

Abstract
Pseudomonas aeruginosa is a leading cause of hospital-acquired pneumonia and severe chronic lung infections in cystic fibrosis patients. The reference strains PA14 and PAO1 have been studied extensively, revealing that PA14 is more virulent than PAO1 in diverse infection models. Among other factors, this may be due to two pathogenicity islands, PAPI-1 and PAPI-2, both present in PA14 but not in PAO1. We compared the global contributions to virulence of PAPI-1 and PAPI-2, rather than that of individual island-borne genes, using murine models of acute pneumonia and bacteremia. Three isogenic island-minus mutants (PAPI-1-minus, PAPI-2-minus, and PAPI-1-minus, PAPI-2-minus mutants) were compared with the wild-type parent strain PA14 and with PAO1. Our results showed that both islands contributed significantly to the virulence of PA14 in acute pneumonia and bacteremia models. However, in contrast to the results for the bacteremia model, where each island was found to contribute individually, loss of the 108-kb PAPI-1 island alone was insufficient to measurably attenuate the mutant in the acute pneumonia model. Nevertheless, the double mutant was substantially more attenuated, and exhibited a lesser degree of virulence, than even PAO1 in the acute pneumonia model. In particular, its ability to disseminate from the lungs to the bloodstream was markedly inhibited. We conclude that both PAPI-1 and PAPI-2 contribute directly and synergistically in a major way to the virulence of PA14, and we suggest that analysis of island-minus strains may be a more appropriate way than individual gene knockouts to assess the contributions to virulence of large, horizontally acquired segments of DNA.
Pseudomonas aeruginosa is an environmentally ubiquitous opportunistic pathogen that causes a wide range of acute life-threatening infections, especially in immunocompromised patients. Hospitalized patients with damaged airways due to mechanical ventilation, injury, or viral infections are at significant risk of acute pneumonia caused by P. aeruginosa (51). Resulting cases of ventilator-associated pneumonia are associated with very high rates of mortality (12). In addition, this bacterium is a common cause of chronic respiratory infection in patients with cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), and non-CF bronchiectasis (43, 50). A large variety of extracellular virulence factors, including proteases, hemolysins, pyocyanin, pili, lipopolysaccharide, alginate, and the type III secretion system (T3SS) effector proteins, ExoS, ExoT, ExoU, and ExoY, are associated with P. aeruginosa (52, 62).
P. aeruginosa PA14 is a fully sequenced, highly virulent wild-type (WT) reference strain that has been used extensively to study the contribution of putative virulence factors to disease (31). Furthermore, as defined through study of a large panel of strains representative of this species, PA14 was found to belong to the most common P. aeruginosa global lineage (61). Another fully sequenced strain, PAO1, is widely regarded as a laboratory strain of P. aeruginosa. PA14 has been shown to be much more virulent than PAO1 in a number of diverse models of infection, leading to the hypothesis that PA14 is a multihost pathogen capable of infecting invertebrate and vertebrate animal species and plant species (29). The genomes of individual P. aeruginosa strains have a highly mosaic structure made up of conserved cores and variable accessory genomes (36, 61), resulting in composite genomes of 5.2 to 7 Mb (36). The majority of large-scale genomic differences arise from the presence or absence of an expanding list of genomic islands within conserved “hot spots” throughout the genome (36). PA14 carries two well-characterized pathogenicity islands: PAPI-1, a 108-kb island integrated within a lysine tRNA gene (PA4541.1), and PAPI-2, a much smaller, 11-kb island inserted into a sequence-identical but distinct tRNALys gene (PA0976.1) (19). In addition to targeting integration sites bearing identical sequences, these two islands share a limited but recognizable degree of synteny and code for factors that have been implicated in pathogenesis in murine, plant, and nematode models of infection (19, 31, 37, 52). Interestingly, islands with significant homology to PAPI-1 and/or PAPI-2 have been identified in many P. aeruginosa strains and several other bacterial species (5, 6, 19, 23, 27, 30, 61, 63) (see Tables S2 and S3 and Fig. S2 in the supplemental material).
PAPI-1 is a member of the increasingly populated pKLC102/PAGI-2 island family (26). PAPI-1 encodes a number of likely virulence factors, including type IVB pili, the cupD1 to cupD5 fimbrial biogenesis gene cluster (39), and PvrR, a two-component response regulator involved in biofilm synthesis and antibiotic resistance (11, 38). However, the large majority of PAPI-1 predicted proteins have no assigned function, and many of these are encoded by homologues on related islands in other P. aeruginosa strains (27, 63). PAGI-5, a hybrid island that shares 79 of 121 predicted open reading frames (ORFs) with PAPI-1, has recently been implicated in acute murine pneumonia (5). Isogenic mutants harboring deletions within NR-I or NR-II, PAGI-5-specific novel regions, were shown to be attenuated in a model of acute murine pneumonia, demonstrating that an island in the pKLC102/PAGI-2 family played a role in this infection.
PAPI-2 is a stably integrated island belonging to the ExoU island family (30). This island encodes the cytotoxin ExoU, a potent phospholipase, and its cognate chaperone SpcU. ExoU has been studied extensively and has been shown to be the most potent of the known T3SS effector proteins in P. aeruginosa (54). The introduction of exoU into strains that lacked this gene resulted in marked increases in virulence in a murine acute pneumonia model (1). Recent work has suggested that ExoU impairs host response in the lung through targeted killing of phagocytes, which play a crucial role during the early phase of infection (10). Additionally, the secretion of ExoU by an infecting strain has been shown to be a marker of high virulence and poor clinical outcome in hospital-acquired pneumonia (52).
Since genomic islands are widely believed to be acquired and/or lost as whole units, and since PAPI-1-like islands are known to be widespread and possibly highly mobile (46), we investigated the contributions of PAPI-1 and PAPI-2 to virulence as single unitary modules rather than defining the roles of individual island-borne genes (5, 19). We postulated that this en bloc deletion approach would reveal combinatorial and/or synergistic effects that would be missed by single-gene knockout approaches. To pursue this goal, we created three isogenic whole-island deletion mutants that lacked either PAPI-1 (PA14ΔPAPI-1), PAPI-2 (PA14ΔPAPI-2), or both (PA14Δ1Δ2) in a PA14 background and examined these relative to their wild-type parent and the laboratory strain PAO1, which naturally lacks both islands. Our results revealed that both PAPI-1 and PAPI-2 contributed individually and synergistically to the virulence of P. aeruginosa strain PA14 in murine models of acute pneumonia and bacteremia.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth media.
The bacterial strains and plasmids used in this study are described in Table 1. P. aeruginosa and Escherichia coli strains were grown routinely at 37°C on LB medium plus 100 μg/ml ampicillin, 30 μg/ml kanamycin, and/or 15 μg/ml gentamicin for E. coli and 200 μg/ml carbenicillin and/or 30 μg/ml gentamicin for P. aeruginosa, as required. The minimal medium used for conjugative transfer was VBMM (57), supplemented when appropriate with antibiotics and sucrose at 5% (wt/vol).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| P. aeruginosa | ||
| PA14 | Wild-type, hypervirulent strain from burn infection | 31 |
| PA14ΔPAPI-1 | PA14 with PAPI-1 deleted | This study |
| PA14ΔPAPI-2 | PA14 with PAPI-2 deleted | This study |
| PA14Δ1Δ2 | PA14 with both PAPI-1 and PAPI-2 deleted | This study |
| PA14ΔexoU | PA14 with exoU deleted | This study |
| PA14ΔPAPI-1ΔexoU | PA14 with PAPI-1 and exoU deleted | This study |
| PAO1 | Laboratory strain, originally from wound infection | 56 |
| E. coli | ||
| SM10 λ-pir | Kmr (thi-1 thr leu tonA lacY supE recA::RP4-2Tc::Mu) | 55 |
| DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ−thi-1 gyrA96 relA1 | 17 |
| One Shot T1-R | F′ proAB lacIqlacZΔM15 Tn10(Tetr) Δ(ccdAB) mcrA Δ(mrr hsdRMS-mcrBC) φ80(lacZ)ΔM15 Δ(lacZYA-argF)U169 endA1 recA1 supE44 thi-1 gyrA96 relA1 tonA panD | Invitrogen |
| Plasmids | ||
| pFLP2 | FLP recombinase-expressing plasmid; Ampr | 21 |
| pPS856 | Source of Gmr cassette; Gmr | 21 |
| pEX18Ap | Suicide vector; Ampr | 53 |
| pUCP24 | E. coli-P. aeruginosa shuttle vector; Gmr | 60 |
| pCR4-Topo | Topo PCR cloning vector; Ampr Kmr; ccdB gene | Invitrogen |
| pEX18ApGW | Gateway suicide destination vector; Ampr | 9 |
| pDONR221 | Gateway cloning entry vector; Kmr CmrattP1 attP2; ccdB gene | Invitrogen |
| pDONR221-ΔPAPI-2 | Gateway entry vector with SOE fragment for deletion of PAPI-2; Kmr Gmr | This study |
| pEXΔPAPI-2 | pEX18ApGW with SOE fragment for deletion of PAPI-2; Ampr Gmr | This study |
| pCR4-ΔSOJ | pCR4-Topo with PCR-generated 49-bp fragment of soj; Kmr | This study |
| pEXΔExoU | Suicide vector with SOE fragment for deletion of exoU; Ampr Gmr | This study |
| pEXΔSOJ | Suicide vector for PAPI-1 deletion containing 498-bp insert into EcoRI; Ampr | This study |
DNA manipulations.
