

Abstract
Previous studies have suggested that both inflammatory monocytes and neutrophils are important for controlling acute toxoplasmosis in the mouse model. To test the role of these cell types, we used monoclonal antibody (MAb) RB6-8C5 to deplete both subsets of cells or MAb 1A8 to selectively remove neutrophils. RB6-8C5 MAb-treated mice succumbed to oral infection with Toxoplasma gondii, similar to Ccr2−/− mice, which are deficient in monocyte recruitment but have normal neutrophils. In contrast, mice treated with MAb 1A8 controlled parasite replication and survived acute infection. Ccr2−/− mice suffered from acute ileitis and inflammation in the spleen that was associated with a lack of inflammatory monocytes and elevated numbers of neutrophils. RB6-8C5 MAb-treated C57BL/6 mice also suffered from intestinal pathology and splenic damage, although this was less extensive due to the reduced numbers of neutrophils. Neutrophil-depleted infected wild-type mice displayed no pathological changes, compared to untreated infected controls. Collectively, these observations demonstrate the critical role of inflammatory monocytes during the acute infection with the parasite T. gondii and reveal that neutrophils are not protective but rather contribute to the pathology.
The intracellular parasite Toxoplasma gondii is an opportunistic pathogen that infects approximately one-third of the human population worldwide (19). Following oral infection, the parasite crosses the intestinal epithelium, disseminates widely, and traverses biological barriers such as the blood-brain barrier to reach immunologically privileged sites, where it causes the most severe pathology (3). Histopathological studies document initial invasion into a variety of cell types in the intestine, including CD11b+ and CD11c+ intraepithelial leukocytes, which have been implicated in dissemination of the parasite (11). We previously demonstrated that the numbers of Gr1+ inflammatory monocytes dramatically increase in the lamina propria of the small intestine in response to T. gondii infection and that these cells play a crucial role in protection against the parasite (14).
Murine monocytes have been classified in two subpopulations based on different surface markers and phenotypic characteristics (17, 18). Inflammatory monocytes (CD115+, Gr1+, F4/80+, CD11b+, and CX3CR1lo) express chemokine receptor 2 (CCR2), emerge from the bone marrow in response to chemokines such as MCP1 (CCL2), and home to sites of inflammation (17, 18). These precursors have the potential to differentiate into dendritic cells (CD11c+) and to populate nonlymphoid tissues such as the lamina propria (33). The other major subset of monocytes (Gr1−, F4/80+, CD11b+, and CX3CR1hi) are primarily distributed as resident tissue macrophages, which provide surveillance functions (2). Following oral challenge with T. gondii, inflammatory monocytes upregulate inducible nitric oxide synthase (iNOS), produce interleukin-12 (IL-12), and secreted tumor necrosis factor alpha (TNF-α) in response to T. gondii infection (23, 24). Collectively, these responses likely contribute to the control of parasite replication at the site of primary infection in the small intestine (14). Mice deleted in the gene for chemokine receptor 2 (CCR2), or the major chemokine ligand (CCL2) for this receptor, fail to recruit inflammatory monocytes to the lamina propria in response to T. gondii infection (14). The resulting inability to control parasite replication within the small intestine is associated with neutrophil efflux, tissue destruction, and death of mice due to intestinal necrosis (14). Damage to the small intestine following oral infection with T. gondii is also decreased by the genetic absence of the IL-17 receptor (IL-17R), implicating a role for neutrophils in this pathology (20). Collectively, these findings establish the critical role of inflammatory monocytes in innate mucosal immunity but suggest that neutrophils may contribute to enhanced pathology.
Neutrophils contain numerous potent antimicrobial effectors, and they rapidly accumulate at the sites of many bacterial infections (21). Neutrophils produce several cytokines and chemokines, such as IL-1β, IL-12, TNF-α, MIP-1, and MIP-2, and hence, they recruit and activate other immune cells (27). There is also evidence that neutrophils influence the T-cell response by enhancing the functions of dendritic cells (32) or inflammatory monocytes (30). Infection of mice by intraperitoneal (i.p.) inoculation with low numbers of a highly virulent strain of T. gondii or with a high inoculum of low-virulence strains results in recruitment of neutrophils to the peritoneal cavity (4, 22). Mouse and human neutrophils challenged in vitro with T. gondii also produce IL-12, TNF-α, MIP-1α, and MIP-1β (6, 7). It has been suggested that neutrophils are protective during acute toxoplasmosis in the mouse based on depletion with MAb RB6-8C5 (5, 26), which results in enhanced susceptibility. However, much more modest effects are observed in mice lacking the major neutrophil chemokine receptor CXCR2, and these animals survive acute infection but develop slightly higher chronic tissue burdens (13). These apparently discordant results might reflect the different genetic backgrounds used in these prior studies, since antibody depletion studies were done with susceptible C57BL/6 mice, while CXCR2−/− mice were in the resistant BALB/c background.
