

Abstract
The current Bacillus anthracis vaccine consists largely of protective antigen (PA), the protein of anthrax toxin that mediates entry of edema factor (EF) or lethal factor (LF) into cells. PA induces protective antibody (Ab)-mediated immunity against Bacillus anthracis but has limited efficacy and duration. We previously demonstrated that activation of CD1d-restricted natural killer-like T cells (NKT) with a CD1d-binding glycolipid led to enhanced Ab titers specific for foreign antigen (Ag). We therefore tested the hypothesis that activation of NKT cells with the CD1d ligand (α-galactosylceramide [α-GC]) at the time of immunization improves PA-specific Ab responses. We observed that α-GC enhanced PA-specific Ab titers in C57BL/6 mice. In CD1d−/− mice deficient in type I and type II NKT cells the anti-PA Ab response was diminished. In Jα281−/− mice expressing CD1d but lacking type I α-GC-reactive NKT cells, α-GC did not enhance the Ab response. In vitro neutralization assays were performed and showed that the Ab titers correlated with protection of macrophages against anthrax lethal toxin (LT). The neutralization capacity of the Ab was further tested in lethal challenge studies, which revealed that NKT activation leads to enhanced in vivo protection against LT. Anti-PA Ab titers, neutralization, and protection were then measured over a period of several months, and this revealed that NKT activation leads to a sustained protective Ab response. These results suggest that NKT-activating CD1d ligands could be exploited for the development of improved vaccines for Bacillus anthracis that increase not only neutralizing Ab titers but also the duration of the protection afforded by Ab.
The current AVA anthrax vaccine administered to U.S. military personnel consists of the Bacillus anthracis protective antigen (PA) and induces PA-specific antibody (Ab) titers sufficient to neutralize anthrax toxin (28, 29). However, the anti-PA Ab titers are not sustained, and individuals require administration of multiple booster vaccines to maintain toxin-neutralizing Ab titers (http://www.anthrax.osd.mil/vaccine/schedule.asp#). Thus, there is a need for improving the efficacy of the current anthrax vaccine.
PA is an 83-kDa protein that forms heptameric pores on the surface of target cells expressing anthrax toxin receptors (capillary morphogenesis protein 2 [CMG-2] and tumor endothelial marker 8 [TEM-8]) (5, 6). PA heptamers interact with lethal factor (LF) and edema factor (EF) to form lethal toxin (LT) and edema toxin (ET), which together are referred to as anthrax toxin (6). The PA heptamer facilitates entry of EF and LF into the target cell. Following cell entry, EF generates supraphysiological levels of cyclic AMP via the protein's calmodulin-dependent adenylate cyclase activity (20). Within the intoxicated cell LF functions as a zinc-dependent metalloprotease and cleaves mitogen-activated protein kinase kinases and has recently been found to disrupt the inflammasome (10). Both toxins are lethal in animal models and cause a broad range of defects in target cells, including altered cell cycle, cell growth, and survival and attenuated inflammatory responses (24). Collectively these activities of anthrax toxin cripple the host immune system and allow B. anthracis to grow to high numbers in the bloodstream (6). Hence, immune neutralization of PA prevents all of the detrimental effects of anthrax edema toxin and lethal toxin, providing a crucial advantage to the host during early stages of disease.
PA-specific Abs effectively neutralize LT and ET in vitro and protect immunized animals in vivo following a lethal challenge with the toxins (1, 4, 25, 26, 30, 33). There is a good correlation between PA-specific Ab titers and toxin neutralization by sera from patients who have survived B. anthracis infection (29). Consequently, there is considerable interest in development of efficacious vaccines which incorporate PA as the immunogen but involve fewer immunizations, boost immunological memory, and prolong neutralizing Ab production while stimulating a minimal inflammatory response (28, 29).
Recent work by our laboratory and others has shown that activation of a specialized subset of T cells known as natural killer-like T cells (NKT cells) improves Ab responses against model and pathogen-derived antigens (3, 8, 13, 16, 17). More recently, we and others have also demonstrated that B-cell memory responses and the duration of plasma cell responses are enhanced as a result of administration of an NKT-activating glycolipid known as α-galactosylceramide (α-GC) (8, 11). The α-GC ligand consists of a galactose headgroup with an α-anomeric linkage to hydrophobic sphingosine and acyl chains. The lipid moiety is loaded into hydrophobic pockets in the CD1d molecule expressed by professional antigen-presenting cells, orienting the galactose headgroup for recognition by NKT cells (14, 32). The predominant NKT subset is represented by the type I NKT cells, which express an invariant Vα14, Jα18 T-cell antigen receptor (TCR) in mice and a Vα24, Jα28 TCR in humans (14, 32). Type II NKT cells have variable Vα usage, and while CD1d restricted they are thought to be stimulated by a variety of glycolipids but are not stimulated by α-GC (14, 32).
In this study we therefore tested the hypothesis that coadministration of α-GC and PA leads to enhanced anti-PA Ab titers and that the Ab produced neutralizes LT in vitro and protects in vivo against a lethal challenge. Our results show that NKT cells are required for generation of PA-specific LT-neutralizing Abs and that activation of NKT cells with exogenous CD1d ligand at the time of immunization boosts the toxin-neutralizing Ab titer and leads to enhanced and sustained in vivo protection against a lethal challenge with anthrax toxin.
MATERIALS AND METHODS
Reagents.
BL21 competent cells were purchased from Invitrogen (Carlsbad, CA). pET15b plasmids encoding PA and LF were a gift from J. Collier (Harvard Medical School) and have been described previously (5). The protein assay kit was purchased from Bio-Rad Laboratories (Hercules, CA). The Pyrogent Ultra Limulus amebocyte lysate kit was purchased from Lonza (Walkersville, MD). α-GC was purchased from Axorra Inc. (San Diego, CA). The purity, structural integrity, and functionality of α-GC have been described by us previously (18). Horseradish peroxidase (HRP)-conjugated anti-IgG1 was purchased from Southern Biotechnology (Birmingham, AL). Fluorochrome-conjugated monoclonal antibodies (MAbs) were purchased from BD Biosciences (San Jose, CA). The FcγR-blocking MAb (2.4G2) was purchased from BioXpres (West Lebanon, NH). CD1d/α-GC tetramers were provided by the NIAID Tetramer Facility (Emory University, Atlanta, GA).
