

Abstract
The human NatA protein Nα-terminal-acetyltransferase complex is responsible for cotranslational N-terminal acetylation of proteins with Ser, Ala, Thr, Gly, and Val N termini. The NatA complex is composed of the catalytic subunit hNaa10p (hArd1) and the auxiliary subunit hNaa15p (hNat1/NATH). Using immunoprecipitation coupled with mass spectrometry, we identified endogenous HYPK, a Huntingtin (Htt)-interacting protein, as a novel stable interactor of NatA. HYPK has chaperone-like properties preventing Htt aggregation. HYPK, hNaa10p, and hNaa15p were associated with polysome fractions, indicating a function of HYPK associated with the NatA complex during protein translation. Knockdown of both hNAA10 and hNAA15 decreased HYPK protein levels, possibly indicating that NatA is required for the stability of HYPK. The biological importance of HYPK was evident from HYPK-knockdown HeLa cells displaying apoptosis and cell cycle arrest in the G0/G1 phase. Knockdown of HYPK or hNAA10 resulted in increased aggregation of an Htt-enhanced green fluorescent protein (Htt-EGFP) fusion with expanded polyglutamine stretches, suggesting that both HYPK and NatA prevent Htt aggregation. Furthermore, we demonstrated that HYPK is required for N-terminal acetylation of the known in vivo NatA substrate protein PCNP. Taken together, the data indicate that the physical interaction between HYPK and NatA seems to be of functional importance both for Htt aggregation and for N-terminal acetylation.
Nα-terminal acetylation is among the most common protein modifications in eukaryotes, occurring on ∼50% of Saccharomyces cerevisiae proteins and ∼80% of human proteins (12). In yeast, four types of Nα-terminal acetyltransferases (NATs) have been defined (NatA-NatD), while a fifth type, NatE, has been hypothesized (21, 32-34, 38). For humans, NatA, NatB, NatC, and NatE were recently presented (2, 4, 18, 39, 40). A revised NAT-subunit nomenclature was recently introduced in order to have identical names for orthologous subunits from different species, and each gene was denoted NAA (Nα-acetyltransferase) followed by a number depending on Nat type and the type of subunit (catalytic/auxiliary) (32). The major human NAT complex, hNatA, is composed of the catalytic subunit hNaa10p (previously named hArd1) and the auxiliary subunit hNaa15p (hNat1/NATH) (4). Human NatA is evolutionarily conserved from the yeast complex in terms of subunit composition and substrate specificity (12, 26, 28). However, in contrast to yeast cells, human cells potentially contain several distinct NatA complexes due to the presence of two genes for each of the two NatA subunits, NAA10 and NAA15 (6, 8). Protein N-terminal acetylation occurs on the ribosome when the nascent polypeptide emerges (21, 29, 30, 41, 42). Proteins with Ser, Thr, Gly, Ala, Val, or Cys N termini are potential substrates of NatA (12), while NatB and NatC potentially acetylate specific classes of substrates that still carry the initiator Met (34). The biological importance of the human NatA complex was evident from knockdown experiments where induction of apoptosis and growth arrest of cells in the G1/G0 phase were the resulting phenotypes (9, 11, 20, 25). The phenotypes induced by hNatA depletion most likely reflect the fact that one or more specific substrate proteins lack proper Nα acetylation, in view of the fact that a large quantitative proteomic analysis of the acetylation status of protein N termini in hNaa15p-hNaa10p knockdown cells revealed a decrease in the level of Nα acetylation of some partially acetylated substrates compared to that in control cells (12).
To further characterize the human NatA complex, we looked for the presence of stable interaction partners of hNaa15p and hNaa10p. Here we present data identifying the Huntingtin (Htt) yeast two-hybrid protein K (HYPK) as a novel factor involved in cotranslational NatA acetylation. HYPK, originally identified in a yeast two-hybrid screen during a search for potential interaction partners for the Huntingtin protein (19), was recently found to reduce Htt polyglutamine (polyQ) aggregation upon overexpression (36). However, the role of the endogenous HYPK protein has yet to be revealed. We demonstrate that endogenous HYPK (i) stably interacts with the hNaa10p-hNaa15p NatA N-terminal-acetyltransferase complex and with ribosomes, (ii) is required for normal N-terminal acetylation of a NatA substrate, (iii) is important for cell survival independent of Htt polyQ, and (iv) is important for the prevention of Htt polyQ aggregation. Furthermore, NatA is essential for the proper expression of HYPK protein and modulates Htt polyQ aggregation.
MATERIALS AND METHODS
Cell culture and transfections.
HEK293 cells (human embryonal kidney) (ATCC CRL-1573) and HeLa cells (human epithelial cervix adenocarcinoma) (ATCC CCL-2) were cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 3% l-glutamine. Plasmid transfections were performed using Fugene6 (Roche) according to the instructions in the manual. RNA interference (RNAi) experiments were performed by using 50 nM gene-specific small interfering RNA (siRNA) (Dharmacon) and Dharmafect 1 according to the supplier's manuals. To specifically inhibit caspase activities, 20 μM ZVAD-fmk (R&D Systems Europe Ltd.) was added whenever indicated. The following siRNA duplex target sequences were used: sihNAA10, CCA GAU GAA AUA CUA CUU C; siNAA15, GGG ACC UUU CCU UAC UAC A; siLamin A/C, GGU GGU GAC GAU CUG GGC U; siGAPDH, UGG UUU ACA UGU UCC AAU AUU; siHYPK-1, AAA UAG AUA UGG CGA CCG A; siHYPK-2, AAG AUC UGG AGC UAA UAA U; siHYPK-3, CUA GGA GCC ACU AUA UAU A; siHYPK-4, GAG AAG CCA CGG AAA CAU GUU; and siHYPK-5, UGG AGCGGG UCA CCG ACU AUU. When siHYPK is indicated, an equal mixture of siHYPK-1, siHYPK-2, siHYPK-4, and siHYPK-5 was used. siCtr represents the nontargeting pool.
RT-PCR.
Isolation of mRNA and reverse transcription-PCR (RT-PCR) were performed as described previously (7). Primers used for detection of specific genes will be provided upon request.
Western blotting.
SDS gel electrophoresis and Western blotting were performed as described previously (7). For HYPK detection, 15% polyacrylamide gels were used and blotting was performed at 250 mA for 20 min. Polyclonal rabbit antibodies (1 μg/μl) against hNaa10p (anti-hARD1) and hNaa15p (anti-NATH) (4) were used at a 1:1,000 dilution; anti-β-tubulin (Sigma) was used at a 1:1,000 dilution. The immunogens correspond to amino acids (aa) 853 to 866 of hNaa15p and aa 204 to 217 of hNaa10p. A rabbit anti-HYPK polyclonal antibody directed toward the HYPK-specific peptide (aa 24 to 37) C-SGPERPPEKPRKHD was generated by Biogenes. The affinity-purified antibody (2 μg/μl) was used at a 1:400 dilution. Horseradish peroxidase-linked anti-mouse and anti-rabbit antibodies were from Amersham Life Science, United Kingdom.