The anti-granulocyte receptor 1 (Gr1) monoclonal antibody (MAb) RB6-8C5 has been used extensively to deplete neutrophils in mice and to investigate the role of these cells in host defense to bacterial and protozoal infections (8-10, 21, 31). RB6-8C5 binds to Ly6G, which is present on the surface of neutrophils, and to Ly6C, which is expressed on neutrophils, dendritic cells, and subpopulations of lymphocytes and monocytes (15). In vivo administration of RB6-8C5 depletes not only neutrophils (CD11b+, F4/80−, Ly6Cint [where int is intermediate], and Ly6Ghi) but also inflammatory monocytes (CD11b+, F4/80+, Ly6Chi, and Ly6G−) (12), thus complicating the interpretation of studies using this antibody for neutralization. The Ly6G-specific MAb 1A8 has recently been reported to be an alternative method to selectively deplete neutrophils (12).
In the present study, we sought to determine the respective roles of inflammatory monocytes and neutrophils in the pathogenesis and control of T. gondii in mice. We compared the outcome of oral challenge with T. gondii in mice depleted of neutrophils and monocytes with MAb RB6-8C5 and that of mice where neutrophils were selectively depleted with MAb 1A8.
MATERIALS AND METHODS
Parasite strains and mouse infections.
The luciferase-expressing PRU-Luc-GFP type II strain (provided by J. Boothroyd, Stanford University, Palo Alto, CA) of T. gondii was used for production of tissue cysts in CD1 outbred mice (Charles River Laboratories, Wilmington, MA). Tissue cysts used in experiments were obtained at 3 months postinfection from mice that were inoculated with 20 cysts by i.p. injection. Animals were sacrificed, the brains were removed and homogenized in 1 ml of phosphate-buffered saline (PBS) at pH 7.2, and tissue cysts were counted by microscopic examination of three or more aliquots of 20 μl. For challenge studies, 8- to 10-week-old female mice were used for all studies. Mice were infected orally (p.o.) with 20 cysts, and survival was followed for 25 days. CD1 mice were obtained from Charles River Laboratories, and C57BL/6 mice were obtained from Jackson Laboratories. Ccr2−/− mice fully backcrossed onto a C57BL/6J background were described previously (24). Animals were maintained in an AAALAC-approved facility overseen by the Institutional Animal Care Committee at Washington University.
Depletion of cell subsets.
Neutrophils were depleted using anti-Ly6G MAb 1A8 (provided by Thomas Malek, University of Miami School of Medicine, Miami, FL). Monocytes and neutrophils were depleted using MAb RB6-8C5 (provided by Robert L. Coffman, DNAX Research Institute, Palo Alto, CA). Antibodies were produced by growing the respective hybridomas in serum-free medium in CELLine flasks (IBA Integra Biosciences). The concentrations of IgG in supernatant fractions were estimated by resolution on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels stained with Coomassie blue in comparison to known standards. Antibodies were administered by i.p. injection starting on day 0 and continuing daily until day 5 postinfection. Injections comprised 500 μg for MAb 1A8 and 250 μg for MAb RB6.
Parasite burdens.
For bioluminescence experiments, animals were imaged at 2-day intervals, following mild anesthesia using isofluorene and i.p. injection of 0.15 mg of luciferin D (Biosynth AG) per kg of body weight. Animals were imaged using a Xenogen IVIS 100 (Caliper Life Sciences). Data are expressed in relative light units and are shown as the mean results. Experiments contained 3 to 5 animals per group (except a single animal was used for the noninfected negative control).
Cytokine levels in serum samples.
Blood samples were obtained from anesthetized animals by heart puncturing at 6 days postinfection and centrifuged, and the sera were stored at −80°C. Sera were diluted and used to detect cytokines and chemokines (IL-12, gamma interferon [IFN-γ], and TNF-α) using the cytokine bead array kit from BD Biosciences (San Jose, CA). Experiments were repeated three or more times, with 3 animals per group.
Cell isolation.
Peritoneal cells were obtained by lavaging with Hanks balanced salt solution (HBSS) containing 10 mM HEPES and 0.1 mM EGTA. Spleen cells were isolated by mechanical disruption with frosted glass slides. Erythrocytes were lysed with erythrocyte (RBC) lysis buffer (eBioscience, San Diego, CA). Cell suspensions were filtered through a 40-μm nylon sieve, washed twice by centrifugation at 300 × g for 10 min, and used immediately for flow cytometric analysis. Circulating leukocytes were obtained from heparinized blood that was diluted in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and layered on top of 60% isotonic Percoll. After centrifugation, the buffy coat was collected and washed in PBS. Experiments were repeated three or more times, with 3 animals per group.
Flow cytometry.