Restriction enzymes and T4 DNA ligase were purchased from Promega. Genomic DNA and plasmid DNA were extracted by following the manufacturer's instructions using the DNeasy blood and tissue kit and the QIAspin miniprep kit (Qiagen, United Kingdom). Standard PCR amplification was carried out using GoTaq (Promega, United Kingdom), while KOD Hotstart (Merck, United Kingdom) was used for high-fidelity amplification. DNA sequencing was performed by MWG Biotech.
Construction of defined PA14 mutants harboring deletions of PAPI-1 and/or PAPI-2.
PAPI-1 was deleted from PA14 using an “island-probing” approach (47). Qiu et al. (46) had reported that mutation of the PAPI-1-encoded soj gene increased the instability of the island. A 498-bp internal fragment of soj was amplified by PCR using primers SOJ-F and SOJ-R (see Table S1 in the supplemental material), cloned into pCR4-TOPO (Invitrogen, United Kingdom), excised as a fractionally larger EcoRI fragment, and ligated into the EcoRI site of pEX18Ap (21) to create pEXΔSOJ. This plasmid was conjugally transferred from E. coli SM10 λ-pir into PA14 (21), and a single-crossover plasmid integrant was identified by the resistance phenotype and PCR. The ΔPAPI-1 mutant was generated from this intermediate strain by counterselection on LB agar medium with 5% sucrose to select for the loss of the sacB-tagged PAPI-1 island. Loss of PAPI-1 was confirmed by reversion to carbenicillin sensitivity, the absence of five PAPI-1-specific regions by PCR using primers PP-1-1 to PP-1-5 (see Table S1 in the supplemental material), and sequencing across the newly vacated attB site.
PAPI-2 was deleted by following the method described by Choi and Schweizer (9). Briefly, upstream and downstream flanking regions of PAPI-2 were amplified by PCR using the primer pairs P2UFFW-Gm-P2UFRS-GWR and P2DFFW-GWL-P2DFRS-Gm, respectively (see Table S1 in the supplemental material). These fragments were joined to a FLP recombination target (FRT)-flanked gentamicin resistance (Gmr) cassette amplified from pPS856 (9) using splicing overlap extension (SOE) PCR. The composite fragment was transferred via pDONR221 into the pEX18ApGW suicide vector using the Gateway site-specific recombination system (Invitrogen, United Kingdom). The resulting plasmid, pEXΔPAPI-2, was then transferred by conjugation into PA14 (21). A merodiploid intermediate strain was resolved by sucrose counterselection, and the Gmr cassette was removed using FLP-mediated excision (9) to generate a marker-free PAPI-2 deletant. The PAPI-2 deletion was confirmed by reversion to gentamicin sensitivity, loss of the exoU gene, and sequencing across the deletion site. A double PAPI-1-PAPI-2 deletion mutant was constructed by deleting PAPI-1, as described above, from the PA14ΔPAPI-2 mutant. The ΔexoU mutants were constructed like the ΔPAPI-2 mutant, with the modification that the SOE PCR fragment was cloned directly into pEX18Ap by inclusion of KpnI restriction sites in primers GW-KPN1-F and GW-KPN1-R, used for amplification of the SOE fragment.
Growth, biofilm, motility, and pyocyanin assays.
Strains were grown for 16 h at 37°C at 200 rpm in LB broth and were subcultured 1/100 into LB broth; M63 medium with glucose (0.2%), MgSO4 (1 mM), and Casamino Acids (0. 5%); and M9 medium with glucose (0.2%), MgSO4 (1 mM), and CaCl3 (1 mM). Two hundred microliters of each was placed in a Honeywell plate, and the optical density (OD) was measured using a Bioscreen apparatus (Life Sciences, United Kingdom) at 37°C with continuous shaking for 24 or 48 h. The sizes of the initial inocula were checked by serial dilutions and plating for CFU. Biofilm assays were performed using the microtiter plate-based biofilm assay described by O'Toole and Kolter (45). Twitching motility was evaluated at the agar-plate interface after 24 h at 37°C on LB medium with 1.5% agar (48). Swimming motility was measured after 20 h at 30°C on LB medium with 0.3% agar. Pyocyanin production was measured at 520 nm in a chloroform acidic-extraction solution using a method described previously (20).
Murine acute pneumonia model.
The murine acute pneumonia model used was adapted from previous work with pneumococci (24) and with P. aeruginosa (54). Six hundred microliters of a 37°C shaken P. aeruginosa culture was removed and centrifuged at 13,000 rpm for 1 min. The pellet was added to 20 ml of tryptone soy broth supplemented with 20% fetal calf serum (FCS) (Sigma, United Kingdom). The culture was then incubated at 37°C and 200 rpm for 4 h before being stored at −70°C as 1-ml aliquots. Stock cultures were left at −70°C for at least 3 days. Eight- to 10-week-old female BALB/c mice (Harlan Olac, Bicester, United Kingdom) were anesthetized with 5% (vol/vol) isoflurane over oxygen at 1.8 liter/min. Mice were intranasally challenged with 2 × 106 CFU of P. aeruginosa and were then allowed to recover. Mouse survival, disease signs, and levels of bacteria in the lungs, nasopharynx, and/or blood were measured. Disease severity was monitored at each time point by one of four trained individuals, two of whom were blinded, using the scheme of Morton and Griffiths (42), which scored the following clinical signs: hunching, a starry coat, and lethargy. Mice were scored as follows: 0, no sign detectable; 1, sign just evident; 2, sign obviously present. The individual scores for the three signs were combined to give a maximum clinical sign score of 6. Mice with a score of 6 were culled immediately, because this was the experimental endpoint defined by our Home Office project license. The remaining mice were culled at 18 h postinfection, and samples were taken for analysis. Blood was taken via cardiac puncture under terminal anesthesia, and the nasopharynx and lungs were removed by dissection, weighed, and homogenized in phosphate-buffered saline (PBS) using an Ultra-Turrax tissue homogenizer. Organ homogenates were serially diluted and cultured on LB agar for the determination of CFU counts. Ten mice were used per strain. For survival experiments, mice were culled when they reached a score of 2 for lethargy. Ten mice were used for each strain, and survival was monitored for 96 h.
Murine bacteremia model.
Mice were inoculated via their dorsal tail veins with 2 × 106 CFU. Blood was collected by tail-bleeding at 2 h and 6 h, and finally via cardiac puncture at 24 h postinfection. Five mice were infected per strain.
Differential leukocyte counts and lung histopathology.