Construction of plasmids.
Plasmids encoding Xpress- and/or V5-tagged HYPK and PCNP (PEST proteolytic signal-containing nuclear protein; GenBank accession no. Q8WW12) were constructed from cDNA made from total RNA isolated from human HEK293 cells. Superscript II reverse transcriptase (Invitrogen) was used to make cDNA. PCR products were inserted into the Topo TA vectors pcDNA 3.1/V5-His Topo and pcDNA4/HisMax-Topo (Invitrogen) according to the instructions in the manufacturer's manual. It should be noted that the HYPK variant used in all our studies is the 129-aa isoform (GenBank accession no. NP_057484) listed as the NCBI reference sequence for the HYPK gene (GeneID 25764). A larger isoform of 175 aa, which encompasses the 129-aa variant plus additional amino acids at the N terminus, has also been reported (GenBank accession no. Q9NX55), but we have found no evidence of its existence in our analyses. Furthermore, a 121-aa variant with an alternative translation start site compared to that of the 129-aa variant (at aa 9; see Fig. S1a [MATE−] in the supplemental material) potentially exists (GenBank accession no. AAF29100). Plasmids encoding V5-tagged hNaa15p, hNaa25p, and hNaa35p were previously described (4, 39, 40). Plasmids encoding Huntingtin polyQ-enhanced green fluorescent protein (EGFP) fusion proteins (pHttQ25-EGFP, pHttQ72-EGFP, and pHttQ103-EGFP) (24, 46) were generously provided by Evan Eisenberg, National Heart, Lung and Blood Institute, Bethesda, MD. Plasmids encoding EGFP-Huntingtin polyQ fusion proteins (pEGFP-HttQ23 and pEGFP-HttQ74) (45) were generously provided by David C. Rubinsztein, University of Cambridge, United Kingdom. These Htt-polyQ-encoding plasmids were chosen for our experiments because they were already functionally tested and available. The differences in polyQ length (Q23 versus Q25 and Q74 versus Q72) between the C-terminal Htt-polyQ fusions and the N-terminal Htt-polyQ fusions were not anticipated to have any significant impact on the results. Plasmids encoding MBP-hNaa15p, MBP-hNaa10p, MBP-hNaa50p, and MBP-HYPK were constructed by subcloning hNAA15, hNAA10, hNAA50, and HYPK from phNAA15-V5, phNAA10-V5, phNAA50-V5, and pHYPK-V5, respectively, into the pETM-41 vector. pETM-41 was generously provided by G. Stier, EMBL, Heidelberg, Germany. Primers used for (sub)cloning will be provided upon request.
E. coli protein expression, purification, and in vitro interaction assay.
The various MBP-hNaa15p fusion proteins, as well as MBP-hNaa10p, MBP-hNaa50p, and MBP-HYPK, were expressed and purified as described previously, using Ni-affinity columns and size-exclusion chromatography (10). Efficient expression of MBP-hNaa15p required coexpression of pDC952 (a plasmid carrying the Escherichia coli argU gene), kindly provided by James Walker, University of Texas, Austin, TX. For the interaction assays, MBP-hNaa15p fusions, MBP-hNaa10p, or MBP-hNaa50p was bound to Ni-affinity beads. HYPK was separated from maltose binding protein (MBP) by AcTEV protease (Invitrogen) treatment and subsequently added to the beads in interaction buffer (5 mM dithiothreitol [DTT], 50 mM Tris-HCl, pH 7.4, 200 mM NaCl). The beads were washed several times with interaction buffer containing decreasing amounts of bovine serum albumin (BSA) (0.5% to 0.1%). Finally, the beads were added to the sample buffer and analyzed by SDS-PAGE and Western blotting, using anti-HYPK.
Immunofluorescence.
Cells grown on coverslips were washed in phosphate-buffered saline (PBS) and fixed for 20 min in 4% formaldehyde. Fixed cells were washed three times in PBS, permeabilized in 0.1% Triton X-100 for 10 min, and then incubated in blocking buffer (10% BSA) for 30 min. Cells were then incubated for 1 h with primary antibody diluted in 2% BSA. The antibodies were anti-Xpress (Invitrogen) and anti-hNaa15p, used at a dilution of 1:200, and anti-HYPK, used at a dilution of 1:100. Cells were washed at least three times for 5 min in wash buffer (0.01% Tween 20 in PBS) and then incubated with the fluorochrome-conjugated secondary antibody for 40 min (Alexa-conjugated antibodies; Molecular Probes, Eugene, OR). Cells were washed in washing buffer and, finally, distilled water (dH2O) before being mounted on glass slides in Vectashield H1200 (Vector Laboratories). Examination was performed using a Leica DMRXA microscope with a Leica DC500 camera and a Leica HCX PL Apo 100× oil immersion objective.
Immunoprecipitation.
HEK293 cells were harvested and lysed in 300 μl lysis buffer (50 mM Tris, pH 8, 50 mM NaCl, 0.5% NP-40, 5 mM EDTA, 5 μM trichostatin A [Sigma], 1 mM Na3VO4, 1 mM Pefabloc [Roche]). Typically, 2 × 106 cells were used. Preclearing of the lysates was performed by the addition of 40 μl protein A/G agarose and subsequent incubation for 1 h at 4°C. After centrifugation at 3,000 × g for 2 min, the supernatants were collected and incubated for 16 h at 4°C with a specific antibody (3 μg). The samples were centrifuged as described above, and 40 μl protein A/G agarose was added to the supernatants. After incubation for another 16 h, centrifugation, and three washing steps using lysis buffer, the samples were subjected to SDS-PAGE and Western blotting.
Mass spectrometry for identification of NatA interactors.
