations in AML, we have demonstrated that 2HG production is a common feature of the spontaneous mutations in these two IDH homologs. By showing that 2HG production can result from mutation at any of three residues which normally stabilize the β-carboxyl of isocitrate, IDH1 R132, IDH2 R172, and IDH2 R140, we demonstrate that it is the truncated, non-carboxylating, reduction of α-ketoglutarate to R(-)-2-hydroxyglutarate that is the common feature linking all cancer-associated IDH mutations.
2HG as a screening and diagnostic marker
With our demonstration that 2HG production is the common feature of IDH1 and IDH2 mutations, the measurement of 2HG levels allowed us to identify additional IDH mutations in AML patients. This is exemplified in this study by the use of 2HG screening to identify two distinct mutations in IDH2, R140Q and R172K, that produce 2HG. These data demonstrate that at least IDH2 neomorphic mutations are not limited to a single arginine residue. Therefore, additional IDH1 and IDH2 neomorphic alleles may still remain to be found in gliomas, hematopoietic malignancies, and in other cancers. The 2HG levels in cancers with IDH mutations are consistently 10–100 fold elevated over levels found in samples lacking mutations of IDH1 or IDH2. Inborn errors of metabolism which lead to 2HG elevation are exceptionally rare and have devastating developmental consequences (Kolker et al., 2002a). Tumors displaying elevated 2HG are unlikely to be false positives. Thus, 2HG appears to be an important marker of IDH1/2-mutated neoplasms. Given the quantities of 2HG that accumulate in the culture medium of cells expressing IDH mutant enzymes, 2HG may even be detectable in the peripheral blood of leukemia patients with IDH mutations at the time of presentation. We are currently testing the potential of such a screening and diagnostic approach.
Maintaining at least one IDH1 and IDH2 wild-type allele may be essential for transformed cells
2HG production is a shared feature of heterozygous mutations in either cytosolic IDH1 or mitochondrial IDH2. There is a marked difference in the enzymatic rate of isocitrate-dependent NADPH production by the wild-type proteins and α-ketoglutarate-dependent NADPH consumption by both IDH1 and IDH2 mutants (Figure 2 and Dang et al., 2009). Nevertheless, both mutant proteins lead to an over 100-fold increase in tumor 2HG accumulation. In addition, it appears that both wild-type IDH1 and IDH2 can play positive and non-redundant roles in cell proliferation. The roles of IDH1 and IDH2 in supporting cell proliferation are likely to be different. IDH1 contributes to cytosolic NADPH production. In the case of IDH1 mutant proteins, it has been reported that a local supply of NADPH and α-ketoglutarate is required to drive 2HG production (Dang et al., 2009). In the context of a wild-type/mutant IDH1 heterodimer, the required substrates for the mutant’s neomorphic activity are most readily provided by the wild-type subunit’s normal activity on NADP+ and isocitrate. However, for the neomorphic activity of mitochondrial IDH2 mutants, the requirement for local generation of substrate is unlikely to be absolute. Mitochondria typically have a high NADPH/NADP+ ratio, supported in part by the ability to interconvert NADPH and NADH in the matrix (Rydstrom, 2006). In addition, mitochondrial α-ketoglutarate is maintained at high levels by a variety of anaplerotic substrates. While the exact role of wild-type IDH2 in supporting cell growth will require further investigation, this requirement correlates with the ability of IDH2 to contribute to the conversion of α-ketoglutarate into citrate via a pathway of reductive carboxylation in the mitochondria of proliferating cells.
2HG as an onco-metabolite
Although 2HG has been proposed to increase ROS levels in patients with inborn errors of 2HG metabolism (Kolker et al., 2002b; Latini et al., 2003), to date we have no evidence that the metabolite acts as a mutagen. This is consistent with data presented here that IDH1-mutated and IDH2-mutated AML samples typically do not harbor other known mutations associated with AML, as well as a prior report that IDH-mutated gliomas often lack other mutations that are commonly acquired early in glioma pathogenesis (Yan et al., 2009). In an AML case with an IDH1 mutation that was sequenced in its entirety (Mardis et al., 2009), there were a relatively small total number of somatic, non-synonymous mutations. However, a possibility by which 2HG might contribute to tumorigenesis emerges from consideration of the tumor subtypes in which IDH mutations are found with high frequency: gliomas with mixed astrocytic and oligodendroglial features, and acute myeloid leukemias. In both cases, proliferation of a relatively undifferentiated cell population is central to the disease, and pathogenesis is marked by a block to differentiation rather than simply by an increase in proliferative rate. Thus, one may speculate that 2HG’s effect in the tumor and its microenvironment is to block cellular differentiation. Whether cells carrying a mutant IDH transgene exhibit a block to differentiation in vivo will need to be examined. The production of lineage-specific mutant IDH transgenic animals or hematopoetic bone marrow reconstitution following retroviral introduction of a mutant IDH will allow future studies to test this hypothesis.