Trypan blue was added to lung homogenates at a 1:1 volume ratio. Mouse leukocyte viability following homogenization and purification of cells had previously been shown to be above 90%, as determined by trypan blue exclusion (24; A. Kadioglu, unpublished data). Five milliliters of stained lung homogenate was filtered through 40-μm-pore size nylon Falcon cell strainers (Becton Dickinson [BD], United Kingdom) into a 50-ml Falcon tube. The homogenate was centrifuged at 300 × g for 5 min. The supernatant was discarded, and the pellet was resuspended in 10 ml BD Pharm Lyse lysing buffer (BD Biosciences, United Kingdom) diluted 1:10 with nanopure water and was left for 10 min at room temperature in the dark. The suspension was recentrifuged as before, and the cells were resuspended in 5 ml PBS. Three cytospin slides were prepared per individual mouse. Fifty microliters of the final cell suspension was centrifuged onto a cytospin slide (Shandon, United Kingdom) using a Cytospin 2 centrifuge (Shandon, United Kingdom) at 1,500 rpm for 3 min. The slides were allowed to dry overnight. The slides were stained using the Reastain Quick-Diff kit (Reagena, Finland). Slides were counted using the 40× objective lens on a light microscope to differentially identify monocytes, macrophages, lymphocytes, and polymorphonuclear leukocytes. Cytospin slides prepared from four mice per P. aeruginosa strain investigated were analyzed. For histopathology, lungs were removed, placed in 5 ml phosphate-buffered saline, fixed in OCT tissue embedding compound using dry ice and isopentane as a heat buffer, and then stored at −70°C. The day before sectioning, lungs were moved to −20°C. Lungs were cut into 15-μm-thick sections at −18°C using a Bright microtome and were subsequently stained using hematoxylin and eosin; photographs were taken at a magnification of ×10. Two lungs were examined per strain.
Galleria mellonella killing assay.
The Galleria mellonella assay used was based on assays described by Miyata et al. and Inglis et al. (22, 41). Test strains were grown overnight in 5 ml LB broth at 37°C with shaking at 200 rpm. The next day, a 1/100 subculture grown under identical conditions for 4 h was centrifuged at 3,000 rpm for 10 min; the supernatant was removed; and the cell pellet was resuspended in 10 mM MgSO4 to an OD at 600 nm (OD600) of 0.2. Bacterial suspensions were then serially diluted to 10−5 using 10 mM MgSO4 supplemented with 0.5 mg/ml rifampin (Sigma, United Kingdom) as the diluent. G. mellonella larvae were inoculated between two rear thoracic segments with 10 μl of the 10−5 suspension containing ∼10 CFU of P. aeruginosa by using a 0.3-ml Myjector U-100 insulin syringe (VWR, United Kingdom). Ten larvae were inoculated with each bacterial strain, and three biological replicates originating from independent bacterial colonies were performed for each strain, resulting in an overall number of 30. Larvae were incubated at 37°C and were scored for death every half hour after 14 h. Larvae were scored dead when caterpillars did not respond to direct mechanical stimulation applied to the head.
Invasion assay.
The invasion assay was based on that described by Luck et al. (34) with modifications for P. aeruginosa. Quantitative assessment of bacterial invasion was performed using A549 human lung carcinoma cells. Cell monolayers were obtained by seeding 500 μl of a solution consisting of 1 × 105/ml A549 cells resuspended in RPMI 1640 plus glutamine supplemented with 10% FCS and 100 μg/ml penicillin-streptomycin in a 24-well tissue culture tray and incubating overnight at 37°C under 5% CO2. Semiconfluent cell monolayers were washed three times in warm PBS, and fresh medium was added. A549 cells were inoculated with approximately 3 × 106 total bacteria per well, and trays were centrifuged at room temperature prior to incubation for 2 h at 37°C under 5% CO2. To eliminate extracellular bacteria, A549 cells were washed three times with PBS, and fresh medium containing 400 μg/ml amikacin was added, before a final 1 h of incubation at 37°C under 5% CO2. Finally, A549 cells were washed three times with PBS and lysed in 0.1% digitonin. Following cell lysis, bacteria were resuspended in LB broth and quantified by plating serial dilutions. Assays were carried out in duplicate, and the results from at least three independent experiments were expressed as the percentage of the total number of inoculated bacteria that was intracellular (mean ± standard deviation). PA14 and its three isogenic mutants were shown to be fully susceptible to 200 μg/ml amikacin when incubated in the absence of A549 cells under conditions identical to those used in the invasion assay.
Cytotoxicity assay.
The cytotoxicity assay was based on that described by Lee et al. (32). A549 cells and P. aeruginosa were grown, prepared, and/or inoculated as described for the invasion assay. After 2 h of incubation, cytotoxicity toward A549 cells was quantified by measuring the amount of lactate dehydrogenase (LDH) released into the culture supernatant by using the CytoTox-ONE kit (Promega, United Kingdom). Assays were carried out in duplicate with three independent biological replicates for each strain. The percentage of cytotoxicity was calculated relative to the value for the maximum-LDH-release control (A549 cells lysed by the addition of a 9% [wt/vol] solution of Triton X-100).
Statistics.
Statistical analysis was performed with GraphPad Prism software (version 5; GraphPad Software, Inc.) and SPSS software. CFU and differential leukocyte counts were analyzed, and cytotoxicity assays were performed, by analysis of variance (ANOVA) with Tukey posthoc analysis. Symptom scores were analyzed using the nonparametric Kruskal-Wallis test and Dunn's posthoc analysis. Survival data were analyzed using the Mantel-Cox log rank test. For the invasion assay, differences in invasion were assessed for significance by using an unpaired, two-tailed t test.
RESULTS
PA14 is more virulent than PAO1 in a murine model of acute pneumonia.
As had been reported previously for a wide range of P. aeruginosa infection models (29), PA14 was found to be more virulent than PAO1 in a mouse model of acute pneumonia. Following intranasal infection with equivalent inocula (2 × 106 CFU), PA14-infected mice had to be culled due to the onset of fulminant symptoms at a significantly earlier time point (18 h) than mice infected with PAO1 (27 h) (P < 0.02) (Fig. 1A). Furthermore, PA14 was found in much higher numbers than PAO1 in nasopharynx, lung, and blood specimens obtained at 18 h postinfection (Fig. 2). Consistent with these data, mean qualitative clinical sign scores at 18 h following infection with PA14 and PAO1 were 5.7 (range, 5 to 6) and 4 (range, 2 to 5), respectively; higher scores denoted greater severity of disease (Fig. 1B).
FIG. 1.

(A) Survival graph for the acute murine pneumonia model. Mice were monitored for 96 h. Ten mice were used for each strain. Data were generated from two independent experiments for each strain, except for the ΔexoU and ΔPAPI-1ΔexoU strains, for which data were generated from a single experiment only. Horizontal lines represent the percentage of mice surviving postinfection for each strain. (B) Acute murine pneumonia symptom scores at 18 h postinfection. Ten mice were used for each strain. Each data point represents a single mouse. All data were generated from two independent experiments for each strain. The asterisk indicates a significant difference from wild-type PA14 (P < 0.05). The bars represent mean symptom scores ± standard deviations.
FIG. 2.

P. aeruginosa bacterial burdens in the nasopharynx, lungs, and blood at 18 h postinfection in the acute murine pneumonia model. Ten mice were used for each strain. All data were generated from two independent experiments for each strain. Asterisks and number symbols indicate significant differences (P < 0.05) from wild-type PA14 and PA14ΔPAPI-2, respectively. The error bars represent the standard errors of the means.
Loss of PAPI-1 and/or PAPI-2 caused no change in in vitro fitness, motility, biofilm formation, or pyocyanin production.
We constructed three island-minus isogenic mutants of PA14 that lacked either PAPI-1 or PAPI-2, or both PAPI-1 and PAPI-2 (PA14ΔPAPI-1, PA14ΔPAPI-2, and PA14Δ1Δ2), as described in Materials and Methods. None of these mutants showed significant growth defects in either rich (LB) or minimal (M9 or M63) medium compared to the growth of WT PA14 (data not shown), suggesting that the deletions had no significant effect on growth kinetics in vitro. In the biofilm assays on two different surfaces (polyvinyl chloride [PVC] and polystyrene), none of the mutants showed consistent differences from wild-type PA14 (data not shown). Similarly, no significant difference in pyocyanin production was detected (data not shown). Furthermore, the three mutants showed no change in swimming or twitching motility (data not shown), even though twitching motility is known to be dependent on type IV pili, and PAPI-1 codes for type IV pilus chaperone genes. Consistent with these findings, He et al. (19) reported no significant difference in biofilm growth, motility, or pyocyanin production in the 23 PAPI-1 and PAPI-2 single-gene mutants screened in their study.
PAPI-1- and/or PAPI-2-minus isogenic mutants retain their abilities to invade A549 cells.