For two-dimensional liquid chromatography-tandem mass spectrometry (2D LC-MS/MS) analysis of tryptic peptides, we used a ThermoFinnigan LTQ-FT hybrid linear-ion-trap-Fourier transform ion cyclotron resonance (FTICR) mass spectrometer or an LCQ Deca XP Plus 3D ion-trap mass spectrometer with an Agilent 1100 Nanoflow high-performance liquid chromatograph (HPLC). Data analysis utilized SEQUEST (16), an upgraded version of the Medusa database and software (22), called MASSIEVE, and support vector machine learning (3). For the preparation of samples for LC-MS/MS analysis, the immunoprecipitation protocol was followed, with the following exceptions: approximately 5 × 107 cells were used per sample, 1 ml lysis buffer was added to the cell pellet, and 100 μl protein A/G-agarose and 10 μg of antibody were used. The pellets of protein A/G agarose-antibody with the protein complexes were washed three times in PBS and two times in TEN buffer (0.02 M Tris-HCl, pH 7.5, 1 mM EDTA, 0.15 M NaCl). The beads were resuspended in 100 μl TEN buffer, and 140 μl extraction solution (8 M urea, 20 mM methylamine, 1 M LiCl, 2 mM EDTA in 100 mM Tris-HCl, pH 8.5) was added. After incubation for 30 min at room temperature, the samples were centrifuged for 2 min at 13,000 rpm. Two microliters of 0.5 M DTT was added to the supernatants, which were then incubated for 2 h at room temperature, after which 6 μl of 0.5 M iodoacetamide was added, followed by an incubation step for another 2 h, with protection from light. Another 2 μl of 0.5 M DTT was added, and after 1 h of incubation, 90 μl dH2O was added to the mixture. LysC endoprotease (Sigma) was added at a concentration ratio of 1:50 (wt/wt) relative to the total protein content of the sample. After 15 h of incubation at 37°C, 200 μl 10 mM CaCl2 (pH 8.5) was added. Trypsin (Sigma) was added at a 1:10 (wt/wt) ratio relative to the total protein content. After 10 h of incubation at 37°C, 2 μl acetic acid was added. Tryptic digests were desalted, and the peptides were purified by solid-phase extraction using C18 reversed-phase cartridges (Vydac Inc., Hesperia, CA) on an Agilent 1100 HPLC system. For 2D LC-MS/MS analysis of tryptic peptides, the ammonium acetate salt steps for the LTQ-FT instrument were 0 mM (spectra collected during sample loading), 25 mM, 100 mM, and 500 mM. Each salt step was followed by elution of the C18 resin with a gradient of acetonitrile. After the last salt step, a final wash of the C18 resin in the 2D column was performed (with data collection), using 90% acetonitrile-0.1% formic acid. Due to the longer duty cycle for the LCQ machine, more salt steps were included in the first-dimension LC separation. The ammonium acetate salt steps were 0 mM, 25 mM, 50 mM, 100 mM, 250 mM, and 500 mM, with a final wash with 90% acetonitrile-0.1% formic acid. Control IgG-affinity extractions were analyzed in the same fashion.
Cell cycle analysis.
After harvesting of approximately 1 × 106 cells by trypsin-EDTA treatment, the cells were processed by fluorescence-activated cell sorter (FACS) analysis as described previously (6).
Isolation of polysomes.
Total ribosome isolation was performed using a modification of previously described methods (31, 43). Approximately 2 × 107 HEK293 cells were used per experiment. Prior to harvest, cells were treated with 10 μg/ml cycloheximide (CHX) for 5 min at 37°C. Cells were harvested, lysed with KCl-containing ribosome lysis buffer (1.1% [wt/vol] KCl, 0.15% [wt/vol] triethanolamine, 0.1% [wt/vol] magnesium acetate, 8.6% [wt/vol] sucrose, 0.05% [wt/vol] sodium deoxycholate, 0.5% [vol/vol] Triton X-100, 0.25% [vol/vol] Pefabloc), and incubated on ice for 15 min. After removing the nuclear and membrane-containing fractions by centrifugation at 400 × g for 10 min, 700 μl cell lysate was ultracentrifuged at 436,000 × g for 25 min on a 0.4-ml cushion of 25% sucrose in KCl ribosome lysis buffer, using an MLA-130 rotor (Beckman, Geneva, Switzerland). Pellets were resuspended in ribosome lysis buffer with the indicated KCl concentrations, followed by ultracentrifugation as described above. Pellets were resuspended in KCl ribosome lysis buffer and prepared for analysis by SDS-PAGE and Western blotting.
Huntingtin aggregation analysis.
HeLa cells were grown on coverslips and transfected with siRNAs as indicated above. After 24 h, cells were transfected with plasmids encoding Huntingtin polyQ-EGFP fusion proteins. After an additional 48 h, Hoechst 33342 was added to the cells, which were then fixed after 15 min of incubation and further processed as described above (see “Immunofluorescence”), with the antibody incubation steps omitted. A final concentration of 20 μM ZVAD-fmk (R&D Systems Europe Ltd.) was added at 24 and 48 h to prevent apoptosis. Cells were analyzed by counting 200 transfected cells from each sample per experiment (at least three independent experiments per sample). The number of transfected cells displaying Htt-EGFP aggregates was counted, and the fraction of transfected cells displaying aggregation was calculated for each sample.
Determination of N-terminal-acetylation status.
SDS-PAGE-separated and Coomassie-stained gel bands were excised, washed for 15 min with water, twice with water-acetonitrile (1/1), and finally with acetonitrile, and then dried in vacuo. Gel samples were rehydrated, and primary amines were trideutero-acetylated by the addition of 1 mg N-hydroxysuccinimide trideutero-acetate (prepared according to the method described in reference 23) to each sample for 1 h at 30°C in 100 mM sodium phosphate buffer, pH 8.5. Samples were subsequently washed for 15 min with water and acetonitrile and then vacuum dried. The trideutero-acetylation, washing, and drying steps were repeated twice. Gel pieces were then washed three times (15 min each) with digestion buffer (10% CH3CN in 50 mM NH4HCO3), and (partial) O-acetylation (Ser, Thr, and Tyr side chains) was reversed by adding 2 μl NH2OH for 15 min at room temperature during the first washing step. Following vacuum drying, the gel pieces were rehydrated in 20 μl trypsin-containing (0.05 μg) digestion buffer, and additional digestion buffer was added until the gel pieces were completely immersed. Trypsin digestion proceeded for 16 h at 37°C, and the resulting peptide mixtures were acidified (0.1% formic acid) and analyzed by LC-MS/MS analysis.
LC-MS/MS analysis was done using an Ultimate 3000 HPLC system (Dionex, Amsterdam, Netherlands) connected in-line to an LTQ Orbitrap XL mass spectrometer (Thermo Electron, Bremen, Germany). Peptides (8 μl loaded) were first trapped on a trapping column (PepMap C18 column; 0.3-mm internal diameter × 5 mm) (Dionex), and following back-flushing from the trapping column, the sample was loaded on a 75-μm-internal-diameter by 150-mm reverse-phase column (PepMap C18; Dionex). Peptides were eluted with a linear gradient of a 1.8% solvent B (0.05% formic acid in water-acetonitrile [2/8 {vol/vol}]) increase per minute at a constant flow rate of 300 nl/min.