Isolated cells were pretreated on ice for 20 min with MAb 2.4G2 (2.0 μg/ml) to block nonspecific binding to Fcγ receptors. Thereafter, cells were incubated on ice for 30 min, using the following fluorescently conjugated antibodies for cell surface markers: Cy5-conjugated anti-Gr1 MAb RB6-8C5 (eBioscience, San Diego, CA), phycoerythrin (PE)- or allophycocyanin (APC)-conjugated anti-F4/80 MAb A3-1 (AbD Serotec, Raleigh, NC), APC-conjugated anti-CD115 MAb (eBioscience, San Diego, CA), APC-conjugated anti-CD11b MAb (eBioscience, San Diego, CA), fluorescein isothiocyanate (FITC)-conjugated anti-Ly6C MAb AL-21 (BD Biosciences), and PE-conjugated anti-Ly6G MAb 1A8 (BD Biosciences). Isotype controls used were PE-conjugated IgG2b, Cy5-conjugated IgG2b (eBioscience), and FITC-conjugated rat IgG (eBioscience). Analysis of stained cells was performed with a FACSCanto flow cytometer (BD Biosciences). Dead cells were gated out on the basis of 7-AAD staining, and isotype controls were used to set appropriate gates. Data were analyzed with FACSDiva (BD Biosciences) and FlowJo 6.4.7 (Tree Star, Ashland, OR). For all samples, approximately 20,000 cells were analyzed to generate scatter plots.
Histological studies.
Animals were sacrificed at 9 days postinfection, and the spleen and ileum were removed and fixed in 10% neutral buffered formalin. Tissues were dehydrated in ethanol and embedded in paraffin, and 5-μm sections were stained with hematoxylin and eosin (H&E). Sections were evaluated blindly to score pathological changes. Experiments were repeated three or more times, with 3 animals per group.
Statistical analysis.
Samples were compared using the unpaired Student t test (two-tailed), with equal variance and unpaired samples, and calculated using Excel (Microsoft, Seattle, WA).
RESULTS
In vivo depletion of neutrophils and/or inflammatory monocytes using specific antibodies.
To establish conditions for selective depletion in vivo, we tested the ability of antibodies to deplete inflammatory cells that were elicited by i.p. infection of T. gondii tachyzoites. Infection with T. gondii elicited both neutrophils (Gr1hi, Ly6Cint, Ly6G+, F4/80−) and inflammatory monocytes (Gr1int, Ly6Chi, Ly6G−, F4/80+) (Fig. 1A), as described previously (23). Mice were given either MAb RB6-8C5 or 1A8 on days 1 and 3, and cell recruitment to the peritoneal cavity was tested on day 4 by flow cytometry. Application of MAb 1A8 depleted only the neutrophil cell population (Gr1hi, Ly6Cint, Ly6G+, F4/80−), while inflammatory monocytes remained unaltered (Fig. 1B). Treatment with MAb RB6-8C5 depleted both inflammatory monocytes and neutrophils from the peritoneal cavity of mice (Fig. 1C). These findings demonstrate that selective depletion of neutrophils can be achieved with MAb 1A8, while MAb BR6-8C5 effectively depletes both neutrophils and monocytes, as reported previously (12).
FIG. 1.

Flow cytometry analysis of peritoneal cells from T. gondii-infected mice following in vivo administration of depleting antibodies. (A) Inflammatory monocytes (Gr1int, Ly6Chi, Ly6G−, and F4/80+) (circular gate = percentage of these cells) and neutrophils (Gr1hi, Ly6Cint, Ly6G+, and F4/80−) (square gate = percentage of these cells) were increased in the peritoneal cavity of control mice at day 4 following T. gondii infection. (B) Monocytes remained elevated, while neutrophils were depleted in the peritoneal cavities of those treated with MAb 1A8. (C) Inflammatory monocytes and neutrophils were absent in the peritoneal cavities of mice after administration of MAb RB6-8C5. CD1 mice were infected i.p. with 200 tachyzoites of type II (PRU-Luc-GFP) T. gondii 4 days before the cell isolation. Antibodies were administered on day 1 and day 3 after the infection. Representative example obtained from four similar experiments, with 2 mice each.
Characterization of circulating blood leukocytes and cytokine levels after depletion of neutrophils and inflammatory monocytes.
To confirm the extent of cellular depletion in antibody-treated mice, blood leukocytes were isolated and characterized by fluorescence staining and flow cytometry. When animals were given neutralizing antibodies every 48 h, the efficiency of depletion by MAb 1A8 was reduced, especially in lymphatic organs such as the spleen (data not shown). Instead, animals were given daily doses of neutralizing antibodies from day 0 to 5, and the efficiency of depletions was monitored in the blood and spleen at day 6 (Fig. 2). To provide more-specific labeling of the respective cell populations, we used MAb AL-21 to Ly6C (high on monocytes and intermediate on neutrophils) and anti-mouse CD115, which is present on blood monocytes but not neutrophils. In infected control mice, elevated levels of inflammatory monocytes (Ly6Chi and CD115+) and neutrophils (Ly6Cint and CD115−) were detected in the blood, compared to noninfected mice (Fig. 2A). Similar results were observed in the spleen, where neutrophils (CD11b+ and Ly6Cint) and inflammatory monocytes (CD11b+ and Ly6Chi) were increased in infected animals relative to noninfected controls (Fig. 2B). In infected C57BL/6 mice treated with MAb 1A8, the numbers of neutrophils were reduced by 3-fold, to levels equivalent to those seen in noninfected animals (Fig. 2A and B). In infected C57BL/6 mice treated with RB6-8C5 MAb, both inflammatory monocytes and neutrophils were efficiently depleted (Fig. 2A and B). Ccr2−/− mice showed elevated numbers of neutrophils, while inflammatory monocytes remained largely absent from the blood (Fig. 2A and B). Treatment of Ccr2−/− mice with MAb 1A8 reduced the level of neutrophils to a level comparable to that of noninfected mice (Fig. 2A and B).