Expression and purification of PA and LF.
Competent Escherichia coli (BL21) cells transformed with the pET15b-rPA plasmid were grown at 37°C in Luria-Bertani broth containing ampicillin (50 μg/ml final concentration) with shaking at 250 rpm in 2-liter baffled flasks. When the absorbance of the culture reached 1.0 at a wavelength of 600 nm, isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 0.1 mM to induce PA expression. Following induction for 18 h at 16°C, BL21 bacteria were harvested by centrifugation at 15,000 × g at 4°C for 20 min. The supernatant was discarded and the pellet was then resuspended gently in binding buffer (20 mM sodium phosphate, 0.5 M NaCl, and 20 mM imidazole; pH 7.4). The resuspended cells were lysed using an Emulsiflex C-3 homogenizer (Avestin, Ottawa, ON, Canada) at 4°C for four complete cycles at 5,000 lb/in2 of pressure. The lysate was centrifuged at 15,000 × g at 4°C for 40 min, and the supernatant was collected and further clarified using a 0.45-μm filter unit.
Filtered lysate was then loaded onto a 5-ml HisTrap FF column (GE LifeSciences, Piscataway, NJ) using a peristaltic pump set at a 5-ml/min flow rate. The column was then washed with 30 ml of wash buffer (20 mM sodium phosphate, 0.5 M NaCl, and 40 mM imidazole; pH 7.4), and the protein was eluted from the column in 5-ml fractions using elution buffer (20 mM sodium phosphate, 0.5 M NaCl, and 0.5 M imidazole; pH 7.4). The eluted fractions were analyzed via SDS-PAGE, and those containing protein of the appropriate size were collected and pooled. To avoid rapid precipitation of PA during desalting, the isolated protein was placed in dialysis tubing with a 14-kDa cutoff and then dialyzed at 4°C against 10 mM sodium phosphate, 2.7 mM KCl, 1.8 mM KH2PO4, 0.25 M NaCl by stepping down the salt as follows: 2 h at 0.4 M NaCl, 2 h at 0.3 M NaCl, 2 h at 0.25 M NaCl, and 0.25 M NaCl overnight. To concentrate purified PA, the dialyzed protein in the dialysis tubing was packed into polyethylene glycol with a 35-kDa cutoff (VWR) for 1 to 2 h. The concentrated PA was centrifuged and filtered to remove debris, and the protein concentration was determined using the Bio-Rad assay. Endotoxin contamination was reduced from purified PA by using an EndoTrap Red column (Lonza, Walkersville, MD) following the manufacturer's instructions. PA was eluted from the column into phosphate-buffered saline (PBS) and subsequently passed through a 0.22-μm sterile filter, and the protein concentration of the sample was determined. The concentration of residual endotoxin was determined using the Limulus assay, which has a sensitivity of 0.06 endotoxin units/ml. PA was then stored in aliquots at −20°C. The amount of lipopolysaccharide (LPS) remaining in the PA samples after the Endotrap column removal procedure was such that no more than 6 ng of LPS was administered to any mouse, an amount insufficient to have adjuvant properties for Ab responses in mice (27). Western blot analysis was used to confirm that sera from PA-immunized mice contained PA-specific Ab (data not shown). Immunizations were also performed to compare PA before LPS removal, PA after LPS removal, and PA after LPS removal and mixing with freshly added LPS. These experiments showed that there was no significant LPS contamination of our PA preparations that could exert adjuvant effects on anti-PA Ab responses (data not shown).
LF was expressed and purified in an identical manner to PA except that the pET15b-rLF plasmid was used for expression and the elution buffer for the affinity column contained 0.2 M NaCl. Endotoxin association with LF was considerably less than that observed for PA, and the concentration in the LF samples following the endotoxin affinity column procedure was below the limit of detection.
Mice.
C57BL/6 mice were purchased from the National Cancer Institute (Bethesda, MD). CD1d−/− mice on the C57BL/6 genetic background have been described previously (9) and were kindly provided by M. Exley (Harvard Medical School). CD1d−/− mice do not express CD1d on antigen-presenting cells and therefore fail to develop either type I or type II NKT cells in the thymus and export them to the periphery. Jα18−/− mice were obtained from M. Kronenberg (La Jolla Institute for Allergy and Immunology, La Jolla, CA) with kind permission from M. Taniguchi (RIKEN Institute, Japan) and are described in reference 7. Jα18−/− mice express CD1d but fail to develop type I NKT cells because of a targeted deletion in the Jα281 gene segment of the TCR. CD1d−/− and Jα18−/− mice were bred in the Animal Resource Center at the University of Oklahoma Health Sciences Center (OUHSC). All procedures were approved by the Institutional Animal Care and Use Committee at OUHSC.
Immunizations and experimental schedule.
Female mice of 6 to 10 weeks of age were used, and five mice per group were immunized unless indicated otherwise. A single subcutaneous (s.c.) immunization was administered over both flanks on day zero immediately following collection of prebleed sera. Unless indicated otherwise, immunizations consisted of 10 μg of PA in 200 μl of sterile endotoxin-free PBS or PA mixed with 2 μg of α-GC. Mice were then bled at day 14 postimmunization and sera were obtained. On day 28 mice were bled and then boosted s.c. with 5 μg of PA in PBS and bled on days 33, 38, and 43 unless indicated otherwise. At the end of the experimental period, mice were either challenged with toxin or euthanized in order to obtain bone marrow and spleen cells.
Retro-orbital eye bleed and serum collection.
Mice were anesthetized using a vaporized 4% isofluorane-96% oxygen mixture, and 100 μl of blood was collected by retro-orbital bleed with heparinized microcapillary tubes (Fisher Scientific, Hampton, NH). Samples were transferred immediately to polypropylene microcentrifuge tubes. Blood samples were incubated for 30 min at room temperature and then allowed to clot overnight at 4°C before centrifugation at 13,000 × g for 15 min at 4°C. Sera were withdrawn with a pipette and stored in aliquots at −20°C.