Whether 2HG fits into the broad class of mutagens or plays a distinct role in carcinogenesis remains to be determined. For now, it appears to represent a highly correlative maker for an emerging class of somatic mutations in the isocitrate dehydrogenase enzymes. Importantly, these mutations appear to define a significant subset of CNS tumors and leukemias. The identification of frequent IDH2 mutations in AML increases the significance of this family of mutations in understanding the pathogenesis of AML. Taken together, IDH mutations occur in 23% of samples analyzed here. All IDH mutations reported to date share the neomorphic ability to produce high levels of tumor 2HG. Future studies are required to understand the role of 2HG in leukemic transformation and to elucidate the role of IDH1/IDH2 in the pathogenesis of AML.
EXPERIMENTAL PROCEDURES
Patient selection and statistical analysis
Patient samples were obtained from either the Stem Cell and Xenotransplantation Core Facility of the University of Pennsylvania or from the tissue collections of Memorial Sloan-Kettering Cancer Institute. Approval was obtained from the institutional review boards at the University of Pennsylvania (IRB protocol 703185) and Memorial Sloan Kettering Cancer Institute (IRB protocols 95–091 and 06–107), and informed consent was provided according to the Declaration of Helsinki. All samples were collected after de-identification for these studies. For assessing clinical and genetic parameters of IDH1/2 wild-type and mutant AML, 78 serial samples from AML patients referred for molecular testing at Memorial Sloan-Kettering Cancer Center were examined. For 2HG assays, patient samples were obtained from the Stem Cell and Xenotransplantation Core Facility of the University of Pennsylvania. The initial 18 samples were selected from de-identified AML patients aged 50 years or older at diagnosis with lesions determined to have normal cytogenetic status. Cells used for these assays were prepared by Ficoll separation of mononuclear cells (MNCs) from peripheral blood or bone marrow. MNCs were frozen as viable cells in 10% DMSO. Student’s t test and Chi-square analysis were used to analyze data. A p value < 0.05 was considered significant.
Sequence Analysis of IDH1 and IDH2
Genomic DNA was extracted from bone marrow mononuclear cells or from sorted leukemic cells; for samples with less than 70% blasts flow cytometric sorting (FACSAria) was used to isolate blast cells according to leukemic blast immunophenotype before DNA isolation. High-throughput DNA sequence analysis was used to screen for IDH1 and IDH2 mutations. All DNA samples were whole genome amplified using Ø29 polymerase and mutations were validated on unamplified DNA to ensure all mutations were present in the diagnostic sample. Sequencing of IDH1 used primers which cover amino acid residues 41–138 (sense, 5’-TGTGTTGAGATGGACGCCTA-3’; antisense, 5’-GGTGTACTCAGAGCCTTCGC-3’). Sequencing of IDH2 used primers which cover amino acid residues 125–226 (sense, 5’-CTGCCTCTTTGTGGCCTAAG-3’; antisense, 5’-ATTCTGGTTGAAAGATGGCG-3’). Sequence analysis was performed using Mutation Surveyor (SoftGenetics, State College PA) and all mutations were validated by repeat PCR and sequencing on unamplified DNA from the archival sample.
Structural modeling
Human IDH2 has 97% homology with pig IDH2, and none of the 13 residues that are different (out of 418 total, excluding the N-terminal mitochondrial signal sequence) are found in the active site. A PDB structure of pig IDH2 is available, with isocitrate in the active site (1LWD) (Ceccarelli et al., 2002). Based on the highly homologous pig structure, conservative structural models of human IDH2 were built with the CHARMM molecular mechanics package using the CHARMM27 force field. To model wild-type IDH2 with isocitrate, the 13 residues of 1LWD differing between pig and human were first changed to the human sequence. The side chains were then rebuilt, hydrogen atoms added to all residues, the substrate, active site Mn2+ ions, and conserved residues were restrained, and the structure minimized allowing only the changed residues to relax. For the α-ketoglutarate complexes, the substrate conformation/pose was first modeled on isocitrate by removing the β-carboxyl group and replacing it with a hydrogen. The wild-type IDH2-α-ketoglutarate complex was then minimized allowing only the active site residues (as defined in entry 1LWD) and substrate to relax. The R140Q mutant IDH2-α-ketoglutarate complex was subsequently modeled by changing arginine 140 to glutamine, and rebuilding the side chain. The structure was then minimized in two stages. First the mutated residue was allowed to relax, followed by a second relaxation of the active site residues, Mn2+, and substrate. All modeling images were generated using PyMOL Viewer (DeLano, 2002).