The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the six most abundant ion peaks per MS spectrum. Full-scan MS spectra were acquired at a target value of 1E6, with a resolution of 30,000. The six most intense ions were then isolated for fragmentation in the linear ion trap. In the LTQ instrument, MS/MS scans were recorded in profile mode at a target value of 5,000. Peptides were fragmented after filling the ion trap, with a maximum ion time of 10 ms and a maximum of 1E4 ion counts. From the MS/MS data in each LC run, Mascot generic files (mgf) were created using Mascot Distiller software (version 2.2.1.0; Matrix Science). In generating these peak lists, grouping of spectra was performed with a maximum intermediate retention time of 30 s and a maximum intermediate scan count of 5, where possible. Grouping was done with a 0.005-Da tolerance on the precursor ion. A peak list was generated only when the MS/MS spectrum contained more than 10 peaks, no de-isotoping was performed, and the relative signal-to-noise ratio limit was set at 2.
The peak lists were then searched with Mascot, using the Mascot Daemon interface (version 2.2.0; Matrix Science). Spectra were searched against the human section of the SwissProt database. Variable modifications were set to methionine oxidation, pyroglutamate formation of N-terminal glutamine, trideutero-acetylation of lysine, and (trideutero)acetylation of the N terminus. Mass tolerance on the precursor ion was set to 10 ppm, and a 0.5-Da mass tolerance was used for peptide fragment ions. The peptide charge was set to 1+, 2+, or 3+, and 1 missed cleavage by trypsin was allowed. Peptide identifications were considered significant when the Mascot ion score was above the identity threshold, set at a 99% confidence interval, and when they were top ranked.
The extent of PCNP Nα acetylation was calculated according to the ratio of in vivo Nα acetylation to in vitro Nα-trideutero acetylation determined from the peptide ion signals observed in the MS spectra. The modified peptide sequences were used to calculate the theoretical isotope peak distribution, using an MS isotope pattern calculator (http://prospector.ucsf.edu). The predicted intensity of the 4th isotope in the isotopic pattern of Nα-acetylated peptide was subtracted from the measured intensity of the monoisotopic peak of the isotope cluster of the Nα-trideutero-acetylated peptide to correct for the overlapping isotopic envelopes.
In vitro Nα-acetyltransferase assay.
Acetylation assays were performed with immunoprecipitated hNatA complex, using 5 × 106 HeLa cells as input, 50 μl protein A/G agarose beads (Santa Cruz), and 2 μg antibodies per sample. Beads were mixed with 5 μl synthetic peptide (1 mM custom-made peptide from Biogenes), 5 μl [14C]acetyl coenzyme A ([14C]acetyl-CoA) (50 μCi, 2.07 GBq/mmol; GE Healthcare), and 250 μl acetylation buffer (50 mM Tris-HCl, pH 8.5, 1 mM DTT, 800 μM EDTA, 10 mM sodium butyrate, 10% glycerol). The mixture was incubated for 2 h at 37°C with rotation and then added to 250 μl SP Sepharose (50% slurry in 0.5 M acetic acid; Sigma) and incubated on a rotor for 5 min. The mixture was centrifuged, and the beads were washed three times with 0.5 M acetic acid and one time with methanol. Radioactivity in the peptide-containing pellet was determined by scintillation counting. The custom-made peptide contains 7 substrate-derived amino acids from the N terminus, since these residues encompass the major determinants for N-terminal acetylation. The next 17 amino acids are identical to the adrenocorticotropin (ACTH) peptide sequence to maintain a positive charge, facilitating peptide solubility and effective isolation by cation-exchange Sepharose beads. The ACTH-derived lysines were replaced by arginines to minimize any potential interference by Nɛ-acetylation. The peptide sequence for the high-mobility-group protein A1 (GenBank accession no. P17096) was H-SESSSKSRWGRPVGRRRRPVRVYP-OH.
Alignment and tree building.
HYPK and nascent polypeptide-associated complex alpha chain (NACA) family sequences were taken from Ensembl, version 40 (13) (sequence accession no. ENSF00000002302 and ENSF00000007398, respectively). Peptide sequences from these families were aligned using Muscle (15) with the default settings. Coding sequences were aligned with reference to the alignment of the corresponding peptide sequences. A tree was built from the coding sequence alignment by use of MrBayes (37), with different rates for transitions and transversions and running for 750,000 generations. The first 250,000 generations were discarded as burn-in, after which the likelihood scores had converged. A consensus tree was built from the remaining 500,000 generations by sampling every 100th generation and summarizing the results as a majority-rule consensus tree. The resultant unrooted tree was rooted at the branch that separates most of the HYPK and NACA sequences.
RESULTS
Huntingtin-interacting protein HYPK coimmunoprecipitates with hNaa10p and hNaa15p.
In order to identify novel endogenous interaction partners of the NatA complex in human cells, potentially functionally involved in NatA-mediated Nα-terminal acetylation, we immunoprecipitated the two NatA subunits, hNaa15p and hNaa10p, using specific antibodies, and analyzed the immunoprecipitated proteins by LC-MS/MS after trypsin digestion. From four independent analyses of both anti-hNaa15p and anti-hNaa10p, we considered only those proteins identified in all eight setups analyzed by at least two distinct peptides to be very likely specific candidate interaction proteins of the hNaa15p-hNaa10p complex. By using these stringent criteria, we identified only one novel putative interaction partner, HYPK, in addition to the known NatA subunits hNaa15p and hNaa10p (4) and the physical partner hNaa50p (hNat5/hSan) (5). HYPK was not present in any of the four negative controls using unspecific antibodies. An overview of the identified HYPK-specific peptides is presented in Table S1 and Fig. S1a in the supplemental material. The MS/MS spectrum of one of these peptides, 54EIQSSNLETAMSVIGDR70, is shown in Fig. S1b in the supplemental material. Furthermore, the hNatA-HYPK interaction was confirmed by several assays, as described below.
Direct physical interaction between HYPK and hNaa15p.
To elucidate the direct physical interaction between HYPK and the components of the hNatA complex, we performed interaction studies using recombinant proteins produced in E. coli. The results demonstrated that purified HYPK physically binds to hNaa15p but not to hNaa10p or hNaa50p. Using domains of the hNaa15p protein, we mapped the interaction to amino acids 500 to 865 (Fig. 1A). This region contains a predicted coiled coil at amino acids 583 to 635. Also, HYPK is predicted to have a coiled-coil region at amino acids 69 to 111 (Fig. 1B). Thus, it is possible that hNaa15p and HYPK associate directly via a coiled-coil interaction. The human HYPK protein contains 129 amino acids and has a predicted molecular mass of 14.7 kDa and a pI of 4.9. Figure 1B schematically shows the regions of the HYPK protein, including an acidic region, a putative SH3 binding domain, and the predicted coiled coil (from ELM [http://elm.eu.org/]). The hNaa15p protein, of 866 amino acids, is also outlined schematically, with tetratricopeptide repeat (TPR) domains and a putative coiled-coil region.