Isolation of splenocytes.
Spleens were harvested into Hanks balanced salt solution buffer, and a single-cell suspension was obtained by mechanical disruption. Erythrocytes were removed by incubation with ammonium chloride lysis buffer (0.16 M NH4Cl, 0.17 M Tris-HCl; pH 7.4) for 2 min at 37°C. After washing in culture medium, cell viability was confirmed as >98% by trypan blue exclusion. Cells were enumerated using a Beckman Coulter Z2 cell/particle counter (Fullerton, CA).
ELISA.
Immulon 4 enzyme-linked immunosorbent assay (ELISA) plates (Dynex Technologies Inc., Chantilly, VA) were coated with PA at 10 μg/ml in binding buffer (0.1 M Na2HPO4; pH 9.0) overnight at 4°C before washing plates and blocking for 2 h at room temperature with 1.0% (wt/vol) bovine serum albumin (BSA) in PBS-0.05% (vol/vol) Tween 20. Sera were diluted 100- or 10,000-fold in PBS-0.05% (vol/vol) Tween and subjected to 2-fold serial dilutions before adding the mixtures to PA-coated, preblocked plates. Plates were incubated overnight at 4°C with diluted sera before washing four times in PBS-0.05% (vol/vol) Tween 20. Plates were incubated for 1 h at room temperature with HRP-conjugated anti-mouse IgG1 at a final concentration of 0.2 μg/ml. Plates were washed and developed for 5 min at room temperature using 90 μl of 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) substrate per well (KPL, Gaithersburg, MD). Reactions were stopped by addition of 110 μl of a 10% (wt/vol) SDS solution. Plates were analyzed using a Dynex MRX Revelation plate reader. Endpoint titers were determined as an optical density (OD) of <0.01 at 405 nm (equivalent to the OD of a 1/200 dilution of prebleed sera). Individual Ab titers were plotted as geometric means using GraphPad Prism software. Differences in Ab titers between two experimental groups were assessed for statistical significance using a nonparametric Mann-Whitney U test. This test was used rather than an analysis of variance because an independent control was performed for each experimental group, as opposed to testing multiple groups against the same control.
Flow cytometry.
Cells were incubated at 4°C or room temperature at a density of 107 cells/ml in RPMI plus 10% fetal calf serum (FCS) with the 2.4G2 MAb at a final concentration of 20 μg/ml. Fluorochrome-conjugated MAbs were added at a 1:100 to a 1:500 dilution as appropriate or with allophycocyanin-conjugated CD1d tetramer at a 1:250 dilution. After 1 h, unbound MAb was removed by washing and centrifugation. Cells were fixed with 1% (wt/vol) paraformaldehyde and analyzed using a Becton-Dickinson FACSCalibur (Palo Alto, CA).
In vitro toxin neutralization assay.
Murine 264.7 macrophages were adjusted to 106 cells/ml in Dulbecco's modified Eagle's medium (DMEM) containing 4 μM l-glutamine, 10% FCS, and 40 μg/ml gentamicin. One hundred thousand cells in a 100-μl volume were then seeded into 96-well culture plates and incubated overnight with 5% CO2. Serial 2-fold dilutions of sera in media were made (1/125 to 1/500) and mixed with lethal toxin at a final concentration of 1 to 16 μg/ml of each subunit. The toxin mixture was then incubated for 1 h at room temperature. Media were then removed from cell cultures and replaced with the serum-toxin-medium mixture. Plates were incubated for a further 2 h at 37°C before addition of cell counting Kit-8 reagents (Dojindo Technologies, Rockville, MD). After 2 h of incubation, plates were read at an absorbance of 450 nm. Differences in Ab titers between two experimental groups were assessed for statistical significance using a nonparametric Mann-Whitney U test. As with the ELISA experiments the Mann-Whitney U test was used because an independent control was performed for each experimental group.
In vivo toxin challenge.
The LF and PA subunits were mixed at a 1:1 molar ratio in PBS. Unless indicated otherwise an amount of lethal toxin equivalent to 200 μg of PA and in a 100-μl volume was then administered by the intravenous (i.v.) paraorbital route to mice. The mice were then monitored daily for the duration of the experiment, and time to death or euthanasia was recorded.
RESULTS
NKT cells regulate anti-PA Ab responses.
C57BL/6, CD1d−/−, and Jα18−/− mice were immunized with PA or PA plus α-GC and later boosted with PA alone before determining anti-PA endpoint titers in serum (Fig. 1). We observed that PA alone stimulated a strong Ab response in C57BL/6 mice and that this was further enhanced 3- to 4-fold by the inclusion of α-GC during the primary immunization (Fig. 1a). Surprisingly, there was a much weaker Ab response in CD1d−/− mice than in C57BL/6 mice immunized with PA alone (Fig. 1a to c). Consistent with the lack of CD1d and NKT cells, α-GC had no effect on the Ab titer. These results were not due to subtly different kinetics of the responses between groups, as the same differences were observed in successive bleeds on days 38 and 43 postimmunization (Fig. 1b). The anti-PA endpoint titers in PA-immunized C57BL/6 and Jα18−/− mice were similar, suggesting that type II cells are responsible for the defect in CD1d−/− mice and confirming that type I cells are responsible for the α-GC-stimulated enhancement (Fig. 1d).
FIG. 1.