Constructing IDH1 and IDH2 mutants
The cDNA clone of human IDH2 (BC009244) was purchased from Invitrogen in pOTB7. Human IDH1 (BC012846.1) was purchased from ATCC in pCMV-Sport6. Standard site-directed mutagenesis techniques were used to generate IDH2 R172K by introducing a g515a change in the IDH2 open reading frame (ORF). IDH1 R132H was made by introducing a g395a base pair change in the IDH1 ORF. Wild-type and mutant sequences were then subcloned into pcDNA3 (Invitrogen) and confirmed by direct sequencing before expression in mammalian cells.
Cell culture, transfection, and metabolic labeling
293T cells and Bcl-xL-transfected SF188 cells (SF188) were cultured in DMEM (Dulbecco’s modified Eagle’s medium; Invitrogen) with 10% fetal bovine serum (CellGro). For expression of wild-type and mutant IDH1 and IDH2, cells were transfected with Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. For IDH siRNA experiments, cells were transfected with Lipofectamine RNAiMax, with oligonucleotides obtained from Sigma-Proligo. For metabolic labeling experiments, cells were cultured in glutamine-free DMEM supplemented with 4 mM [13C-U]-L-glutamine (Cambridge Isotope) for the 3h prior to metabolite extraction.
Cell lysate based enzyme assays
For IDH2 enzymatic assays, cells were lysed 48 h following transfection, using mammalian protein extraction reagent (Pierce) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktails 1 and 2 (Sigma). Lysates were sonicated and centrifuged at 14,000g at 4 °C. Supernatants were then collected and normalized for total protein concentration. To measure IDH oxidative activity, 0.3 µg of lysate protein was added to 200 µl of an assay solution containing 100 mM Tris-HCl buffer (pH 7.5), 1.3 mM MnCl2, 0.33 mM EDTA, 0.1 mM β-NADP+, and 0.1 mM D-(+)-threo-isocitrate. The increase in 340 nm absorbance (OD340) as a measure of NADPH production was measured every 20 s for 30 min on a SpectraMax 190 spectrophotometer (Molecular Devices). Data are plotted as the mean activity of 3 replicates per lysate averaged among 5 time points centered at every 5 minutes. To measure IDH reductive activity, 3 µg of lysate protein was added to 200 µl of an assay solution containing 100 mM Tris-HCl (pH 7.5), 1.3 mM MnCl2, 0.01 mM β-NADPH, and 0.5 mM α-ketoglutarate. NADPH consumption was measured as the decrease in OD340, with 3 replicates per lysate. For all experiments, OD340 changes in assay buffer lacking lysate protein were measured and subsequently subtracted from the OD340 changes measured in lysate replicates to arrive at final values.
Western blot
To measure IDH2 levels in cell lysates used for enzymatic assays, aliquots of the same lysates used in activity measurements were separated by SDS-PAGE, transferred to nitrocellulose, probed with IDH2 mouse monoclonal antibody (Abcam, ab55271), and then detected with HRP-conjugated anti-mouse antibody (GE Healthcare, NA931V). For assessing IDH knockdown in siRNA experiments, cells treated in parallel with those used for labeling or proliferative studies were lysed 48 h following transfection in standard RIPA buffer (1% NaDOC, 0.1% SDS, 1% Triton X-100, 0.01 M Tris pH 8.0, 0.14 M NaCl), and then probed with IDH2 antibody as above, IDH1 goat polyclonal antibody (Santa Cruz Biotechnology, sc49996), or IDH3A rabbit polyclonal antibody (Abcam, ab58641). Actin antibody (Santa Cruz, sc1616) was also used to assess equal protein loading of Western blots.