FIG. 1.

The C terminus of hNaa15p interacts directly with HYPK. (A) Purified MBP-hNaa15p fusion proteins containing N-terminal MBP and a His tag fused to a segment of the hNaa15p protein, as indicated, were purified and bound to Ni-affinity beads. HYPK was separately fully purified and added to the beads containing different MBP-hNaa15p fusions. After incubation with rotation for 10 min and several washing steps, the MBP-hNaa15p beads were analyzed for HYPK binding by SDS-PAGE and Western blotting using anti-HYPK. A positive signal indicates that purified HYPK binds to the MBP-hNaa15p fusion protein. Equal amounts of the different MBP-hNaa15p fusions used were confirmed by SDS-PAGE and Coomassie staining. (B) Schematic model of the predicted domains of hNaa15p and HYPK and the potential regions involved in the hNaa15p-HYPK interaction. TPR, tetratricopeptide repeat.
HYPK is a cytoplasmic protein associated with ribosomes.
To study the expression of the HYPK protein in human cells, we generated a rabbit polyclonal antibody to HYPK, using a HYPK-specific peptide. Western blotting of several human cell lines demonstrated that the antibody recognized a protein of the expected size of approximately 15 kDa (see Fig. S2a in the supplemental material). Western blotting using anti-HYPK with cell lysates overexpressing Xpress-HYPK or HYPK-V5 gave a strong signal at the expected size of ∼19 kDa, demonstrating that the antibody recognizes the HYPK protein (see Fig. S2b in the supplemental material). In order to study the subcellular localization of HYPK, Xpress-HYPK was transfected into HeLa cells. Immunofluorescence analysis revealed that Xpress-HYPK localized mainly to the cytoplasm (Fig. 2A). Endogenous HYPK also localized primarily to the cytoplasm, although some staining was observed in the nucleus (Fig. 2B). The cytoplasmic localization is in agreement with a possible functional connection to the NatA complex. We further isolated polysomes from HeLa cells and identified HYPK in the polysomal fractions along with hNaa15p, indicating that the protein is associated with ribosomes (Fig. 3). This observation suggests that the interaction between HYPK and hNaa15p-hNaa10p may occur at the ribosome. Since HYPK was coimmunoprecipitated with both anti-hNaa15p and anti-hNaa10p, it is likely that HYPK interacts directly or indirectly with both proteins simultaneously, and thus HYPK is most likely associated with the hNaa15p-hNaa10p complex. hNaa15p-hNaa10p cotranslationally acetylates the N termini of nascent polypeptides, and the hNatA complex is associated with ribosomes (4). If HYPK is a component of the hNaa15p-hNaa10p complex per se and not just associated with ribosomes, then HYPK should interact with hNaa15p and hNaa10p independently of translation. We therefore analyzed the interaction between hNaa15p and HYPK proteins in HeLa cells after treatment with cycloheximide, which inhibits translation (17). The amount of hNaa15p-V5 protein coimmunoprecipitated by anti-Xpress-HYPK was not significantly affected by the cycloheximide treatment (Fig. 4). This indicates that even though the HYPK-hNatA interaction may occur at the ribosome, active translation is not obligatory for the interaction to occur. Thus, HYPK stably associates with hNatA, and their common binding to the ribosomes strongly indicates a common function during protein translation.
FIG. 2.

Subcellular localization of HYPK. (A) (Left) HeLa cells were transfected with a plasmid encoding Xpress-HYPK, and at 48 h posttransfection, cells were fixed and anti-Xpress antibodies and Alexa 488-conjugated anti-mouse antibodies were used to visualize Xpress-HYPK. (Middle) DAPI (4′,6-diamidino-2-phenylindole) staining was used to visualize the nuclei of the cells. (Right) Overlay of DAPI and Xpress-HYPK signals. More than 200 transfected cells were inspected, and representative cells are shown. (B) Twenty-four hours after being seeded, HeLa cells were fixed and anti-HYPK and Alexa 488-conjugated anti-rabbit antibodies were used to visualize endogenous HYPK. Rabbit IgG was used as an unspecific negative control. DAPI staining is presented next to each sample to visualize nuclei and verify the presence of cells. Pictures are representative of three independent experiments in which at least 100 cells were taken into account.
FIG. 3.

HYPK cosediments with polysomal fractions in a salt-sensitive manner. Polysomal pellets from HeLa cells were resuspended in buffer containing increasing concentrations of KCl. Cell lysate and polysomal pellets after KCl treatment were analyzed by SDS-PAGE and Western blotting. The membrane was incubated with anti-HYPK, anti-hNaa15p, anti-L26 (ribosomal protein), and anti-CytC antibodies. Molecular mass markers (in kDa) are indicated on the left. Results shown are representative of three independent experiments.
FIG. 4.

HYPK-hNaa15p interaction is independent of active translation. HEK293 cells were cotransfected with plasmids expressing Xpress-HYPK and hNaa15p-V5. At 48 h posttransfection, cells were treated with 50 μg/ml cycloheximide (CHX) or solvent (control) for 30 min. The cells were then harvested and subjected to immunoprecipitation (IP), using anti-Xpress or negative-control antibodies. The presence of hNaa15-V5 in complex with Xpress-HYPK was analyzed by SDS-PAGE and Western blotting, using anti-V5. The inhibitory effect of CHX on protein translation was verified in parallel samples detecting the reduction of unstable proteins. The results shown are representative of three independent experiments.
HYPK is a specific factor of the NatA complex.
The human hNaa15p-hNaa10p NatA complex is homologous to the yeast NatA complex. In yeast and humans, there are three major NAT complexes performing cotranslational N-terminal acetylation, namely, NatA, NatB, and NatC. The human homologues of the large auxiliary subunits of the NatB and NatC complexes, hNaa25p (hMDM20) and hNaa35p (hMAK10), respectively, probably perform functions in the human NatB and NatC complexes similar to that of the large auxiliary hNaa15p subunit in the human NatA complex (39, 40). To investigate if HYPK specifically interacts with the human NatA complex or if it more generally contacts NAT complexes, we tested the interaction between Xpress-HYPK and the hNaa25p and hNaa35p proteins. Xpress-HYPK did not interact with hNaa25p or hNaa35p-V5, in contrast to what was observed for hNaa15p-V5 (Fig. 5). Thus, HYPK appears to be a specific factor for the hNatA complex, not interacting with the other two major human NATs, hNatB and hNatC.