FIG. 2.

Depletion of monocytes and neutrophils in the peripheral blood and spleen of mice treated with neutralizing antibodies and infected with T. gondii. (A) Characterization of circulating blood leukocyte subsets by flow cytometry. Inflammatory monocytes (Ly6Chi and CD115+) and neutrophils (Ly6Cint and CD115−) were defined by the indicated boxes (numbers represent percentages of cells). (B) Characterization of leukocyte subsets from the spleen using flow cytometry. Inflammatory monocytes (Ly6Chi and CD11b+) and neutrophils (Ly6Cint and CD11b+) were defined by the indicated boxes (numbers represent percentages of cells). Elevated levels of inflammatory monocytes and neutrophils were present in the blood and spleen of infected control mice (wild-type [WT] control infected) compared to those of noninfected mice (WT control noninfected). 1A8 treatment of control (WT + 1A8) and Ccr2−/− (Ccr2−/− + 1A8) mice reduced neutrophil numbers by 3-fold, resulting in levels similar to those of noninfected controls. RB6-8C5 treatment (WT + RB6) efficiently depleted both inflammatory monocytes and neutrophils. Infected Ccr2−/− mice (Ccr2−/−) lacked inflammatory monocytes, but neutrophils were present in higher numbers, similar to those of infected control mice. Antibodies were applied every day from day 0 to 5 after infection, and cells were isolated at day 6. Experiments were repeated three times, with 2 animals in each group. Plots are the results obtained from representative animals from one experiment.
Next, we examined the effects of alterations in inflammatory monocytes and neutrophils on cytokine production. To this end, serum cytokines were analyzed from T. gondii-infected mice treated with neutralizing antibodies from day 0 to 5 and tested 6 days postinfection. Cytokine levels were elevated in all infected mice, indicating that the cause of death was not due to failure to mount a type 1 cytokine response. The sera obtained from Ccr2−/− mice showed elevated levels of IFN-γ, IL-12, and TNF-α, although only the IFN-γ levels were significantly higher than those of control infected mice (Fig. 3). Treatment of Ccr2−/− mice with MAb 1A8 to deplete neutrophils resulted in diminished levels of all three cytokines, although only the difference in the level of IL-12 was statistically significant (Fig. 3).
FIG. 3.

Serum cytokine levels of mice after infection with T. gondii. Significantly elevated levels of IFN-γ were measured in Ccr2−/− mice (Ccr2−/−) compared to those in infected control mice (WT). IL-12 and TNF-α levels were also elevated in Ccr2−/− mice although not significantly. Administration of MAb 1A8 to Ccr2−/− mice reduced IFN-γ and TNF-α levels and significantly reduced IL-12 levels. Neutralizing antibodies (shown beneath the bars) were applied daily from day 0 to 5, and sera were collected on day 6 after p.o. infection with T. gondii. Three or more experiments were combined. Values represent the means ± SEM. *, P < 0.05; **, P < 0.01.
Susceptibility of C57BL/6 mice to T. gondii infection following depletion of neutrophils and/or inflammatory monocytes.
Having established efficient conditions to deplete cells, we directly compared the survival of C57BL/6 mice that were depleted of leukocyte subsets and the survival of those that were orally challenged with T. gondii. Ccr2−/− mice on a C57BL/6 background were used for a comparison, as they are deficient in inflammatory monocyte recruitment to the site of the infection (14). Infected wild-type mice and the majority of MAb 1A8-treated mice survived the infection (Fig. 4A). In contrast, all RB6-8C5 MAb-treated mice and Ccr2−/− mice succumbed to the infection within 14 days, indicating a requirement for inflammatory monocytes (Fig. 4A). Parasite replication also was monitored in living mice by luciferase imaging (Fig. 4B). Ccr2−/− mice and MAb RB6-8C5-treated mice were unable to control parasite replication, and parasite burdens reached very high levels prior to death (Fig. 4B). In contrast, low parasite numbers were observed in mice treated with MAb 1A8 and in control mice, indicating their ability to control parasite replication (Fig. 4B).
FIG. 4.