Metabolite extraction
Cellular organic acids were extracted as previously described (Bennett et al., 2008). Briefly, after gentle removal of culture medium from proliferating cells, or freezing medium from frozen viable AML samples, cells were rapidly quenched with 80% methanol, chilled to −80°C, and then incubated at −80°C for 15 min. Extracts were subsequently transferred and centrifuged at 14,000g for 20 min. at 4°C. The organic acid pool in the supernatant was further purified by drying under nitrogen gas, redissolving in deionized water, and then elution from an AG-1 ×8 100–200 anion exchange resin (Bio-Rad) in 3 N HCl after washing with five column volumes. For media analysis, culture medium from transfected cells 24–48 h following transfection was collected and diluted five-fold with methanol. After centrifugation at 14,000g for 20 min at 4 °C to remove precipitated protein, supernatants were dried under nitrogen gas, and organic acids were purified as described above.
Gas chromatography-mass spectrometry (GC-MS) analysis
After drying the HCl eluate, samples were redissolved in a 1:1 mixture of acetonitrile and N-methyl-N-tert-butyldimethylsilyltrifluoroacetamide (MTBSTFA; Regis) and heated for 1 h at 60°C to derivatize prior to GC-MS analysis. Samples were injected into an Agilent 7890A GC with an HP-5MS capillary column, connected to an Agilent 5975C Mass selective detector operating in splitless mode using electron impact ionization with ionizing voltage of −70 eV and electron multiplier set to 1060 V. GC temperature was started at 100 °C for 3 min, ramped to 230 °C at 4 °C/min and held for 4 min, then ramped to 300 °C and held for 5 min. Mass range of 50–500 amu was recorded at 2.71 scans/sec. Isotopic enrichment in citric acid was monitored using ions at m/e− 463 and 464 for citrate +4 and citrate +5 (containing 4 and 5 13C-enriched atoms, respectively), formed through loss of a t-butyl (−57 amu) and t-butyldimethylsilanol (−132 amu) from the molecular ion tetra-TBDMS-citric acid (648 amu). Isotopomer distributions were simultaneously corrected for naturally occurring heavy isotopes of all elements in each mass fragment using a correction matrix as previously described (Weckwerth, 2007). Identification of the 2HG metabolite peak was confirmed using standards obtained from Sigma. 2HG and glutamate signal intensities were quantified by integration of peak areas.
Liquid chromatography-mass spectrometry (LC-MS)
Organic acids from cellular extracts were purified as described above, followed by evaporation to dryness under nitrogen. After redissolving samples in deionized water, citrate was detected on two different LC-MS approaches, both of which gave comparable results. In both cases, LC separation was by reversed phase chromatography using tributylamine as an ion pairing agent (Lu et al., 2008; Luo et al., 2007) with ionization by negative electrospray at 23 kV. The first MS approach used a Thermo Discovery Max triple quadrupole mass spectrometer in multiple reaction monitoring mode, with citrate quantified using the reaction 191 → 87 at 20 eV. Additional reactions for every possible labeled form of citrate were also monitored using variations of the same transition. Reactions used to monitor other TCA components have been described previously (Bajad et al., 2006). The second MS approach used a Thermo Exactive Orbitrap mass spectrometer operated at 100,000 mass resolving power, with citrate and its isotope-labeled forms quantified based on extracted ion chromatograms at their exact masses.
SIGNIFICANCE
Most cancer-associated enzyme mutations result in either catalytic inactivation or constitutive activation. Here we report that the common feature of IDH1 and IDH2 mutations observed in AML and glioma is the acquisition of an enzymatic activity not shared by either wild-type enzyme. The product of this neomorphic enzyme activity can be readily detected in tumor samples and we show that tumor metabolite analysis can identify patients with tumor-associated IDH mutations. Using this method, we discovered a 2HG-producing IDH2 mutation, IDH2 R140Q, that was present in 9% of serial AML samples. Overall, IDH1 and IDH2 mutations were observed in over 23% of AML patients.
HIGHLIGHTS
All IDH mutations reported in cancer share a common neomorphic enzymatic activity.
Both wild-type IDH1 and IDH2 are required for cell proliferation.
IDH2 R140Q mutations occur in 9% of AML cases.