FIG. 5.
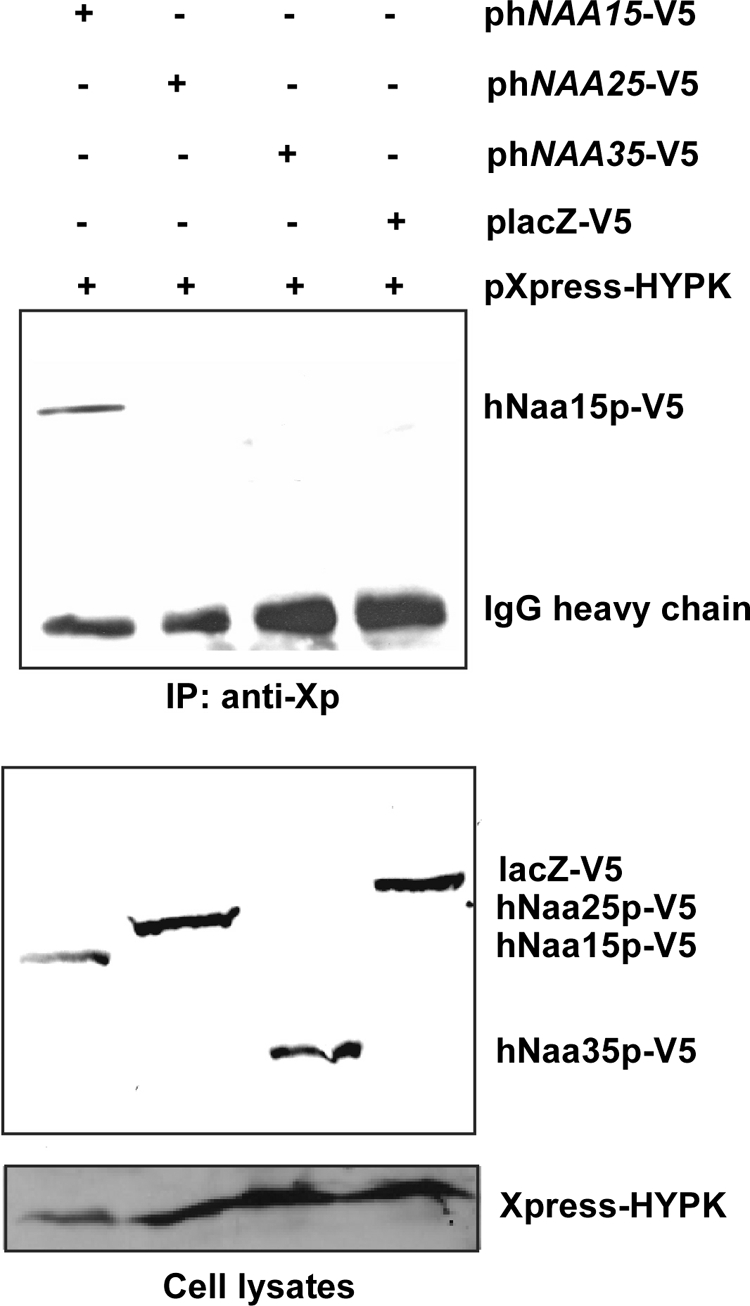
HYPK interacts specifically with hNaa15p of hNatA, not with hNaa25p of hNatB and hNaa35p of hNatC. HEK293 cells were cotransfected with plasmids encoding Xpress-HYPK plus hNaa15p-V5, hNaa25p-V5, hNaa35p-V5, or lacZ-V5, as indicated. At 48 h posttransfection, cells were harvested and subjected to immunoprecipitation using anti-Xpress (anti-Xp) antibodies. The presence of hNaa15p-V5, hNaa25p-V5, and hNaa35p-V5 in complex with Xpress-HYPK was investigated by SDS-PAGE and Western blotting, using anti-V5. (Top) Western blot of immunoprecipitates. (Bottom) Western blot of cell lysates prior to immunoprecipitation. The results shown are representative of three independent experiments.
HYPK knockdown results in cell death and accumulation of cells in the G1/G0 phase.
Different siRNAs were tested for the ability to knock down HYPK expression. RT-PCR demonstrated that 4 of 5 tested siRNAs significantly reduced the level of HYPK mRNA in HeLa cells at 24 h posttransfection (Fig. 6A). The most efficient siRNAs, siHYPK-1, -2, -4, and -5, were used as a mixed pool in subsequent experiments. RT-PCR demonstrated efficient and specific knockdown of all three of the NAA10, NAA15, and HYPK genes by gene-specific siRNAs (Fig. 6B). Western blotting of cell lysates at 72 h post-siRNA transfection, using anti-HYPK, demonstrated efficient knockdown at the protein level (Fig. 6C). These results further proved that the ∼15-kDa band detected by Western blotting using anti-HYPK was indeed HYPK.
FIG. 6.

siRNA-mediated knockdown of HYPK, hNAA10, and hNAA15. (A) Five individual siRNAs potentially targeting HYPK expression (H1 to H5) were tested by siRNA transfection of HeLa cells and subsequent RT-PCR analysis at 48 h posttransfection, using primers specific for HYPK and β-actin. G, siGAPDH (negative control). (B) siRNA-mediated knockdown in HeLa cells of the specific genes HYPK, hNAA10, and hNAA15, as indicated. siLamin and siGAPDH were used as negative controls. siHYPK is an equal mixture of the four effective siHYPK siRNAs shown in panel A (H1, H2, H4, and H5; 50 nM total concentration). RT-PCR analysis was performed at 72 h posttransfection. (C) SDS-PAGE and Western blotting, using anti-HYPK, anti-hNaa10p, anti-hNaa15p, and antiactin, of HeLa samples treated identically to those described for panel B. Protein levels were quantified using a Fuji Film IR LAS 1000 documentation system and Image Gauge 3.45. Protein levels in siLamin- and siGAPDH-treated cells (the mean for these two negative controls) were set to 1.0, and protein levels in sihNAA10-, sihNAA15-, and siHYPK-treated cells were estimated relative to this and normalized to actin levels. HYPK appeared as one or two specific bands, and the sum of the two was used for quantification. All results are representative of at least three independent experiments.