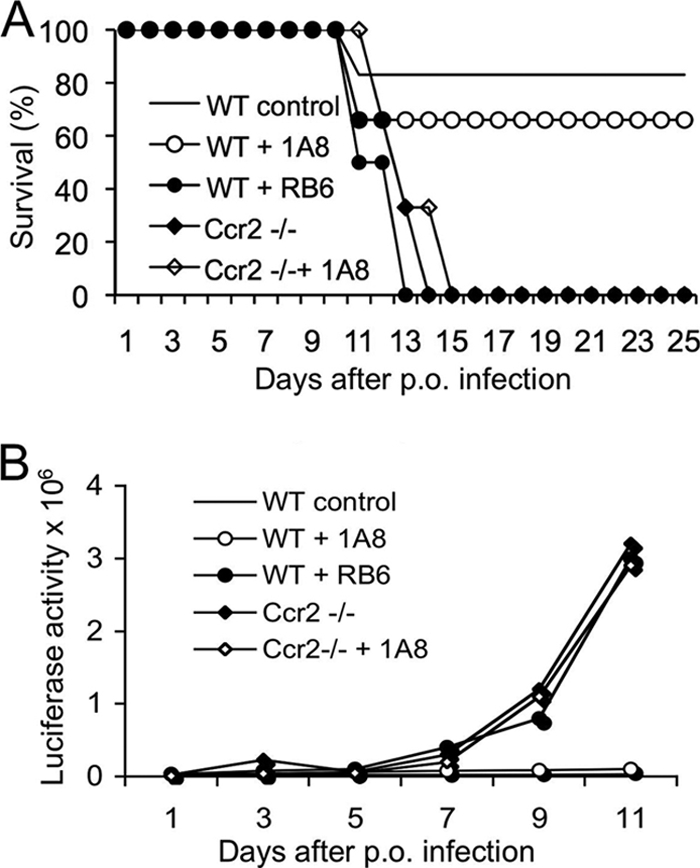
Survival of mice following oral infection with T. gondii. (A) Mice were infected with 20 cysts of type II gondii (PRU-Luc-GFP), and survival was monitored for 25 days. The majority of infected control mice (WT control) and MAb 1A8-treated mice (WT + 1A8) survived. Ccr2−/− mice, Ccr2−/− mice treated with MAb 1A8 (Ccr2 −/− + 1A8), and MAb RB6-8C5-treated mice (WT + RB6) all died within 14 days after infection. Antibodies were applied daily from days 0 to 5. Data are combined from five individual experiments, with 12 to 16 total animals per group. (B) Bioluminescence tracking of parasite dissemination. Luciferase imaging revealed control mice (WT control) and mice treated with 1A8 MAb (WT + 1A8) controlled the infection. In contrast, Ccr2−/− mice, Ccr2−/− mice treated with MAb 1A8 (Ccr2−/− + 1A8), and MAb RB6-8C5-treated mice (WT + RB6) were not able to control parasite replication. This figure shows one representative experiment (mean ± SD; 3 to 4 animals per group) from five similar experiments.
Ccr2−/− mice and RB6-8C5 MAb-treated C57BL/6 mice suffer from ileitis and inflammation of the spleen following infection with T gondii.
To investigate whether lethal T. gondii infection was associated with pathological changes in the ileum and spleen, mice were examined at day 9 after infection with T. gondii. In mice treated with MAb 1A8, the small intestine appeared similar to that in control infected mice and was typified by an absence of cellular pathology (Fig. 5A). RB6-8C5-treated mice had abundant apoptotic foci in the lamina propria and edema in the overlying epithelium, although damage was less extensive (Fig. 5A). Ccr2−/− mice suffered from extensive necrosis of the small intestine, massive lymphoid depletion, and infiltration of neutrophils in the lamina propria (Fig. 5A). Pathological changes seen in Ccr2−/− mice were partially offset by the administration of MAb 1A8, resulting in reduced neutrophil influx and overall less damage to the villus architecture (Fig. 5A).
FIG. 5.

Histological changes in the intestine and spleen of mice infected with T. gondii. (A) Pathological changes were not observed in the ileum of infected control mice (WT Control). 1A8-treated mice (WT + 1A8) displayed small numbers of apoptotic foci in the lamina propria. RB6-8C5-treated mice (WT + RB6) showed frequent apoptotic foci in the lamina propria and edema in the overlying epithelium. Ccr2−/− mice (Ccr2−/−) showed massive necrosis in the ileum, with destruction of the villi and infiltrating neutrophils. Ccr2−/− mice treated with MAb 1A8 (Ccr2−/− + 1A8) showed fewer infiltrating neutrophils and less overall damage to the villi. The sections shown are representative results for three mice in each group of animals examined at 9 days postinfection. Scale bar = 200 μm. (B) Histological changes in the spleens of mice infected with T. gondii. Spleens of infected control mice (WT Control) displayed no significant lesions. Small numbers of apoptotic foci with infiltrating macrophages were observed in the spleens of 1A8-treated mice (WT + 1A8). RB6-8C5-treated mice (WT + RB6) showed fewer infiltrating neutrophils but still contained lesions with parasites. Histological examination of the spleens of Ccr2−/− mice (Ccr2−/−) revealed foci with parasites and neutrophils. Ccr2−/− mice treated with MAb 1A8 (Ccr2−/− + 1A8) showed reduced influx of neutrophils, but foci of parasites remained. The sections shown are representative results for three mice in each group of animals examined at 9 days postinfection. Scale bar = 200 μm.