Invasion assays performed with PA14 and its three isogenic mutants suggested that PA14ΔPAPI-2 and PA14Δ1Δ2 showed significant apparent increases (P < 0.05) in the ability to invade A549 cells over that of PA14 (Fig. 3A). However, the significant reduction in cytotoxicity (P < 0.05) exhibited by the two mutants that lacked PAPI-2 relative to the cytotoxicity of PA14 or PA14ΔPAPI-1, as shown in Fig. 3B, suggested that much, if not all, of this altered invasion phenotype was due to reduced cytotoxicity. A similar apparent increase in the ability of a ΔexoU strain to invade corneal epithelial cells had been reported previously in a different strain background (13). Nevertheless, the equivalent invasion assay and cytotoxicity data noted for PA14 relative to PA14ΔPAPI-1 and for PA14ΔPAPI-2 relative to PA14Δ1Δ2 (Fig. 3) strongly suggested that PAPI-1 alone played little or no role in the invasion of, or cytotoxicity toward, A549 cells under the conditions used in the assay.
FIG. 3.
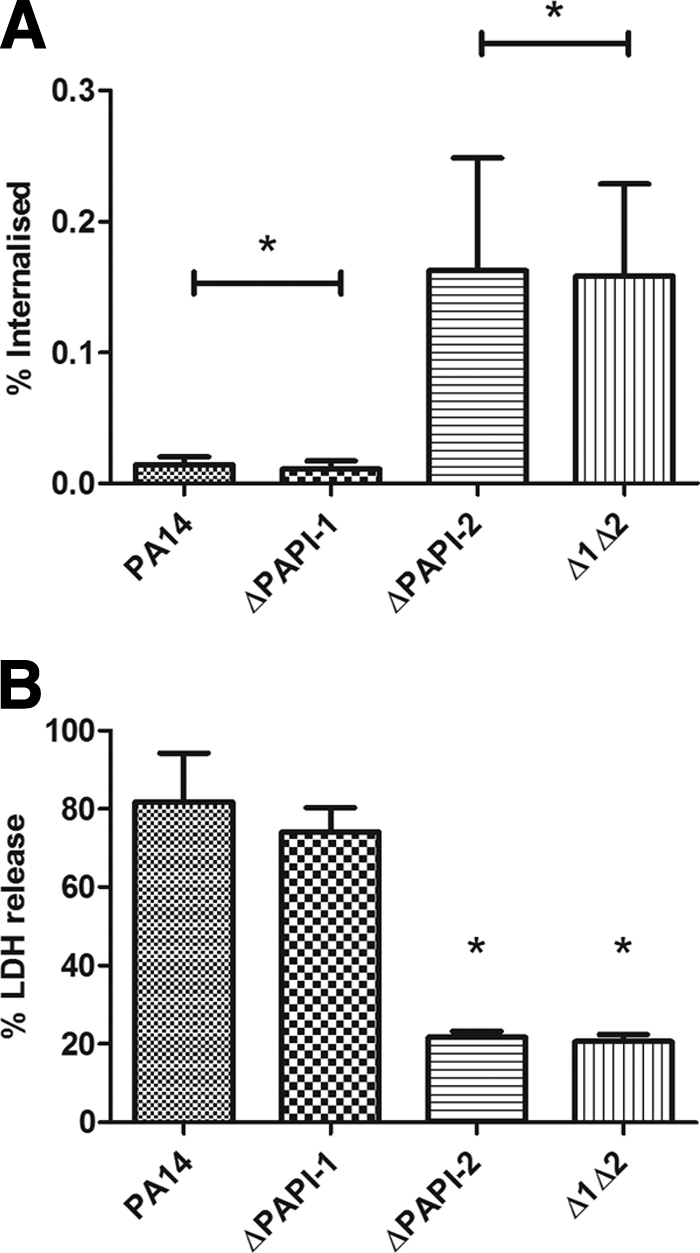
(A) Comparison of the invasive ability of wild-type P. aeruginosa PA14 with those of isogenic mutants lacking PAPI-1 and/or PAPI-2 using an A459 human lung carcinoma cell-based assay. The percentages of internalized bacteria protected from amikacin killing that were recovered after lysis of the remaining intact A459 cells relative to the original inocula are shown. Strains that are highly cytotoxic for A459 cells would result in an apparent reduction in invasive ability. Bars represent means and standard deviations. The asterisks indicate no significant difference between the strains compared. (B) Comparison of the cytotoxicity of P. aeruginosa strain PA14 with those of its isogenic mutants lacking PAPI-1 and/or PAPI-2 against A459 human lung carcinoma cells. The percentage of cytotoxicity was calculated relative to that of a maximum-LDH-release control. Bars represent means and standard deviations. Asterisks indicate significant differences from PA14 (P < 0.05).
Both PAPI-1 and PAPI-2 have roles in the full virulence of PA14 in a murine acute pneumonia model.
Comparison of wild-type PA14 and the PA14ΔPAPI-1 mutant in the murine acute pneumonia model demonstrated that there was no significant difference between the strains in the ability to cause disease as measured by clinical sign scores (Fig. 1B) or CFU counts in the nasopharynx, lungs, and blood (Fig. 2). Indeed, there was an apparent marginal increase in the number of mutant bacteria over that of PA14 in the nasopharynx samples (Fig. 2A). Furthermore, the survival times of mice infected with PA14ΔPAPI-1 were the same as those of mice infected with PA14 (Fig. 1A). Conversely, deletion of PAPI-2 resulted in a strain (PA14ΔPAPI-2) that was clearly attenuated compared to PA14 based on a 1- to 2-log-unit reduction in the burden of viable bacteria in nasopharynx, lung, and blood specimens (P < 0.05). This conclusion was also supported by a slight reduction in clinical sign scores for PA14ΔPAPI-2 (mean, 4.7; range, 3 to 5) compared to PA14 (mean, 5.7; range, 5 to 6) (Fig. 1B). Indeed, PA14ΔPAPI-2-infected mice had survival kinetics, clinical sign scores, and blood and tissue bacterial loads similar to those of PAO1-infected mice. Despite the fact that the loss of PAPI-1 alone contributed insignificantly to attenuation in this model, the double mutant PA14Δ1Δ2 was even further attenuated than the PA14ΔPAPI-2 mutant. Ninety percent of mice infected with the double mutant survived to 48 h, with 25% survival rates by the endpoint of the experiment (96 h), compared with 100% mortality by 24 h for PA14 and the two single island deletant strains (Fig. 1A). Marked reductions in viable bacterial counts in nasopharynx, lung, and blood specimens obtained at 18 h were also observed for the double mutant PA14Δ1Δ2 compared to PA14, PA14ΔPAPI-1, and even PA14ΔPAPI-2 (P < 0.05) (Fig. 2). Interestingly, only 1 of 10 mice infected with PA14Δ1Δ2 had detectable CFU in the blood (Fig. 2C), demonstrating that either the ability of the double mutant to disseminate from the lung to the blood was significantly impaired or its ability to survive in the bloodstream was reduced.
exoU is not the only virulence determinant encoded on PAPI-2.
In the murine acute pneumonia model, the PA14ΔexoU, PA14ΔPAPI-1ΔexoU, and PA14ΔPAPI-2 mutants exhibited similar survival times, suggesting that the further attenuation observed with the double island-minus mutant, PA14Δ1Δ2, resulted from the loss of part or all of PAPI-2, and not just exoU, in combination with the deletion of PAPI-1 (Fig. 1A). Comparison of the Kaplan-Meier survival plots obtained using the G. mellonella model showed that PA14ΔPAPI-2 was significantly attenuated (P, <0.001 by the log rank test) compared to PA14, while PA14ΔexoU was not statistically different from its wild-type parent (P, 0.228) (Fig. 4). These findings support the existence of an additional, non-exoU virulence gene(s) on PAPI-2 that is unmasked following the deletion of PAPI-1.
FIG. 4.