HYPK knockdown did not significantly affect the levels of hNaa15p and hNaa10p protein (Fig. 6C). In contrast, knockdown of hNAA10 and hNAA15 appeared to reduce the level of HYPK protein, in addition to a reduction of the proteins encoded by the respective target genes. As previously observed, sihNAA15 also decreased the level of the hNaa10p protein (7). In contrast to HYPK protein levels, levels of HYPK mRNA were quite stable after sihNAA10 or sihNAA15 treatment. In all setups analyzed, the pan-caspase inhibitor ZVAD-fmk was added to the cells to avoid indirect effects of hNAA10 and hNAA15 knockdown due to the induction of apoptosis (9), further indicating that the observed reduction of HYPK protein might be due to destabilization of the HYPK protein.
RNAi-mediated knockdown of HYPK was performed to investigate the biological importance of HYPK. Cells treated with siRNAs targeting HYPK, hNAA10, hNAA15, or controls were subjected to FACS cell cycle analysis at 72 h posttransfection (Fig. 7). The data demonstrated that siHYPK-treated cells underwent cell death, to a comparable extent (∼22%) to that observed for knockdown of hNAA10 or hNAA15 (∼25%). Addition of the pan-caspase inhibitor ZVAD-fmk inhibited cell death, indicating that it was caspase dependent. Furthermore, there was an accumulation of cells in the G1/G0 phase after siHYPK treatment, similar to that observed for sihNAA10 and sihNAA15 treatment. Similar effects were also observed when individual siRNAs targeting HYPK, siHYPK-1, and siHYPK-2 were used, demonstrating the specificity of the observed effects (data not shown). In conclusion, knockdown of both HYPK and hNatA induces cell death in HeLa cells, potentially via a common function.
FIG. 7.

HYPK knockdown induces cell death and G1/G0 cell cycle arrest. Flow cytometric cell cycle analysis of HYPK, hNAA10, and hNAA15 knockdown cells was performed at 72 h posttransfection. siLamin- and siGAPDH-treated samples were used as negative controls. (Top panels) No ZVAD treatment. (Bottom panels) ZVAD treatment to inhibit caspase activity and, thereby, induction of caspase-mediated cell death. Experiments were performed three times, and representative values are given.
Knockdown of HYPK and hNAA10 increases Huntingtin Q72 and Q103 aggregation.
To investigate the possible functional relationship between HYPK and the hNatA complex, we wanted to test whether the hNatA complex is required for the function of HYPK relating to Htt aggregation. HYPK was previously shown to exhibit chaperone-like activity, interacting with the N terminus of Htt, and overexpression of HYPK reduced Htt polyQ aggregation and toxicity in Neuro2A cells (36). In this study, we wanted to investigate endogenous HYPK in relation to Htt polyQ. Knockdown of HYPK significantly increased the level of aggregation of HttQ72-EGFP and HttQ103-EGFP compared to that in control HeLa cells (Fig. 8). This indicates that endogenous HYPK reduces polyQ-mediated protein aggregation in nonneuronal cells. Furthermore, since HYPK physically interacts with the hNatA complex and both hNaa10p and hNaa15p are required for the stability of the HYPK protein, we analyzed the effects of hNAA10 knockdown on Htt polyQ aggregation. Indeed, hNAA10 knockdown also increased the number of cells with HttQ72-EGFP and HttQ103-EGFP aggregates (Fig. 8). We were not able to detect any aggregates of HttQ25-EGFP, even when knocking down HYPK and hNAA10. In order to elucidate the importance of a native N terminus in this process, since this potentially will be exposed to hNatA and HYPK during translation, we used another set of plasmids expressing Htt polyQ with an N-terminal EGFP tag: EGFP-HttQ23 and EGFP-HttQ74. Interestingly, even though EGFP-HttQ74 demonstrated a significant number of aggregates in control cells, HYPK or hNAA10 knockdown did not increase the number of cells with EGFP-HttQ74 aggregates (Fig. 8). To visualize HYPK and the NatA complex in cells expressing different Htt proteins and to determine whether HYPK and/or hNatA associates with Htt aggregates, we performed immunofluorescence colocalization experiments. Aggregates with different appearances were observed for the Htt proteins with long polyQ stretches (Q72, Q74, and Q103), while a general and diffuse staining was observable for HttQ23 and HttQ25 (Fig. 9). Importantly, we were not able to detect significant colocalization between any of the Htt proteins and HYPK or hNaa15p. Both HYPK and hNatA prevent Htt polyQ aggregation, most likely by contacting and/or acetylating the nascent Htt polyQ N-terminal polypeptide during translation.
FIG. 8.

HYPK and hNAA10 knockdown increases Huntingtin aggregation. HeLa cells were transfected with the indicated siRNAs, and at 24 h posttransfection, the medium was replaced and the cells were transfected with plasmids encoding the indicated Htt-EGFP fusion proteins. Twenty-four hours after plasmid transfection, the cells were fixed, cells displaying Htt-EGFP aggregates were counted, and the percentage of cells with aggregates among the transfected cells was calculated. At least 200 transfected cells were counted per sample, and values presented are means ± standard deviations (SD) for three to six independent experiments. P values for independent t tests for samples versus control are indicated with asterisks (P < 0.05).
FIG. 9.

HYPK and hNaa15p do not colocalize with Htt aggregates. HeLa cells expressing various EGFP-tagged polyQ versions (left column) were incubated with HYPK- or hNaa15p-specific antibodies, as indicated in the figure (second column from left). Alexa 594-conjugated anti-mouse antibodies were used to visualize HYPK and hNaa15p. The third column from left presents an overlay of polyQ-EGFP and HYPK or hNaa15p. DAPI staining was used to visualize DNA, as included in the overlay shown in the right column. Images were processed by deconvolution (Leica 4000 software). Bar, 25 μm.
Knockdown of HYPK decreases Nα-terminal acetylation of a known NatA substrate.