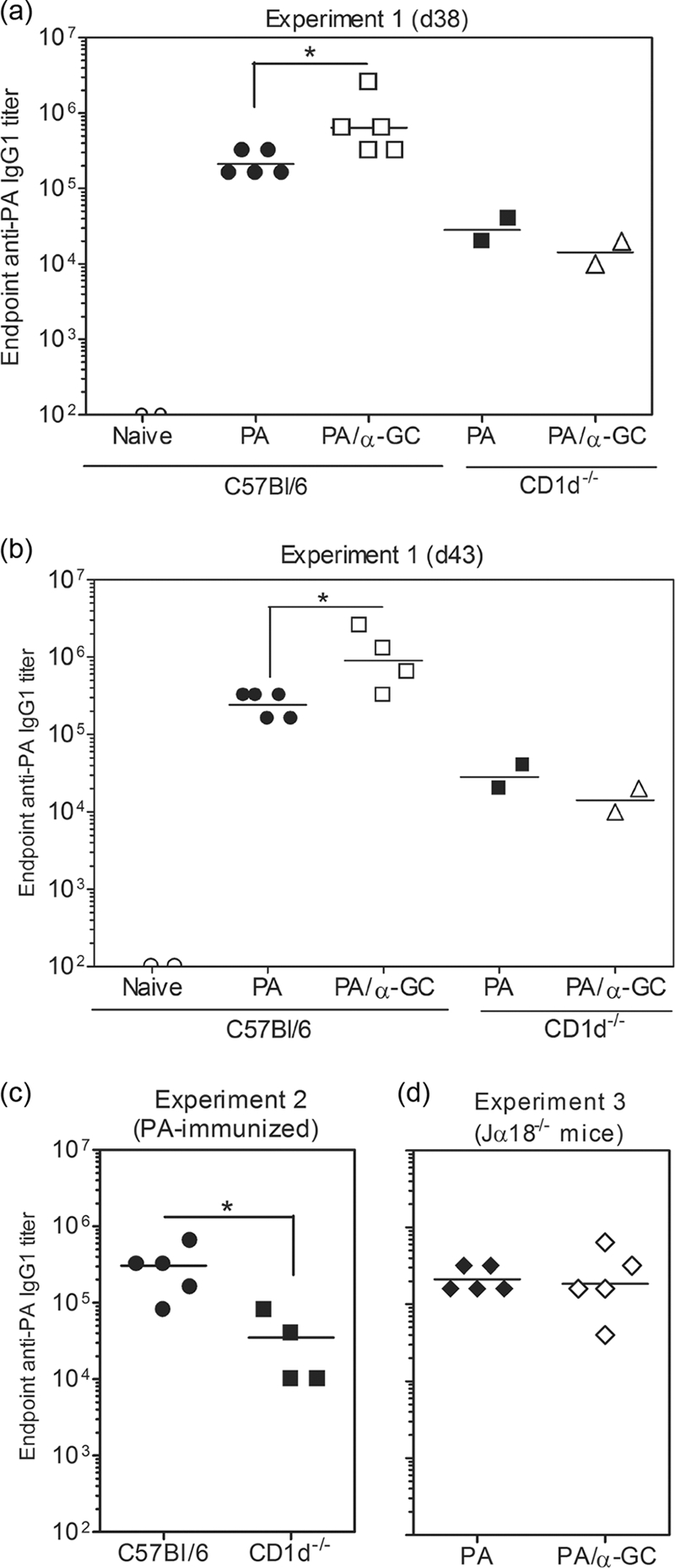
CD1d expression and NKT cells are required for optimal production of PA-specific Abs. C57BL/6, CD1d−/−, and Jα18−/− mice were immunized s.c. with 10 μg of PA or 10 μg of PA plus 2 μg of α-GC in a 100-μl volume of sterile endotoxin-free PBS. Sera were collected before immunization and on day 28 (data not shown). On day 28 mice were then boosted with half the original dose of PA and sera collected again on day 38 (d38) (a) and day 43 (b). ELISAs were performed to determine the endpoint anti-PA IgG1 titer. Data points represent the endpoint titers for individual mice. (c) Since only two CD1d−/− mice were used in the first experiment, additional groups of C57BL/6 and CD1d−/− mice were immunized with PA and analyzed at day 43. (d) Responses in Jα18−/− mice are shown in a third experiment (also day 43 bleeds). Geometric means are shown. Statistically significant differences between experimental groups are indicated by asterisks.
The predominant anti-PA Ab subclass produced was IgG1, and we therefore considered the possibility that the lack of anti-PA IgG1 in CD1d−/− mice could be reflected by an increase in other Ig classes or subclasses. Significant IgG2b and IgG2c responses did not occur in C57BL/6, CD1d−/−, or Jα18−/− mice immunized with PA alone or PA plus α-GC (data not shown). Our results showed that CD1d and type I and/or type II NKT cells contribute to the vaccination-induced anti-PA Ab response. In further controls, we determined if PA directly modulated the activity of NKT cells in our system. Although PA is not generally thought to impact cell physiology, studies have shown that under specific circumstances PA can be cytotoxic (31). At the dosage and routes of immunization described, we did not observe any deleterious effects on NKT cells, B cells, T cells, granulocytes, or dendritic cells (data not shown).
NKT cells contribute to and enhance Ab-mediated LT neutralization.
Sera from the immunized mice were then tested for the ability to neutralize LT in vitro (Fig. 2). We observed across several serum concentrations that type I NKT activation with α-GC led to an enhanced neutralization capacity (Fig. 2a). Sera from CD1d−/− mice immunized with PA plus α-GC had a poor neutralization capacity (Fig. 2a). We also observed in a further experiment that sera from α-GC-treated mice were able to neutralize higher doses of toxin (Fig. 2b). When a final toxin concentration of 16 μg/ml was used in the assay (4- to 8-fold higher than in a typical assay), a greater separation of the PA versus PA plus α-GC groups was observed. There was no apparent neutralization of the higher dose of the toxin by sera from PA-immunized mice, whereas four of five sera from mice immunized with PA plus α-GC showed substantial toxin neutralization.
FIG. 2.

CD1d-dependent NKT activation enhances in vitro lethal toxin neutralization. Sera from experiments with results shown in Fig. 1 were used for in vitro neutralization assays as described in Materials and Methods. Briefly, RAW267.4 macrophages were untreated (none) or incubated with toxin alone or toxin mixed with sera from naïve and immunized mice before assessing viability. Each data point represents the percent cell viability conferred by serum from individual mice. All samples were analyzed in duplicate. Asterisks indicate statistically significant differences between PA-immunized versus PA plus α-GC-immunized groups. (a) Effect of serum dilution on resistance to the toxin. (b) Effect of increasing toxin concentration on neutralization.