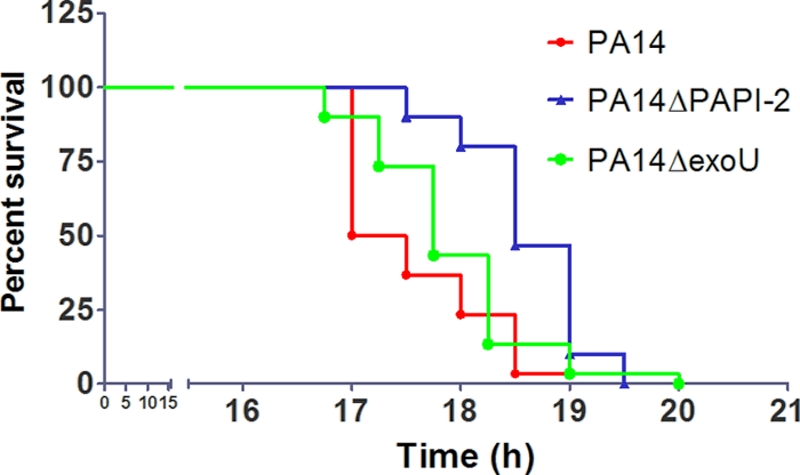
Survival graph for Galleria mellonella killing assay. A total of 30 larvae were used for each strain. Horizontal lines represent the percentage of G. mellonella larvae surviving after inoculation with each bacterial strain at the indicated time point.
Both PAPI-1 and PAPI-2 have a role to play in a murine model of bacteremia.
The set of P. aeruginosa strains discussed above was also studied using a murine bacteremia model initiated through intravenous inoculation of 2 × 106 P. aeruginosa organisms. After 6 h, all PA14-infected mice were moribund and had to be culled; hence, no data were available for PA14 beyond this time point. However, mice infected with PAO1 or any of the three PA14 mutants survived until the endpoint of 24 h (Fig. 5A), demonstrating that in a murine bacteremia model, both PAPI-1 and PAPI-2 make direct and individual contributions to the full virulence of the wild-type parent, PA14. Surprisingly, though, at 6 h postinfection, CFU counts in the blood of mice infected with PA14 were similar to those for the other four strains investigated. Furthermore, PA14ΔPAPI-1-infected mice seemed to have slightly higher blood CFU counts at 6 h and 24 h than mice infected with PAO1, PA14ΔPAPI-2, or PA14Δ1Δ2. Although not statistically significant, this result may imply reduced susceptibility of PA14ΔPAPI-1 to clearance from the bloodstream (Fig. 5A). The clinical sign scores at 2 h postinfection (Fig. 5B) revealed no hint of disease, except for a solitary mouse in the PA14 group that exhibited a score of 2. At 6 h, all 4 PA14-infected mice were moribund (clinical sign scores, ≥5), while only 4 other mice (all infected with PA14ΔPAPI-1) out of the 16 in the four other arms of the experiment displayed even minimal signs of disease, with a maximum clinical sign score of 1 (Fig. 5C). These 6-h-time-point data clearly highlighted a very major difference in virulence phenotype in the murine bacteremia model between PA14 and the other four strains examined. However, although there was no or minimal evidence of disease at 6 h in mice infected with PAO1 or any of the three PA14 mutants, by 24 h (Fig. 5C and D) all 16 mice in these groups exhibited clinical sign scores of 5 or 6. Examination of the 6-h and 24-h clinical sign score data suggested that PA14ΔPAPI-1 was subtly more virulent than either PA14ΔPAPI-2 or PA14Δ1Δ2; infection with either of the latter two strains resulted in equivalent clinical sign scores at these two time points. In addition, these results demonstrated that despite a marked difference in the rate of progression of disease between PA14-infected mice and mice infected with any of the isogenic mutants, none of the mutants were impaired in their ability to survive in the blood, with CFU counts equivalent to or even higher than that of PA14 at 6 h. However, the loss of one or both islands did markedly reduce disease severity and rate of progression at the 6-h time point. Importantly, these results also demonstrated that the ability of the double mutant PA14Δ1Δ2 to survive and/or replicate in the blood following intravenous inoculation was not impaired relative to that of PAO1 or the two single island-minus mutants, thus strongly supporting the earlier suggestion that the very low numbers of PA14Δ1Δ2 found in the blood following respiratory tract infection (Fig. 2C) were due to a markedly reduced ability of this mutant to disseminate from the lungs into the bloodstream.
FIG. 5.

(A) P. aeruginosa bacterial burdens in blood over a 24-h period following intravenous infection with 2 × 106 CFU. A total of five mice were used for each strain, and the same five mice were used for each time point. Each data point represents the mean CFU/ml of blood. All data were generated from a single experiment for each strain. The standard errors of the means are indicated. No data were available for the 24-h time point for mice infected with PA14, because severe signs of disease required culling of these mice at 6 h postinfection. (B, C, and D) Intravenous infection symptom scores at 2, 6, and 24 h postinfection, respectively. A total of five mice were used for each strain. No symptom score data were available at 24 h for PA14. Each symbol represents a single mouse. The bars represent mean symptom scores ± standard deviations. Asterisks mark strains exhibiting significant differences from PA14 (P < 0.05).
Combined deletion of PAPI-1 and PAPI-2 has a marked effect on the lung immune response and a subtle effect on lung histopathology.
Groups of mice infected intranasally with PAO1, PA14, or one of the three PA14 island-minus strains were sacrificed at 18 h postinfection. The lungs and nasopharynx were removed and processed for immunological cell counts and/or histology. Surprisingly, despite marked differences in disease severity, at the 18-h time point there were no significant differences between any two strains in the numbers of neutrophils (Fig. 6A) or lymphocytes (Fig. 6B) that had infiltrated lung tissue; all five strains showed a 1- to 2-log-unit increase in cell numbers over those for uninfected controls. In addition, with the exception of the double mutant PA14Δ1Δ2, none showed significant differences in either macrophage (Fig. 6C) or monocyte (Fig. 6D) cell numbers. Lung tissue from mice infected with PA14Δ1Δ2 showed significantly fewer (P < 0.05) macrophages and monocytes (Fig. 6C and D) than lungs from PA14-infected mice. Furthermore, unlike infection with other strains, PA14Δ1Δ2 infection resulted in lung tissues with numbers of macrophages equal to those in uninfected controls. Of particular note was the finding that monocyte numbers in the lungs of PA14Δ1Δ2-infected mice were about 1 log unit lower than those for uninfected controls (Fig. 6D). At 18 h postinfection, two mice per strain were culled for lung histology, and the histological specimens were scored qualitatively. The lung sections were given an overall score for damage and inflammation. The score ranged from 1 to 5 based on the levels of hypertrophy of bronchial walls, general consolidation, and cellular infiltration. A score of 1 represented negligible tissue damage or inflammation, and a score of 5 represented severe tissue damage and inflammation. The lung histology scores obtained with the different strains, in order of decreasing severity by subjective assessment, were as follows: PA14, 4.5; PA14ΔPAPI-1, 3.5; PAO1, 3.0; PA14ΔPAPI-2, 3.0; PA14Δ1Δ2, 3.0. Furthermore, as with other measures of attenuation reported above, there was a suggestion that loss of PAPI-2 had a greater effect than deletion of PAPI-1 on the histological damage in the lungs at 18 h postinfection.
FIG. 6.

Numbers of neutrophils (A), lymphocytes (B), macrophages (C), and monocytes (D) in lung tissue obtained 18 h postinfection in the murine acute pneumonia model. Four mice were used for each strain. The measurements refer to the number of target cells per milliliter of lung homogenate. Asterisks indicate significant differences from wild-type PA14 (P < 0.05). The standard errors of the means are shown.
DISCUSSION
This study has shown that both PAPI-1 and PAPI-2 contributed significantly to the full virulence of P. aeruginosa PA14 in murine acute pneumonia and bacteremia infection models. Consistent with these findings, PA14 was also found to be significantly more virulent in an acute pneumonia model than PAO1, a clonally distinct P. aeruginosa strain that naturally lacked both islands, strongly supporting comparative virulence data derived from other models of infection (31).