We also analyzed pathological changes in the spleens of mice at day 9 after infection with T. gondii. The spleens of 1A8-treated mice showed only mild reduction of lymphocytes and low numbers of parasites, similar to those shown in control infected mice (Fig. 5B). RB6-8C5-treated mice also showed foci of parasites, depletion of lymphocytes, and infiltration of macrophages (Fig. 5B). Foci of parasites, marked lymphocyte depletion, and significant accumulation of neutrophils were observed in the spleens of infected Ccr2−/− mice (Fig. 5B). Treatment of Ccr2−/− mice with MAb 1A8 resulted in less influx of neutrophils, although lesions containing parasites persisted.
DISCUSSION
Previous studies have suggested that neutrophils (5, 13) and inflammatory monocytes (14, 23, 24) are important for control of toxoplasmosis in the mouse. However, differences in experimental models have made it difficult to directly compare these results and to determine the relative importance of these cell types during acute infection. Here we used selective depletion of neutrophils using MAb 1A8 that reacts specifically to Ly6G (12) and compared this to depletion of both inflammatory monocytes and neutrophils using the previously characterized MAb RB6-8C5, which reacts to both Ly6G and Ly6C (15). Because Ly6C is found on monocytes and neutrophils, treatment with this antibody removes both subsets efficiently. Our studies revealed a strong phenotype when both cell types were depleted yet surprisingly little change when neutrophils alone were depleted. Combined with the results of infection in mice with a genetic defect in Ccr2−/−, which results in an inability to recruit inflammatory monocytes (28), these results reveal that inflammatory monocytes are required for control of acute toxoplasmosis in the murine model. In contrast, neutrophils do not appear to be essential for control of acute infection and instead may increase pathology. Since RB6-8C5 has been used extensively to study other bacterial and protozoal infections, the relative importance of inflammatory monocytes versus neutrophils in these other systems should also be reconsidered.
Mouse monocytes have been divided into two main populations based on phenotypic markers, migratory patterns, and their cellular phenotypes (18). Inflammatory monocytes exit from the bone marrow and are recruited to sites of inflammation (18). These cells express Gr1 and also the chemokine receptor CCR2, which responds to MCP1 (CCL2). We previously reported that i.p. oral infection with type II strains of T. gondii results in recruitment of inflammatory monocytes (CCR2+, Gr1int, Ly6Chi, Ly6G−, F4/80+, and CD11b+) to the peritoneal cavity (23, 24). Following oral infection, similar inflammatory monocytes are specifically recruited to the lamina propria (14). Inflammatory monocytes recruited in response to T. gondii express iNOS and TNF-α (14, 23), which are essential mediators for the control of parasite replication by activated macrophages (1, 29). Consistent with this, inflammatory monocytes are able to prevent replication of T. gondii in vitro (23), suggesting they are also involved in the control of parasite expansion in vivo. Mice lacking the chemokine receptor CCR2 are unable to mobilize inflammatory monocytes from the blood (28) and, hence, are highly susceptible to acute toxoplasmosis (23, 24). Collectively, these studies indicate that inflammatory monocytes are critical for the control of acute infection, presumably acting as a first line of defense following i.p. (24) or oral (14) challenge.
Neutrophils are primary responder cells that home to sites of inflammation in response to chemokines such as KC (CXCL1) and MIP-2 (CXCL2) that signal through CXCR2 (16). They are phagocytic and express potent antimicrobial responses, including defensins, proteases, hydrolases, and reactive oxygen products (21). Neutrophils have been implicated in the control of a variety of bacterial infections and inherited genetic disorders that affect neutrophil function, resulting in profound susceptibility to bacterial infections. The role for neutrophils in combating infection has partially been established by depletion with MAb RB6-8C5, which recognizes the myeloid differentiation antigen 1 (Gr1) that is abundant on mature granulocytes (15). Previously, studies demonstrated that treatment of mice with the MAb RB6-8C5 increased susceptibility to the parasite T. gondii, and it was suggested that this might reflect an important role of neutrophils, which strongly express Ly6G (5, 26). RB6-8C5 has also been used for depletion studies to explore the role of cellular immunity to Leishmania infantum, Leishmania major, Plasmodium berghei ANKA, and Trypanosoma cruzi (8-10, 25). However, later studies revealed that inflammatory monocytes are also stained by RB6 (Gr1) due to expression of Ly6C (18). Hence, these depletion studies would be more accurately interpreted as demonstrating a possible role for neutrophils and/or inflammatory monocytes.