Although it is well established that neutralizing Ab is responsible for protection against LT, we performed additional controls to determine if NKT activation was causing any protective effects not attributable to Ab. We immunized B-cell-deficient mice (μMT strain), collected sera, and performed ELISAs, neutralization assays, and in vivo challenges. As expected, there was no measurable Ig or anti-PA IgG or in vitro neutralization activity (data not shown). There was also no in vivo protection against LT, which demonstrated similar lethality as that for naïve C57BL/6 mice (data not shown).
NKT activation enhances in vivo protection against lethal toxin.
We initially determined the lethal dose of LT for our studies, because the C57BL/6 mouse strain is more resistant than some other strains (1). We observed that a 200-μg dose (of each subunit) was sufficient to kill 100% of naïve C57BL/6 mice within 3 to 5 days (Fig. 3a). We therefore challenged C57BL/6 mice and CD1d−/− mice that had previously been immunized and boosted with PA alone (Fig. 3b). Surprisingly, although the anti-PA Ab titers and neutralization capacities of the sera were lower in CD1d−/− and C57BL/6 mice, using death as an end point did not distinguish between the two experimental groups, since all immunized mice survived. However, with daily observations it was clear that C57BL/6 mice appeared much healthier than CD1d−/− mice. The C57BL/6 mice had ruffled fur, a hunched posture, and slower movement for the first 24 h and then proceeded to recover and regain a normal appearance over the next 48 h. The C57BL/6 mice then appeared healthy until euthanasia at the end of the experiment. In contrast, the CD1d−/− mice had similar symptoms that were noticeably worse and also displayed curled forelimbs and a slight tremor. The CD1d−/− mice made a partial recovery over the course of the experiment, but by day 12 they still had slower movement, hunched posture, tremor, and ruffled fur. These data indicate that NKT cells make substantial contributions to Ab responses that maintain the health of the animal following a lethal challenge with toxin.
FIG. 3.

CD1d-dependent activation of NKT cells enhances in vivo protection against lethal toxin. Naïve and immunized mice were challenged i.v. with PA plus LF at the times indicated by arrows. Survival was then monitored daily for the times indicated. (a) Naïve mice, treated with 0, 50, 200, or 300 μg of each toxin subunit. (b) Survival in C57BL/6 and CD1d−/− mice, previously immunized with PA and challenged with 200 μg of each toxin subunit. (c) Survival in C57BL/6 previously immunized with PA alone or PA plus α-GC and challenged with the 200-μg dose on day 0 and then the 100-μg dose on day 1. Numbers of mice per group are indicated.
Given that a fairly low Ab titer correlated with survival but not good health in the CD1d−/− mice, it was anticipated that in vivo challenge with the 200-μg dose would not reveal survival differences between C57BL/6 mice immunized with PA versus PA plus α-GC. In light of this observation and a recent report elegantly demonstrating that multiple doses of toxin could be used to tease apart genetic differences in susceptibility in vivo (22), we performed a challenge experiment using a 200-μg dose of LT on day 0 followed with a second 100-μg dose on day 2 (Fig. 3c). We observed that PA-immunized mice died rapidly, while three of five mice immunized with PA plus α-GC survived. The surviving mice were initially symptomatic and then appeared healthy within 48 h of the second toxin challenge. These data are consistent with the in vitro challenge data (Fig. 2), and this indicates that α-GC leads to increased resistance to higher amounts of LT.
NKT activation leads to a sustained protective antibody response.
We performed experiments to determine if α-GC-activated NKT cells play a role in the development of a sustained protective anti-PA Ab response. We immunized C57BL/6 mice with PA or PA plus α-GC and administered a PA booster on day 28 as described in Materials and Methods. We then collected sera at different time points over a period of 141 days (Fig. 4). Consistent with the results shown in Fig. 1, shortly after the booster vaccine there was a lower anti-PA Ab titer in C57BL/6 mice immunized with PA alone than with PA plus α-GC (Fig. 4a). In both groups the total anti-PA IgG1 titer was sustained for the duration of the experiment. Of note, the absolute Ab titers and neutralization titers were lower than in other experiments because a lower dose of PA (8 μg) was used and a higher dose of α-GC was used (4 μg). This allowed us to better measure the adjuvant effect of α-GC. Furthermore, we had observed a fairly well sustained Ab titer following immunization with PA alone at higher doses (data not shown) and reasoned that a lower dose might lead to a less sustained anti-PA Ab titer that could be boosted with α-GC.
FIG. 4.

α-GC leads to sustained protective anti-PA Ab responses. C57BL/6 mice were immunized with 8 μg of PA alone or PA plus 4 μg of α-GC and then boosted 28 days later with 4 μg of PA alone. Sera were collected at the times indicated before and after booster administration. (a) Endpoint anti-PA IgG1 titers were determined. Data show geometric mean titers (± the 95% confidence interval) for nine mice per group. (b) In vitro neutralization of toxin was assessed at the times shown. Data show the mean percent neutralization for nine mice per group. (c) After 160 days, mice were challenged i.v. with 200 μg of PA and LF on day 0 and 100 μg of PA and LF on day 1 (as indicated by arrows). Survival was then monitored daily. Five mice per group encompassing the range of endpoint anti-PA titers were selected for the challenge experiment. Of the original nine mice, the remaining four mice per group were used for analysis of bone marrow PA-specific plasma cells. However, the sensitivity of the assay was low and the data are not reported here.
There was a gradual decline in the in vitro neutralization of LT by the sera collected over the 141-day period of the experiment (Fig. 4c). However, the samples from the PA/α-GC-immunized mice consistently had a higher neutralizing Ab titer than the samples from the PA-immunized mice. We performed additional analyses on our neutralization assay data, whereby the rate of decay of the neutralizing Ab responses was measured. The mean times required for a 50% drop in neutralization activity were 22 days and 60 days for the PA and PA/α-GC groups, respectively (data not shown). This reinforces the concept that NKT activation leads to a more sustained protective anti-PA Ab response.