In the acute pneumonia model, PA14ΔPAPI-2 was significantly less virulent than PA14, resulting in lower viable bacterial numbers in the nasopharynx, lungs, and blood, reduced lung pathology, and delayed time to death. Loss of the PAPI-2-encoded exoU gene and its cognate chaperone gene spcU are likely to have contributed substantially in the acute pneumonia model. However, other factors encoded on PAPI-2 appear to play an important role in PA14 virulence, since PA14ΔPAPI-2 was even more attenuated than PA14ΔexoU in the G. mellonella model, and PA14Δ1Δ2 was significantly more crippled than PA14ΔPAPI-1ΔexoU in the acute murine pneumonia model. Indeed, a mutant carrying an in-frame deletion of the PAPI-2 gene PA14_5160, which is predicted to code for a 94-amino-acid membrane lipoprotein, has been shown to be significantly attenuated in an Arabidopsis model but not the burnt-skin mouse model (19). ExoU has previously been shown to play a major role in severe pneumonia in murine models. Comparison of a pair of isogenic strains secreting either ExoU or ExoS had revealed that the ExoU-secreting strain had a 50% lethal dose (LD50) 8-fold lower than that of its ExoS-secreting “sibling” (54). Furthermore, for patients with acute P. aeruginosa pneumonia, disease severity and mortality have been shown to correlate positively with the presence of ExoU-secreting isolates (10, 18). Our data are further supported by those of Shaver and Hauser (54), who have observed that ExoU was important for the dissemination of bacteria from the lungs, and by more-recent work by Diaz et al. (10) showing that ExoU allowed P. aeruginosa strains to persist in the lung by targeted killing of recruited phagocytes, particularly neutrophils. However, we observed no change in neutrophil numbers at 18 h postinfection, possibly due to the different strain backgrounds studied. The bacteremia model data also demonstrated a major role for PAPI-2 in the full virulence of PA14 in bloodstream infections, in particular with regard to disease severity and rate of progression at the 6-h time point. This was entirely consistent with the recent observation that the exoS-negative, exoU-positive genotype dominates among bloodstream infection-associated isolates in humans (59). Further investigations are required to identify the other PAPI-2 genes implicated in virulence in this study and to dissect the mechanistics of the newly demonstrated PAPI-1-PAPI-2 synergistic interaction.
The effects of PAPI-1 were more subtle; strain PA14ΔPAPI-1 was as virulent as PA14 in the murine pneumonia model, as judged by the 100% mortality rate at 18 h. Nevertheless, the PAPI-1-minus strain probably exhibited minor attenuation, since it resulted in slightly lower disease severity scores on average and marginally less lung histological damage. In contrast, in the bacteremia model, the PA14ΔPAPI-1 mutant was substantially impaired: all mice infected with this strain survived up to the 24-h endpoint, while PA14-infected mice universally succumbed at 6 h. As would be expected with these findings, the rate of progression of disease severity scores paralleled the mortality figures.
The most dramatic result was the marked reduction in virulence of the double mutant PA14Δ1Δ2 in the murine acute pneumonia model. This strain showed a highly significant reduction in virulence compared to the wild-type strain PA14 and even compared to each of the single-island deletion mutants, PA14ΔPAPI-1 and PA14ΔPAPI-2, clearly demonstrating that both PAPI-1 and PAPI-2 played key roles in acute pneumonia. Despite the minimal differences observed in qualitative lung histological scores, the broader set of data obtained was perfectly consistent with PA14 being the most virulent strain and PA14Δ1Δ2 being at the other end of the spectrum in the acute pneumonia model. Twenty-two genes borne on PAPI-1 and PAPI-2 have previously been implicated in virulence by using defined PA14 single-gene knockouts and the Arabidopsis leaf infiltration, murine burnt-skin, G. mellonella, and Caenorhabditis elegans slow-killing infection models (8, 19, 31, 41), but our findings are the first data to clearly demonstrate a role for PAPI-1 in acute pneumonia.
The markedly lower numbers of monocytes and macrophages in the lungs of mice infected with the double mutant than those with the other strains that caused more-severe disease could be attributed to apoptosis of monocytes and/or macrophages as part of a successful modulation of the immune response to PA14Δ1Δ2 infection; such a defense has previously been hypothesized to occur during P. aeruginosa lung infection (65). This fits with data derived from P. aeruginosa infections in humans and a rabbit sepsis model (2, 16), which showed that early apoptosis of blood monocytes was correlated with positive resolution of infection. This association was postulated to be due to a reduced inflammatory response and a consequent dampening of the sepsis cascade. A similar phenomenon has been described for pneumococcal pneumonia and the likelihood of its progression to bacteremia (35). It is possible that the host is able to mount a protective immune response merely as a result of the marked attenuation of PA14Δ1Δ2 or that the recruitment of monocytes and macrophages is caused by a different antigenic profile. However, an alternative hypothesis would be that loss of PAPI-1 and PAPI-2 resulted in a strain that was specifically unable to modulate the immune response in its favor. Previous studies have shown that P. aeruginosa, like Salmonella (28), is capable of preventing Kupffer cell (3) and epithelial cell (64) apoptosis by secretion of unknown antiapoptotic factors. The ability of P. aeruginosa to prevent the apoptosis of monocytes and macrophages and to shift the immune response toward inflammation would constitute a significant virulence factor.
The PA14 PAPI-1 island encodes more than 30 putative virulence genes that are also present on several other sequenced P. aeruginosa genomes (see Table S2 and Fig. S1 in the supplemental material). The RscBC two-component system is an excellent candidate, since counterparts in E. coli, Salmonella, Erwinia, and Yersinia have been shown to play essential roles in the regulation of T3SSs and many other virulence factors. A recently study published by Mikkelsen and coworkers demonstrated that the PAPI-1-encoded RcsBC and PvrRS systems antagonistically regulated the expression of the PAPI-1-borne fimbrial cupD operon (40), affecting the ability of the bacteria to attach and form biofilms. However, cupD regulation was not directly demonstrable under laboratory growth conditions, suggesting that specific, as yet undefined stimuli were required to engage the two-component systems. Reduced levels of cell fimbriation may contribute to the lower numbers of monocytes and macrophages present in the lungs of mice infected with the double mutant, since E. coli fimbriae have previously been reported to be potent stimulators of the Toll-like receptor 4 immune cell recruitment pathway (3). Other work, by Nicastro and Baldini (44), demonstrated that a P. aeruginosa rscC rscB double mutant exhibited an altered lipopolysaccharide O-antigen profile, as has been reported previously for Salmonella (25). The length and structure of O-antigen chains are a key factor in serum resistance, type III secretion, and humoral immunogenicity (4, 49), potentially explaining the finding that PA14ΔPAPI-1 appeared to defy clearance from the blood, relative to that of PA14 and the other strains, despite being found to be significantly attenuated in the murine bacteremia model.
Synergistic relationships between pathogenicity islands have been described for E. coli (PAI-I and PAI-II) (7) and Salmonella enterica (SPI-1 and SPI-4) (14, 15). However, to the best of our knowledge, this phenomenon has not been described previously for P. aeruginosa or any other bacterial species. Clearly, the mechanistic basis of the synergistic relationship between PAPI-1 and PAPI-2 would be of key interest. Besides potential contributions of PAPI-1 to bacterial fimbriation and/or biofilm formation, another plausible hypothesis would be that loss of PAPI-1 leads to aberrant T3SS activity as a result of an altered O-antigen profile, which has been implicated in T3SS function (4), and/or loss of the Rcs system, which has been shown to regulate the expression of T3SS in Yersinia enterocolitica and S. enterica (33, 58). Despite the dominant role of ExoU, it is likely that other T3SS effectors encoded elsewhere on the genome also play key roles in the fine tuning of the pathogenesis process; the deployment of multiple, specifically targeted effectors is a common theme for several bacterial pathogens.
Our findings with regard to PAPI-1 complement those of Battle et al. (5), who demonstrated that novel non-PAPI-1 sequences encoded on PAGI-5, a PAPI-1-like hybrid structure, played an important role in murine acute pneumonia. As far as we are aware, this is the first report that clearly demonstrates a substantial role in virulence for PAPI-1 by using in vivo models. Further work is clearly required to identify the combinations and temporal activation patterns of island-borne genes brought into play during Pseudomonas infections and to further dissect their interactions with the genes of the core genome. The widely recognized presence of PAPI-1-like islands in diverse environmental and clinical isolates (27, 61, 63), including those causing acute pneumonia, chronic cystic fibrosis, and burn-associated infections, suggests that these islands may be subjected to considerable positive selection. This is particularly so given the known spontaneous instability of these elements.