The recent description of MAb 1A8 that specifically recognizes Ly6G has allowed the role of neutrophils to be studied using more precise depletion protocols (12). As in a previous report (12), we observed that RB6-8C5 depleted both inflammatory monocytes and neutrophils, while 1A8 selectively depleted neutrophils in mice. Daily treatment with MAb 1A8 was required to effectively deplete neutrophils from both circulation and lymphatic organs. To evaluate the role of inflammatory monocytes and neutrophils in acute infection, mice were orally treated with T. gondii, and then antibodies were used to selectively deplete cellular subsets. Depletion with MAb RB6-8C5 greatly increased susceptibility for T. gondii infection. In contrast, depletion of neutrophils with MAb 1A8 had little effect on survival. Ccr2−/− mice suffered from extensive necrosis of the small intestine and spleen, and marked infiltration of neutrophils and parasites were observed. Pathological changes were associated with extensive neutrophil infiltration, suggesting that they contribute more to tissue damage than to host protection. Consistent with this interpretation, treatment of Ccr2−/− mice with MAb 1A8 reduced neutrophil influx, reduced tissue damage, and reduced inflammatory cytokine levels. Similarly, damage to the small intestine following oral infection with T. gondii is decreased by the genetic absence of IL-17R, implicating a role for neutrophils in this pathology (20). Our findings support the conclusion that inflammatory monocytes are essential for survival of mice infected with T. gondii, while the lack of neutrophils had no effect of the susceptibility but rather contribute to pathology.
Acknowledgments
This work was supported in part by grants from the NIH (grants AI071299 and AI036629 to L.D.S.). I.R.D. was partially supported by a fellowship from the DFG, Germany.
Editor: J. F. Urban, Jr.
Footnotes
Published ahead of print on 9 February 2010.
REFERENCES
- 1.Adams, L. B., J. B. Hibbs, R. R. Taintor, and J. L. Krahenbuhl. 1990. Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii: role for synthesis of inorganic nitrogen oxides from L-arginine. J. Immunol. 144:2725-2729. [PubMed] [Google Scholar]
- 2.Auffray, C., D. Fogg, M. Garfa, G. Elain, O. Join-Lambert, S. Kayal, S. Sarnacki, A. Cumano, G. Lauvau, and F. Geissmann. 2007. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317:666-670. [DOI] [PubMed] [Google Scholar]
- 3.Barragan, A., and L. D. Sibley. 2003. Migration of Toxoplasma gondii across biological barriers. Trends Microbiol. 11:426-430. [DOI] [PubMed] [Google Scholar]
- 4.Bliss, S. K., B. A. Butcher, and E. Y. Denkers. 2000. Rapid recruitment of neutrophils containing prestored IL-12 during microbial infection. J. Immunol. 165:4515-4521. [DOI] [PubMed] [Google Scholar]
- 5.Bliss, S. K., L. C. Gavrilescu, A. Alcaraz, and E. Y. Denkers. 2001. Neutrophil depletion during Toxoplasma gondii infection leads to impaired immunity and lethal systemic pathology. Infect. Immun. 69:4898-4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bliss, S. K., A. J. Marshall, Y. Zhang, and E. Y. Denkers. 1999. Human polymorphonuclear leukocytes produce IL-12, TNF-α, and the chemokines macrophage-inflammatory protein-1α and -1β in response to Toxoplasma gondii antigens. J. Immunol. 162:7369-7375. [PubMed] [Google Scholar]
- 7.Bliss, S. K., Y. Zhang, and E. Y. Denkers. 1999. Murine neutrophil stimulation by Toxoplasma gondii antigen drives high level production of IFN-γ-independent IL-12. J. Immunol. 163:2081-2088. [PubMed] [Google Scholar]
- 8.Chen, L., Z. Zhang, and F. Sendo. 2000. Neutrophils play a critical role in the pathogenesis of experimental cerebral malaria. Clin. Exp. Immunol. 120:125-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, L., Z. H. Zhang, T. Watanabe, T. Yamashita, T. Kobayakawa, A. Kaneko, H. Fujiwara, and F. Sendo. 2005. The involvement of neutrophils in the resistance to Leishmania major infection in susceptible but not in resistant mice. Parasitol. Int. 54:109-118. [DOI] [PubMed] [Google Scholar]
- 10.Chen, L. A., T. Watanabe, H. Watanabe, and F. Sendo. 2001. Neutrophil depletion exacerbates experimental Chagas' disease in BALB/c but protects C57BL/6 mice through modulating the Th1/Th2 dichotomy in different directions. Eur. J. Immunol. 31:265-275. [DOI] [PubMed] [Google Scholar]