We then determined whether PA/α-GC immunization could lead to sustained protection against LT in vivo (Fig. 4c). In this experiment, 160 days after the initial immunization, PA/α-GC-immunized mice did not succumb to the lethal challenge, whereas every mouse in the PA-immunized group died within 4 days of receiving LT. These data show that activation of type I NKT cells with α-GC at the time of the primary immunization leads to the development of a sustained protective Ab response in vivo.
DISCUSSION
We have demonstrated for the first time that CD1d-restricted NKT cells are important for the generation of specific protective Ab responses against B. anthracis LT. Our data showed that coadministration of a CD1d-binding glycolipid (α-GC) which activates type I NKT cells led to a 4-fold increase in the endpoint PA-specific serum Ab titer. The α-GC molecule was unable to increase Ab titers in CD1d−/− mice lacking type I and type II NKT cells and Jα18−/− mice lacking type I NKT cells. These data indicate that foreign CD1d-binding glycolipids may have utility as adjuvants for inducing protective Ab titers against LT. This may be particularly important for the current anthrax vaccine, for which multiple boosters are required to maintain protection. In our study, one immunization and one booster elicited sustained in vivo protection against a lethal challenge with LT.
Efforts are under way to understand the mechanisms by which NKT cells enhance Ab responses and to understand how this differs from the stimulation provided by CD4+ helper T cells (Th). PA immunization leads to Th-dependent Ab responses (15, 25). Th cells utilize cytokine- and CD154-dependent mechanisms to drive the differentiation of B cells into Ab-secreting cells via cytokine receptor and CD40 signaling, respectively (23). This process is known broadly as B-cell help. We have demonstrated that vaccination with PA/α-GC leads to a superior and durable PA-protective Ab response compared to PA immunization, suggesting that there could be mechanistic differences between NKT- and Th-mediated B-cell help. At present, researchers do not sufficiently understand the mechanisms by which α-GC-stimulated type I NKT cells help B cells to be able to make an adequate comparison with Th cells.
However, we and others have shown that cognate interactions between CD1d+/+ B cells and NKT cells are required for α-GC-enhanced Ab responses (2, 16, 19). Therefore, B cells that present PA-derived peptides on MHC class II likely have to copresent α-GC on CD1d to provide a coordinated activation of CD4+ Th cells and NKT cells. This potentially explains why α-GC/NKT-enhanced Ab responses are Ag specific and indicates that α-GC could be a useful adjuvant for boosting PA-specific Ab responses.
One group has also suggested that NKT-derived gamma interferon (IFN-γ) and interleukin-4 (IL-4) could be important for α-GC-enhanced Ab responses (11). However, NKT-derived cytokines were not examined specifically, because knockout mice in which all T cells lacked IFN-γ and IL-4 were used. Nonetheless, this raises the intriguing possibility that NKT cells provide direct help to PA-specific B cells via cytokine signals. There is also evidence that CD154-expressing NKT cells mediate help for Ab production via cognate interactions with B cells expressing CD40. In our previous studies we showed that a combination of T-dependent Ag, α-GC, and an agonistic anti-CD40 MAb could induce a limited specific Ab response in class II null mice (17). Another group recently confirmed our findings (11) but as yet, NKT-specific CD154 signals have not been examined.
The studies herein demonstrate that NKT activation programs the B-cell response to lead to durable protective Ab responses. Although there is likely some commonality in the mechanism of B-cell help with that provided by Th cells, there are logically some differences, given the distinct immunological outcomes. Future studies will determine how Th-derived B-cell help differs from Th/NKT-derived B-cell help, and use of PA as the immunizing Ag may help determine the mechanisms.
When no adjuvant was added to the vaccine, PA induced a robust specific Ab response, in accordance with the findings of several other investigators (1, 21, 25, 26, 28, 33). While the Ab response against PA alone was defective in CD1d−/− mice, in Jα18−/− mice the Ab response was comparable to the C57BL/6 control strain. This implicates type II NKT cells in the induction of specific Ab responses against PA. Since no foreign CD1d ligand was added, a self-CD1d Ag could be directing anti-PA Ab responses via type II NKT cells. Few self-Ags for CD1d have been identified but include sulfatide, which can activate type II NKT cells (12). However, a mechanism by which type II NKT cells contribute to humoral immunity is presently unclear. An alternative explanation for the defect in anti-PA Ab responses in CD1d−/− mice is that type II NKT cells make a contribution to the development and/or function of the B-cell compartment. Compared to C57BL/6 mice we have observed up to 50% fewer numbers of a bone marrow-resident cell population in CD1d−/− mice that is consistent with plasma cell precursors (H. B. Shah and M. L. Lang, unpublished observation). However, two observations argue against the reduced plasma cell precursor population as an explanation for our present results. First, the anti-PA Ab response is 10-fold lower in CD1d−/− mice than in C57BL/6 mice. Secondly, our earlier findings showed that if the NKT pathway is bypassed by adsorption of foreign Ags to alum adjuvant, then good Ab titers can be achieved in CD1d−/− mice (8). Future studies will therefore explore the contribution of type II NKT cells to anti-PA Ab responses.
Importantly, the sera obtained from C57BL/6 mice previously immunized with PA plus α-GC were able to neutralize higher concentrations of lethal toxin in vitro than sera from mice previously immunized with PA alone. This shows that an enhanced protective Ab response is induced by immunization with PA and CD1d-binding glycolipid Ags. Conversely, sera from CD1d−/− mice did not provide good in vitro neutralization of LT. Therefore, the total anti-PA Ab titer was generally indicative of in vitro neutralization titers. Since sera from CD1d−/− mice did not effectively neutralize LT in vitro, this suggests that as well as lower amounts of anti-PA Ab, the affinity of the Ab could be important. While we previously reported that CD1d−/− mice had a lower proportion of high-affinity Ab than C57BL/6 controls when a model antigen (nitrophenol-conjugated keyhole limpet hemocyanin) was used (8), we did not observe differences in the proportion of high-affinity anti-PA Ab by ELISA (data not shown). NKT cells could play a role in anti-PA Ab affinity maturation that is not detectable in our assay, and investigation of Ab hypermutation in future studies is warranted.