We propose that the en bloc deletion strategy that we have used for the first time to study the role of genomic islands in P. aeruginosa offers several major advantages. It allows for rapid targeted virulence-related screening of large blocks of DNA in their “native” genomic backgrounds, addresses the key evolutionary question of the impact of the en bloc acquisition of genomic islands, potentially reveals subtle but important contributions to virulence that may be masked by dominant traits encoded by other targeted islands, and greatly facilitates the detection of synergistic and/or combinatorial effects between any of the numerous genes collectively deleted from the mutant strains. The data obtained from these whole-island deletion experiments could then be investigated further in a better-targeted fashion by single-gene mutation, combination gene mutation, transcriptomic, and/or other detailed genetic studies to pinpoint the precise mechanisms involved. This study has clearly demonstrated the key role of the horizontally acquired PAPI-1 and PAPI-2 islands in murine acute pneumonia and bacteremia models, thus firmly cementing their designation as genetic modules of virulence in a major pathogenic species.
Supplementary Material
Acknowledgments
We thank James Lonnen, Barbara Rieck, and Sarah Smeaton for helpful advice and discussions and Craig Winstanley and Herbert Schweizer for providing P. aeruginosa PA14 and key mutagenesis plasmids, respectively. We are especially grateful to Eliana Drenkard for PA14-related strains and for prompt assistance with the generation of the mutants lacking exoU. Thanks also go to Marialuisa Crosatti for help with the Galleria assay and to Hannah Brewin for murine experiments.
This work was funded by an Action Medical Research grant to K.R., A.K., and C.O. E.M.H. was funded by a Medical Research Council/University of Leicester Ph.D. Studentship. H.-Y.O. was funded by a grant from the National Natural Science Foundation of China (30700013/C010103) and the 863 Program, Ministry of Science and Technology, China (2006AA02Z328). K.R. and Z.D. were supported by a Royal Society-National Natural Science Foundation of China International Joint Project grant (IJP2007/R3).
Editor: B. A. McCormick
Footnotes
Published ahead of print on 1 February 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Allewelt, M., F. T. Coleman, M. Grout, G. P. Priebe, and G. B. Pier. 2000. Acquisition of expression of the Pseudomonas aeruginosa ExoU cytotoxin leads to increased bacterial virulence in a murine model of acute pneumonia and systemic spread. Infect. Immun. 68:3998-4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonopoulou, A., M. Raftogiannis, E. J. Giamarellos-Bourboulis, P. Koutoukas, L. Sabracos, M. Mouktaroudi, T. Adamis, I. Tzepi, H. Giamarellou, and E. E. Douzinas. 2007. Early apoptosis of blood monocytes is a determinant of survival in experimental sepsis by multi-drug-resistant Pseudomonas aeruginosa. Clin. Exp. Immunol. 149:103-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashkar, A. A., K. L. Mossman, B. K. Coombes, C. L. Gyles, and R. Mackenzie. 2008. FimH adhesin of type 1 fimbriae is a potent inducer of innate antimicrobial responses which requires TLR4 and type 1 interferon signalling. PLoS Pathog. 4:e1000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Augustin, D. K., Y. Song, M. S. Baek, Y. Sawa, G. Singh, B. Taylor, A. Rubio-Mills, J. L. Flanagan, J. P. Wiener-Kronish, and S. V. Lynch. 2007. Presence or absence of lipopolysaccharide O antigens affects type III secretion by Pseudomonas aeruginosa. J. Bacteriol. 189:2203-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Battle, S. E., F. Meyer, J. Rello, V. L. Kung, and A. R. Hauser. 2008. Hybrid pathogenicity island PAGI-5 contributes to the highly virulent phenotype of a Pseudomonas aeruginosa isolate in mammals. J. Bacteriol. 190:7130-7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Battle, S. E., J. Rello, and A. R. Hauser. 2009. Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 290:70-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brzuszkiewicz, E., H. Bruggemann, H. Liesegang, M. Emmerth, T. Olschlager, G. Nagy, K. Albermann, C. Wagner, C. Buchrieser, L. Emody, G. Gottschalk, J. Hacker, and U. Dobrindt. 2006. How to become a uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc. Natl. Acad. Sci. U. S. A. 103:12879-12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi, J. Y., C. D. Sifri, B. C. Goumnerov, L. G. Rahme, F. M. Ausubel, and S. B. Calderwood. 2002. Identification of virulence genes in a pathogenic strain of Pseudomonas aeruginosa by representational difference analysis. J. Bacteriol. 184:952-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi, K. H., and H. P. Schweizer. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz, M. H., C. M. Shaver, J. D. King, S. Musunuri, J. A. Kazzaz, and A. R. Hauser. 2008. Pseudomonas aeruginosa induces localized immunosuppression during pneumonia. Infect. Immun. 76:4414-4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drenkard, E., and F. M. Ausubel. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740-743. [DOI] [PubMed] [Google Scholar]
- 12.El Solh, A. A., M. E. Akinnusi, J. P. Wiener-Kronish, S. V. Lynch, L. A. Pineda, and K. Szarpa. 2008. Persistent infection with Pseudomonas aeruginosa in ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 178:513-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans, D. J., D. W. Frank, V. Finck-Barbancon, C. Wu, and S. M. Fleiszig. 1998. Pseudomonas aeruginosa invasion and cytotoxicity are independent events, both of which involve protein tyrosine kinase activity. Infect. Immun. 66:1453-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerlach, R. G., N. Claudio, M. Rohde, D. Jackel, C. Wagner, and M. Hensel. 2008. Cooperation of Salmonella pathogenicity islands 1 and 4 is required to breach epithelial barriers. Cell. Microbiol. 10:2364-2376. [DOI] [PubMed] [Google Scholar]
- 15.Gerlach, R. G., D. Jackel, N. Geymeier, and M. Hensel. 2007. Salmonella pathogenicity island 4-mediated adhesion is coregulated with invasion genes in Salmonella enterica. Infect. Immun. 75:4697-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giamarellos-Bourboulis, E. J., C. Routsi, D. Plachouras, V. Markaki, M. Raftogiannis, D. Zervakis, V. Koussoulas, S. Orfanos, A. Kotanidou, A. Armaganidis, C. Roussos, and H. Giamarellou. 2006. Early apoptosis of blood monocytes in the septic host: is it a mechanism of protection in the event of septic shock? Crit. Care 10:R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 18.Hauser, A. R., E. Cobb, M. Bodi, D. Mariscal, J. Valles, J. N. Engel, and J. Rello. 2002. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit. Care Med. 30:521-528. [DOI] [PubMed] [Google Scholar]
- 19.He, J., R. L. Baldini, E. Deziel, M. Saucier, Q. Zhang, N. T. Liberati, D. Lee, J. Urbach, H. M. Goodman, and L. G. Rahme. 2004. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. U. S. A. 101:2530-2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendrickson, E. L., J. Plotnikova, S. Mahajan-Miklos, L. G. Rahme, and F. M. Ausubel. 2001. Differential roles of the Pseudomonas aeruginosa PA14 rpoN gene in pathogenicity in plants, nematodes, insects, and mice. J. Bacteriol. 183:7126-7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 22.Inglis, R. F., A. Gardner, P. Cornelis, and A. Buckling. 2009. Spite and virulence in the bacterium Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 106:5703-5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juhas, M., D. W. Crook, I. D. Dimopoulou, G. Lunter, R. M. Harding, D. J. Ferguson, and D. W. Hood. 2007. Novel type IV secretion system involved in propagation of genomic islands. J. Bacteriol. 189:761-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kadioglu, A., N. A. Gingles, K. Grattan, A. Kerr, T. J. Mitchell, and P. W. Andrew. 2000. Host cellular immune response to pneumococcal lung infection in mice. Infect. Immun. 68:492-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kintz, E., and J. B. Goldberg. 2008. Regulation of lipopolysaccharide O antigen expression in Pseudomonas aeruginosa. Future Microbiol. 3:191-203. [DOI] [PubMed] [Google Scholar]
- 26.Klockgether, J., O. Reva, K. Larbig, and B. Tummler. 2004. Sequence analysis of the mobile genome island pKLC102 of Pseudomonas aeruginosa C. J. Bacteriol. 186:518-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klockgether, J., D. Wurdemann, O. Reva, L. Wiehlmann, and B. Tummler. 2007. Diversity of the abundant pKLC102/PAGI-2 family of genomic islands in Pseudomonas aeruginosa. J. Bacteriol. 189:2443-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knodler, L. A., and B. B. Finlay. 2001. Salmonella and apoptosis: to live or let die? Microbes Infect. 3:1321-1326. [DOI] [PubMed] [Google Scholar]