Overall, IDH2 mutations appear more common than IDH1 mutations in AML.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tullia Lindsten, Anthony Mancuso, Scott Olejniczak, and other members of the Thompson Laboratory for technical help, valuable discussion, and critical reading of the manuscript. We thank Cezary Swider and Joy Cannon at the University of Pennsylvania’s Stem Cell and Xenograft Core for their assistance in obtaining patient samples, and Jesse Platt for help with structural modeling. We also thank Adriana Heguy of the Geoffrey Beene Translational Oncology Core Facility and Kelly Harris for assistance with sequence analysis. R.L.L. is an Early Career Award recipient of the Howard Hughes Medical Institute and is the Geoffrey Beene Junior Chair at Memorial Sloan-Kettering Cancer Center. This work was supported in part by grants from the NCI and NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
NOTE ADDED IN PROOF
While this manuscript was in review, two cases of IDH2 mutations were reported in leukemias that arose in patients with myeloproliferative disease (Green and Beer, 2010). Thus, IDH2 mutations can occur in both de novo cases of AML and in AML arising secondary to myeloproliferative disorders.
REFERENCES
- Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J Chromatogr A. 2006;1125:76–88. doi: 10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Yuan J, Kimball EH, Rabinowitz JD. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat Protoc. 2008;3:1299–1311. doi: 10.1038/nprot.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli C, Grodsky NB, Ariyaratne N, Colman RF, Bahnson BJ. Crystal structure of porcine mitochondrial NADP+-dependent isocitrate dehydrogenase complexed with Mn2+ and isocitrate. Insights into the enzyme mechanism. J Biol Chem. 2002;277:43454–43462. doi: 10.1074/jbc.M207306200. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. 2002 (DeLano Scientific) [Google Scholar]
- Ehrlich RS, Colman RF. Influence of substrates and coenzymes on the role of manganous ion in reactions catalyzed by pig heart triphosphopyridine nucleotide-dependent isocitrate dehydrogenase. Biochemistry. 1976;15:4034–4041. doi: 10.1021/bi00663a018. [DOI] [PubMed] [Google Scholar]
- Green A, Beer P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med. 2010;362:369–370. doi: 10.1056/NEJMc0910063. [DOI] [PubMed] [Google Scholar]
- Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- Kamerling JP, Duran M, Gerwig GJ, Ketting D, Bruinvis L, Vliegenthart JF, Wadman SK. Determination of the absolute configuration of some biologically important urinary 2-hydroxydicarboxylic acids by capillary gas--liquid chromatography. J Chromatogr. 1981;222:276–283. doi: 10.1016/s0378-4347(00)81061-0. [DOI] [PubMed] [Google Scholar]
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ, Lee SH. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125:353–355. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- Kolker S, Mayatepek E, Hoffmann GF. White matter disease in cerebral organic acid disorders: clinical implications and suggested pathomechanisms. Neuropediatrics. 2002a;33:225–231. doi: 10.1055/s-2002-36741. [DOI] [PubMed] [Google Scholar]
- Kolker S, Pawlak V, Ahlemeyer B, Okun JG, Horster F, Mayatepek E, Krieglstein J, Hoffmann GF, Kohr G. NMDA receptor activation and respiratory chain complex V inhibition contribute to neurodegeneration in d-2-hydroxyglutaric aciduria. Eur J Neurosci. 2002b;16:21–28. doi: 10.1046/j.1460-9568.2002.02055.x. [DOI] [PubMed] [Google Scholar]
- Latini A, Scussiato K, Rosa RB, Llesuy S, Bello-Klein A, Dutra-Filho CS, Wajner M. D-2-hydroxyglutaric acid induces oxidative stress in cerebral cortex of young rats. Eur J Neurosci. 2003;17:2017–2022. doi: 10.1046/j.1460-9568.2003.02639.x. [DOI] [PubMed] [Google Scholar]
- Lu W, Bennett BD, Rabinowitz JD. Analytical strategies for LC-MS-based targeted metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;871:236–242. doi: 10.1016/j.jchromb.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Groenke K, Takors R, Wandrey C, Oldiges M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography-mass spectrometry. J Chromatogr A. 2007;1147:153–164. doi: 10.1016/j.chroma.2007.02.034. [DOI] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa S. Biosynthesis of tricarboxylic acids by carbon dioxide fixation; enzymatic mechanisms. J Biol Chem. 1948;174:133–157. [PubMed] [Google Scholar]
- Rydstrom J. Mitochondrial NADPH, transhydrogenase and disease. Biochim Biophys Acta. 2006;1757:721–726. doi: 10.1016/j.bbabio.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Siebert G, Carsiotis M, Plaut GW. The enzymatic properties of isocitric dehydrogenase. J Biol Chem. 1957;226:977–991. [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Weckwerth W, editor. Metabolomics: Methods and Protocols. Totowa, NJ: Humana Press; 2007. [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.