For reasons that are not entirely clear, the total anti-PA Ab was sustained over a 141-day period, yet in vitro neutralization showed a moderate decline. We suggest that a small decline in anti-PA Ab that is not detectable in the ELISA is more detectable in the in vitro assay. The in vivo protection against lethal challenge with LT was sustained over at least the same period. This suggests that anti-PA Ab that recruits effector functions in vivo contributes to sustained protection. A small decline in the amount of Ab may be more important for the in vitro assay, which does not utilize effector functions to prevent toxin activity on target cells.
Our most striking finding was that type I NKT activation with α-GC led to sustained protection in vivo against LT. While there was some resistance among PA-immunized C57BL/6 and CD1d−/− mice challenged with a single dose of LT, a second dose of LT revealed better protection in PA/α-GC-immunized C57BL/6 mice. When we used this method, PA-immunized mice succumbed rapidly to LT, while PA/α-GC-immunized mice were resistant. This effect was apparent 7 days after the vaccination course was complete but even more apparent on day 141 postimmunization. This represents a significant advance in the understanding of how to generate more sustained Ab-mediated protection against LT and may contribute to overcoming the major limitation of the current vaccine. This could be important in the clinical setting. Apart from practical considerations, a shorter vaccination protocol among at-risk individuals will likely increase compliance. More-sustained protection against B. anthracis and fewer vaccination-associated inflammatory responses may also be achieved.
Acknowledgments
This work was supported by Defense Threat Reduction Agency (DTRA) contract HDTRA1-08-C-0013 to M.L.L.
We acknowledge generous support from the NIAID Tetramer Facility (Emory University, Atlanta, GA) for supplying CD1d tetramers. We thank D. Farris and M. Nguyen of the Oklahoma Medical Research Foundation (Oklahoma City, OK) for advice on the neutralization assays. We are grateful to M. Kronenberg of the La Jolla Institute for Allergy and Immunology (La Jolla, CA) for the kind gift of the Jα18−/− mice and to M. Taniguchi of the RIKEN Institute (Riken, Japan) for providing permission to obtain and use the mice.
L.A., T.S.D., G.A.L., and S.K.J. performed experiments and analyzed data. J.D.B. directed and supervised the expression and purification of PA and LF by L.A., analyzed data, and contributed to writing the paper. M.L.L. designed and directed the project, performed experiments, analyzed data, and wrote the paper.
We declare no competing financial interests.
Editor: R. P. Morrison
Footnotes
Published ahead of print on 1 February 2010.
REFERENCES
- 1.Abboud, N., and A. Casadevall. 2008. Immunogenicity of Bacillus anthracis protective antigen domains and efficacy of elicited antibody responses depend on host genetic background. Clin. Vaccine Immunol. 15:1115-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barral, P., J. Eckl-Dorna, N. E. Harwood, C. De Santo, M. Salio, P. Illarionov, G. S. Besra, V. Cerundolo, and F. D. Batista. 2008. B cell receptor-mediated uptake of CD1d-restricted antigen augments antibody responses by recruiting invariant NKT cell help in vivo. Proc. Natl. Acad. Sci. U. S. A. 108:8345-8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belperron, A. A., C. M. Dailey, and L. K. Bockenstedt. 2005. Infection-induced marginal zone B cell production of Borrelia hermsii-specific antibody is impaired in the absence of CD1d. J. Immunol. 174:5681-5686. [DOI] [PubMed] [Google Scholar]
- 4.Boyaka, P. N., A. Tafaro, R. Fischer, S. H. Leppla, K. Fujihashi, and J. R. McGhee. 2003. Effective mucosal immunity to anthrax: neutralizing antibodies and Th cell responses following nasal immunization with protective antigen. J. Immunol. 170:5636-5643. [DOI] [PubMed] [Google Scholar]
- 5.Bradley, K. A., J. Mogridge, M. Mourez, R. J. Collier, and J. A. Young. 2001. Identification of the cellular receptor for anthrax toxin. Nature 414:225-229. [DOI] [PubMed] [Google Scholar]
- 6.Collier, R. J., and J. A. Young. 2003. Anthrax toxin. Annu. Review Cell Dev. Biol. 19:45-70. [DOI] [PubMed] [Google Scholar]
- 7.Cui, J., T. Shin, T. Kawano, H. Sato, E. Kondo, I. Toura, Y. Kaneko, H. Koseki, M. Kanno, and M. Taniguchi. 1997. Requirement for Vα14 NKT cells in IL-12-mediated rejection of tumors. Science 278:1623-1626. [DOI] [PubMed] [Google Scholar]
- 8.Devera, T. S., H. B. Shah, G. A. Lang, and M. L. Lang. 2008. Glycolipid-activated NKT cells support the induction of persistent plasma cell responses and antibody titers. Eur. J. Immunol. 38:1001-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Exley, M. A., N. J. Bigley, O. Cheng, A. Shaulov, S. M. Tahir, Q. L. Carter, J. Garcia, C. Wang, K. Patten, H. F. Stills, F. W. Alt, S. B. Snapper, and S. P. Balk. 2003. Innate immune response to encephalomyocarditis virus infection mediated by CD1d. Immunology 110:519-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fink, S. L., T. Bergsbaken, and B. T. Cookson. 2008. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. U. S. A. 105:4312-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galli, G., P. Pittoni, E. Tonti, C. Malzone, Y. Uematsu, M. Tortoli, D. Maione, G. Volpini, O. Finco, S. Nuti, S. Tavarini, P. Dellabona, R. Rappuoli, G. Casorati, and S. Abrignani. 2007. Invariant NKT cells sustain specific B cell responses and memory. Proc. Natl. Acad. Sci. U. S. A. 104:3984-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jahng, A., I. Maricic, C. Aguilera, S. Cardell, R. C. Halder, and V. Kumar. 2004. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J. Exp. Med. 199:947-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ko, S. Y., H. J. Ko, W. S. Chang, S. H. Park, M. N. Kweon, and C. Y. Kang. 2005. α-Galactosylceramide can act as a nasal vaccine adjuvant inducing protective immune responses against viral infection and tumor. J. Immunol. 175:3309-3317. [DOI] [PubMed] [Google Scholar]