- 29.Kukavica-Ibrulj, I., A. Bragonzi, M. Paroni, C. Winstanley, F. Sanschagrin, G. A. O'Toole, and R. C. Levesque. 2008. In vivo growth of Pseudomonas aeruginosa strains PAO1 and PA14 and the hypervirulent strain LESB58 in a rat model of chronic lung infection. J. Bacteriol. 190:2804-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulasekara, B. R., H. D. Kulasekara, M. C. Wolfgang, L. Stevens, D. W. Frank, and S. Lory. 2006. Acquisition and evolution of the exoU locus in Pseudomonas aeruginosa. J. Bacteriol. 188:4037-4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee, D. G., J. M. Urbach, G. Wu, N. T. Liberati, R. L. Feinbaum, S. Miyata, L. T. Diggins, J. He, M. Saucier, E. Deziel, L. Friedman, L. Li, G. Grills, K. Montgomery, R. Kucherlapati, L. G. Rahme, and F. M. Ausubel. 2006. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 7:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee, V. T., R. S. Smith, B. Tummler, and S. Lory. 2005. Activities of Pseudomonas aeruginosa effectors secreted by the type III secretion system in vitro and during infection. Infect. Immun. 73:1695-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin, D., C. V. Rao, and J. M. Slauch. 2008. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J. Bacteriol. 190:87-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luck, S. N., L. Badea, V. Bennett-Wood, R. Robins-Browne, and E. L. Hartland. 2006. Contribution of FliC to epithelial cell invasion by enterohemorrhagic Escherichia coli O113:H21. Infect. Immun. 74:6999-7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marriott, H. M., P. G. Hellewell, S. S. Cross, P. G. Ince, M. K. Whyte, and D. H. Dockrell. 2006. Decreased alveolar macrophage apoptosis is associated with increased pulmonary inflammation in a murine model of pneumococcal pneumonia. J. Immunol. 177:6480-6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathee, K., G. Narasimhan, C. Valdes, X. Qiu, J. M. Matewish, M. Koehrsen, A. Rokas, C. N. Yandava, R. Engels, E. Zeng, R. Olavarietta, M. Doud, R. S. Smith, P. Montgomery, J. R. White, P. A. Godfrey, C. Kodira, B. Birren, J. E. Galagan, and S. Lory. 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U. S. A. 105:3100-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMorran, B., L. Town, E. Costelloe, J. Palmer, J. Engel, D. Hume, and B. Wainwright. 2003. Effector ExoU from the type III secretion system is an important modulator of gene expression in lung epithelial cells in response to Pseudomonas aeruginosa infection. Infect. Immun. 71:6035-6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meissner, A., V. Wild, R. Simm, M. Rohde, C. Erck, F. Bredenbruch, M. Morr, U. Romling, and S. Haussler. 2007. Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ. Microbiol. 9:2475-2485. [DOI] [PubMed] [Google Scholar]
- 39.Michel-Briand, Y., and C. Baysse. 2002. The pyocins of Pseudomonas aeruginosa. Biochimie 84:499-510. [DOI] [PubMed] [Google Scholar]
- 40.Mikkelsen, H., G. Ball, C. Giraud, and A. Filloux. 2009. Expression of Pseudomonas aeruginosa CupD fimbrial genes is antagonistically controlled by RcsB and the EAL-containing PvrR response regulators. PLoS One 4:e6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyata, S., M. Casey, D. W. Frank, F. M. Ausubel, and E. Drenkard. 2003. Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect. Immun. 71:2404-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morton, D. B., and P. H. Griffiths. 1985. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet. Rec. 116:431-436. [DOI] [PubMed] [Google Scholar]
- 43.Murphy, T. F., A. L. Brauer, K. Eschberger, P. Lobbins, L. Grove, X. Cai, and S. Sethi. 2008. Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 177:853-860. [DOI] [PubMed] [Google Scholar]
- 44.Nicastro, G. G., and R. L. Baldini. 2007. Functional characterization of Pseudomonas aeruginosa virulence-related genes located in a pathogenicity island, abstr. F-20. Abstr. XXXVI Annu. Meet. Braz. Soc. Biochem. Mol. Biol. (SBBq)-10th IUBMB Conf., Salvador da Bahia, Brazil, 2007. http://sbbq.iq.usp.br/arquivos/2007/cdlivro/resumos/R8586.pdf.
- 45.O'Toole, G. A., and R. Kolter. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449-461. [DOI] [PubMed] [Google Scholar]
- 46.Qiu, X., A. U. Gurkar, and S. Lory. 2006. Interstrain transfer of the large pathogenicity island (PAPI-1) of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 103:19830-19835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajakumar, K., C. Sasakawa, and B. Adler. 1997. Use of a novel approach, termed island probing, identifies the Shigella flexneri she pathogenicity island which encodes a homolog of the immunoglobulin A protease-like family of proteins. Infect. Immun. 65:4606-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rashid, M. H., and A. Kornberg. 2000. Inorganic polyphosphate is needed for swimming, swarming, and twitching motilities of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 97:4885-4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rocchetta, H. L., L. L. Burrows, and J. S. Lam. 1999. Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 63:523-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryall, B., J. C. Davies, R. Wilson, A. Shoemark, and H. D. Williams. 2008. Pseudomonas aeruginosa, cyanide accumulation and lung function in CF and non-CF bronchiectasis patients. Eur. Respir. J. 32:740-747. [DOI] [PubMed] [Google Scholar]
- 51.Sadikot, R. T., T. S. Blackwell, J. W. Christman, and A. S. Prince. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 171:1209-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulert, G. S., H. Feltman, S. D. Rabin, C. G. Martin, S. E. Battle, J. Rello, and A. R. Hauser. 2003. Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa isolates obtained from patients with hospital-acquired pneumonia. J. Infect. Dis. 188:1695-1706. [DOI] [PubMed] [Google Scholar]
- 53.Schweizer, H. P. 1992. Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol. 6:1195-1204. [DOI] [PubMed] [Google Scholar]
- 54.Shaver, C. M., and A. R. Hauser. 2004. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect. Immun. 72:6969-6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon, R., U. Priefer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1:784-791. [Google Scholar]
- 56.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 57.Vogel, H. J., and D. M. Bonner. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97-106. [PubMed] [Google Scholar]
- 58.Walker, K. A., and V. L. Miller. 2009. Synchronous gene expression of the Yersinia enterocolitica Ysa type III secretion system and its effectors. J. Bacteriol. 191:1816-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wareham, D. W., and M. A. Curtis. 2007. A genotypic and phenotypic comparison of type III secretion profiles of Pseudomonas aeruginosa cystic fibrosis and bacteremia isolates. Int. J. Med. Microbiol. 297:227-234. [DOI] [PubMed] [Google Scholar]
- 60.West, S. E., H. P. Schweizer, C. Dall, A. K. Sample, and L. J. Runyen-Janecky. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 148:81-86. [DOI] [PubMed] [Google Scholar]
- 61.Wiehlmann, L., G. Wagner, N. Cramer, B. Siebert, P. Gudowius, G. Morales, T. Kohler, C. van Delden, C. Weinel, P. Slickers, and B. Tummler. 2007. Population structure of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 104:8101-8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolfgang, M. C., B. R. Kulasekara, X. Liang, D. Boyd, K. Wu, Q. Yang, C. G. Miyada, and S. Lory. 2003. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:8484-8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wurdemann, D., and B. Tummler. 2007. In silico comparison of pKLC102-like genomic islands of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 275:244-249. [DOI] [PubMed] [Google Scholar]
- 64.Zhang, J., H. Li, J. Wang, Z. Dong, S. Mian, and F. S. Yu. 2004. Role of EGFR transactivation in preventing apoptosis in Pseudomonas aeruginosa-infected human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 45:2569-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang, Y., X. Li, A. Carpinteiro, and E. Gulbins. 2008. Acid sphingomyelinase amplifies redox signaling in Pseudomonas aeruginosa-induced macrophage apoptosis. J. Immunol. 181:4247-4254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.