- 11.Courret, N., S. Darche, P. Songio, G. Milon, D. Buzoni-Gatel, and I. Tardieux. 2005. CD11c and CD11b expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107:309-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daley, J. M., A. A. Thomay, M. D. Connolly, J. S. Reichner, and J. E. Albina. 2007. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83:64-70. [DOI] [PubMed] [Google Scholar]
- 13.Del Rio, L., S. Bennouna, J. Salinas, and E. Y. Denkers. 2001. CXCR2 deficiency confers impaired neutrophil recruitment and increased susceptibility during Toxoplasma gondii infection. J. Immunol. 167:6503-6509. [DOI] [PubMed] [Google Scholar]
- 14.Dunay, I. R., R. A. DaMatta, B. Fux, R. Presti, A. Greco, M. Colonna, and L. D. Sibley. 2008. Gr1+ inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29:306-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleming, T. J., M. L. Fleming, and T. R. Malek. 1993. Selective expression of Ly-6G on myeloid lineage cells n mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J. Immunol. 151:2399-2408. [PubMed] [Google Scholar]
- 16.Furze, R. C., and S. M. Rankin. 2008. Neutrophil mobilization and clearance in the bone marrow. Immunology 125:281-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geissmann, F., C. Auffrey, R. Palframan, C. Wirrig, A. Ciocca, L. Campisis, E. Narni-Manichelli, and G. Lauvau. 2008. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol. Cell Biol. 86:398-408. [DOI] [PubMed] [Google Scholar]
- 18.Geissmann, F., S. Jung, and D. R. Littman. 2003. Blood monocytes consist of two principle subsets with distinct migratory properties. Immunity 19:71-82. [DOI] [PubMed] [Google Scholar]
- 19.Joynson, D. H., and T. J. Wreghitt (ed.). 2001. Toxoplasmosis: a comprehensive clinical guide. Cambridge University Press, Cambridge, United Kingdom.
- 20.Kelly, M. N., J. K. Kolls, K. Happel, J. D. Schwartzman, P. Schwarzenberger, C. Combe, M. M. Moretto, and I. A. Khan. 2005. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infect. Immun. 73:617-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer-Scholl, A., P. Averhoff, and A. Zychlinsky. 2004. How do neutrophils and pathogens interact. Curr. Opin. Microbiol. 7:62-66. [DOI] [PubMed] [Google Scholar]
- 22.Mordue, D. G., F. Monroy, M. La Regina, C. A. Dinarello, and L. D. Sibley. 2001. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J. Immunol. 167:4574-4584. [DOI] [PubMed] [Google Scholar]
- 23.Mordue, D. G., and L. D. Sibley. 2003. A novel population of Gr-1+ activated macrophages induced during acute toxoplasmosis. J. Leukoc. Biol. 74:1015-1025. [DOI] [PubMed] [Google Scholar]
- 24.Robben, P. R., M. Laregina, W. A. Kuziel, and L. D. Sibley. 2005. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 201:1761-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rousseau, D., S. Demartino, B. Ferrua, J. F. Michiels, F. Anjuère, K. Fragaki, Y. Le Fichoux, and J. Kubar. 2001. In vivo involvement of polymorphonuclear neutrophils in Leishmania infantum infection. BMC Microbiol. 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sayles, P. C., and L. L. Johnson. 1996. Exacerbation of toxoplasmosis in neutrophil-depleted mice. Nat. Immun. 15:249-258. [PubMed] [Google Scholar]
- 27.Scapini, P., J. A. Lapinet-Vera, S. Gasperini, F. Calzetti, and M. A. Cassatella. 2000. The neutrophil as a cellular source of chemokines. Immunol. Rev. 177:195-203. [DOI] [PubMed] [Google Scholar]
- 28.Serbina, N. V., and E. G. Pamer. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7:311-317. [DOI] [PubMed] [Google Scholar]
- 29.Sibley, L. D., L. B. Adams, Y. Fukutomi, and J. L. Krahenbuhl. 1991. Tumor necrosis factor-α triggers antitoxoplasmal activity of IFN-γ primed macrophages. J. Immunol. 147:2340-2345. [PubMed] [Google Scholar]
- 30.Soehnlein, O., A. Zernecke, E. E. Eriksson, S. G. Rothfuchs, C. T. Pham, H. Herwald, K. Bidzhekov, M. E. Rottenberg, C. Weber, and L. Linbom. 2008. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 112:1461-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tateda, K., T. A. Moore, M. W. Newstead, W. C. Tsai, X. Zeng, J. C. Deng, G. Chen, R. Reddy, K. Yamaguchi, and T. J. Standiford. 2001. Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect. Immun. 69:2017-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Gisbergen, K. P., T. B. Geijtenbeek, and Y. van Kooyk. 2005. Close encounters of neutrophils and DCs. Trends Immunol. 26:626-631. [DOI] [PubMed] [Google Scholar]
- 33.Varol, C., L. Lamdsman, D. K. Fogg, L. Grennshtein, B. Gildor, R. Margalit, V. Kalchenko, F. Geissmann, and S. Jung. 2007. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 204:171-180. [DOI] [PMC free article] [PubMed] [Google Scholar]