- 14.Kronenberg, M. 2005. Toward an understanding of NKT cell biology: progress and paradoxes. Annu. Rev. Immunol. 23:877-900. [DOI] [PubMed] [Google Scholar]
- 15.Kwok, W. W., J. Yang, E. James, J. Bui, L. Huston, A. R. Wiesen, and M. Roti. 2008. The anthrax vaccine adsorbed vaccine generates protective antigen (PA)-specific CD4+ T cells with a phenotype distinct from that of naive PA T cells. Infect. Immun. 76:4538-4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang, G. A., T. S. Devera, and M. L. Lang. 2008. Requirement for CD1d expression by B cells to stimulate NKT cell-enhanced antibody production. Blood 111:2158-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lang, G. A., M. A. Exley, and M. L. Lang. 2006. The CD1d-binding glycolipid alpha-galactosylceramide enhances humoral immunity to T-dependent and T-independent antigen in a CD1d-dependent manner. Immunology 119:116-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang, G. A., S. D. Maltsev, G. S. Besra, and M. L. Lang. 2004. Presentation of alpha-galactosylceramide by murine CD1d to natural killer T cells is facilitated by plasma membrane glycolipid rafts. Immunology 112:386-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leadbetter, E. A., M. Brigl, P. Illarionov, N. Cohen, M. C. Luteran, S. Pillai, G. S. Besra, and M. B. Brenner. 2008. NK T cells provide lipid antigen-specific cognate help for B cells. Proc. Natl. Acad. Sci. U. S. A. 105:8339-8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leppla, S. H. 1984. Bacillus anthracis calmodulin-dependent adenylate cyclase: chemical and enzymatic properties and interactions with eucaryotic cells. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 17:189-198. [PubMed] [Google Scholar]
- 21.Little, S. F., B. E. Ivins, P. F. Fellows, M. L. Pitt, S. L. Norris, and G. P. Andrews. 2004. Defining a serological correlate of protection in rabbits for a recombinant anthrax vaccine. Vaccine 22:422-430. [DOI] [PubMed] [Google Scholar]
- 22.Liu, S., D. Crown, S. Miller-Randolph, M. Moayeri, H. Wang, H. Hu, T. Morley, and S. H. Leppla. 2009. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. U. S. A. 106:12424-12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McHeyzer-Williams, L. J., and M. G. McHeyzer-Williams. 2005. Antigen-specific memory B cell development. Annu. Rev. Immunol. 23:487-513. [DOI] [PubMed] [Google Scholar]
- 24.Mock, M., and T. Mignot. 2003. Anthrax toxins and the host: a story of intimacy. Cell. Microbiol. 5:15-23. [DOI] [PubMed] [Google Scholar]
- 25.Park, Y. S., J. H. Lee, C. F. Hung, T. C. Wu, and T. W. Kim. 2008. Enhancement of antibody responses to Bacillus anthracis protective antigen domain IV by use of calreticulin as a chimeric molecular adjuvant. Infect. Immun. 76:1952-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peachman, K. K., M. Rao, C. R. Alving, R. Burge, S. H. Leppla, V. B. Rao, and G. R. Matyas. 2006. Correlation between lethal toxin-neutralizing antibody titers and protection from intranasal challenge with Bacillus anthracis Ames strain spores in mice after transcutaneous immunization with recombinant anthrax protective antigen. Infect. Immun. 74:794-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Persson, U., and E. Moller. 1975. The effect of lipopolysaccharide on the primary immune response to the hapten NNP. Scand. J. Immunol. 4:571-581. [DOI] [PubMed] [Google Scholar]
- 28.Pittman, P. R., S. F. Leitman, J. G. Oro, S. L. Norris, N. M. Marano, M. V. Ranadive, B. S. Sink, and K. T. McKee, Jr. 2005. Protective antigen and toxin neutralization antibody patterns in anthrax vaccinees undergoing serial plasmapheresis. Clin. Diagn. Lab. Immunol. 12:713-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinn, C. P., P. M. Dull, V. Semenova, H. Li, S. Crotty, T. H. Taylor, E. Steward-Clark, K. L. Stamey, D. S. Schmidt, K. W. Stinson, A. E. Freeman, C. M. Elie, S. K. Martin, C. Greene, R. D. Aubert, J. Glidewell, B. A. Perkins, R. Ahmed, and D. S. Stephens. 2004. Immune responses to Bacillus anthracis protective antigen in patients with bioterrorism-related cutaneous or inhalation anthrax. J. Infect. Dis. 190:1228-1236. [DOI] [PubMed] [Google Scholar]
- 30.Rivera, J., A. Nakouzi, N. Abboud, E. Revskaya, D. Goldman, R. J. Collier, E. Dadachova, and A. Casadevall. 2006. A monoclonal antibody to Bacillus anthracis protective antigen defines a neutralizing epitope in domain 1. Infect. Immun. 74:4149-4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salles, I. I., D. E. Voth, S. C. Ward, K. M. Averette, R. K. Tweten, K. A. Bradley, and J. D. Ballard. 2006. Cytotoxic activity of Bacillus anthracis protective antigen observed in a macrophage cell line overexpressing ANTXR1. Cell. Microbiol. 8:1272-1281. [DOI] [PubMed] [Google Scholar]
- 32.Taniguchi, M., M. Harada, S. Kojo, T. Nakayama, and H. Wakao. 2003. The regulatory role of Vα14 NKT cells in innate and acquired immune response. Annu. Rev. Immunol. 21:483-513. [DOI] [PubMed] [Google Scholar]
- 33.Welkos, S., S. Little, A. Friedlander, D. Fritz, and P. Fellows. 2001. The role of antibodies to Bacillus anthracis and anthrax toxin components in inhibiting the early stages of infection by anthrax spores. Microbiology 147:1677-1685. [DOI] [PubMed] [Google Scholar]