To further elucidate the functional importance of the HYPK-hNaa15p-hNaa10p interaction, we tested whether HYPK was important for NatA-mediated acetylation. Recently, the first in vivo hNatA substrates were identified. Upon siRNA-mediated knockdown of hNAA10 and hNAA15 in HeLa cells, we observed a partial but significant reduction in N-terminal acetylation for a subset of suboptimal hNatA substrates (12). We therefore cloned one of the genes encoding an sihNatA-regulated substrate protein, namely, PCNP, and coexpressed this protein while simultaneously knocking down HYPK or hNAA10. After PCNP isolation and SDS-PAGE-based separation, in-gel stable isotope labeling was performed by in vitro trideutero acetylation. The extent of PCNP Nα acetylation was assessed following trypsinization and mass spectrometric analysis of the N-terminal peptide 2ADGKAGDEKPEKSQR16. The sihNAA10 sample confirmed the hNatA substrate dependency of PCNP, and interestingly, PCNP was also found to be less acetylated in the siHYPK sample (Fig. 10). This demonstrated that HYPK is important, directly or indirectly, for the Nα-terminal acetylation of PCNP. A contaminating protein, the 40S ribosomal protein S3, with the N terminus 2AVQISKKR9, was picked up in all three samples. Interestingly, the N-terminal-acetylation status of S3 was unaffected by sihNAA10 and siHYPK treatment, in further agreement with our previous large-scale analysis of hNatA knockdown cells (12). Thus, most likely, only the hNatA substrates sensitive to hNatA knockdown are also sensitive to HYPK knockdown in terms of a reduced N-terminal-acetylation pattern. Furthermore, we analyzed the in vitro capacity of the NatA complex immunoprecipitated from control or HYPK knockdown cells to N-terminally acetylate a known substrate peptide. Indeed, the capacity of NatA to acetylate a Ser-starting peptide (SESS) diminished when HYPK levels were reduced (Fig. 11). The N terminus of the SESS peptide is derived from a protein found to be acetylated fully in HeLa cells unaffected by sihNatA knockdown (12). In summary, HYPK is required for optimal hNatA N-terminal-acetylation activity.
FIG. 10.

HYPK knockdown reduces NatA-mediated N-terminal acetylation. HeLa cells were transfected with siRNAs targeting hNAA10, HYPK, or a control. At 24 h posttransfection, the medium was replaced and cells transfected with pPCNP-V5 plasmid. Forty-eight hours after plasmid transfection, the cells were harvested and lysed, and PCNP-V5 was immunoprecipitated and processed as described in Materials and Methods. ZVAD was added every 24 h to prevent induction of apoptosis. MS spectra of doubly charged peptide ions originating from the N terminus of PCNP (GenBank accession no. Q8WW12) are shown. The peptide was identified as 2-ADGKAGDEKPEKSQR-16. The extent of PCNP Nα acetylation, as determined by an MS isotope pattern calculator (http://prospector.ucsf.edu), was calculated as being 97%, 73%, and 90% in the control, sihNAA10, and siHYPK samples, respectively. The ellipsoids indicate the relative increases of the in vivo unacetylated N terminus of PCNP in the sihNAA10- and siHYPK-treated samples compared to that in the control sample. The data are representative of two independent experiments.
FIG. 11.
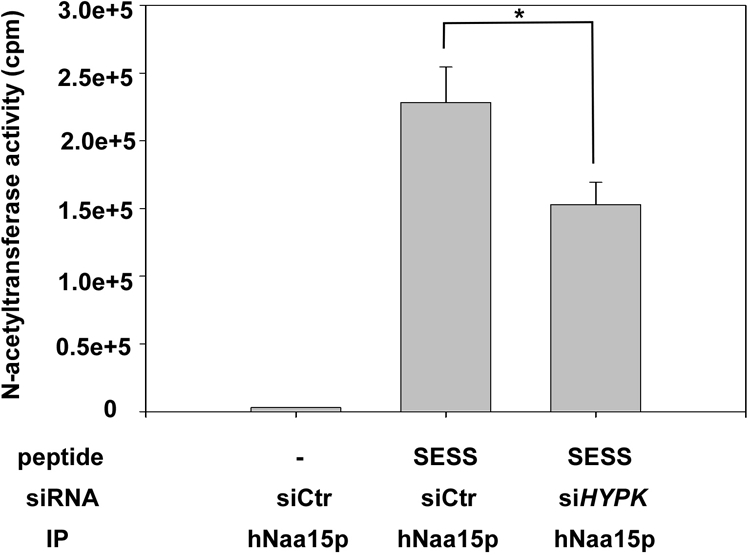
hNatA immunoprecipitated from HYPK knockdown cells displays reduced in vitro acetylation activity. HeLa cells (approximately 5 × 106 per sample) were transfected with the indicated siRNAs, and at 24 h posttransfection, ZVAD was added to prevent induction of apoptosis. After 48 h, the cells were harvested and lysed, and the lysate was subjected to immunoprecipitation (IP) using an anti-hNaa15p specific antibody. The beads containing functional hNatA complexes were analyzed for Nα-acetyltransferase activity, using [14C]acetyl-CoA and a Ser-Asp-Ser-Ser (SESS)-starting 24-mer peptide known to be acetylated by NatA in vitro. The amount of acetyl incorporation was determined by isolation of the peptides followed by scintillation counting. Verification of knockdown and the presence of equal levels of hNaa15p in the immunoprecipitates were routinely confirmed. Experiments were performed three times, and values are means ± SD. P values for independent t tests for siHYPK samples versus control are indicated with an asterisk (P < 0.05).
DISCUSSION
The functional characterization of the NatA Nα-terminal acetyltransferase revealed that two components, Naa10p (Ard1p) and Naa15p (Nat1p), are required for catalytic activity (26). These two partners interact in a physical complex (28), and the complex is associated with ribosomes (21). The large auxiliary subunit Naa15p mediates contact between the ribosome and nascent polypeptide and the catalytic subunit Naa10p (21). The system appears to be conserved in humans, where hNaa10p and hNaa15p interact with each other and with ribosomes to perform N-terminal acetylation in vitro (4). Recent functional studies demonstrated that expression of hNaa10p and hNaa15p can complement a yeast strain lacking yeast NatA by rescuing NatA-deletion phenotypes. Furthermore, when expressed in yeast, hNaa10p and hNaa15p could N-terminally acetylate the same set of substrates as the yeast NatA complex in vivo (12). A third subunit, Naa50p (Nat5p), physically associated with NatA but was not required for NatA activity, suggesting a distinct function (21).
There are several ribosome-associated protein biogenesis factors (RPBs) potentially associated with nascent polypeptides. In yeast, these include the chaperones Ssb1 and Ssb2 and the ribosome-associated complex (RAC), the nascent polypeptide-associated complex (NAC), the signal recognition particle (SRP), methionine aminopeptidases (MAPs), and Nα-terminal acetyltransferase (NATs). A quantitative analysis of the different RPBs relative to the number of ribosomes per cell indicated a dynamic association between RPBs and ribosomes and also suggested an ordered interplay between the RPBs (35). However, direct interactions between NATs and other RPBs have not been investigated. In the identification of the mammalian RAC, HYPK was copurified with RAC and the NatA complex, but without any further distinction between the two (27). Our current data strongly suggest that in human cells, HYPK and the NatA complex cooperate at the ribosome. HYPK is required for efficient N-terminal acetylation of known NatA substrates in vivo and in vitro (Fig. 10 and 11), and NatA is required for HYPK stability and Htt polyQ aggregation (Fig. 6 and 8). The similar knockdown phenotypes also suggest similar functional roles for HYPK and NatA (Fig. 7), but at present it is not known whether these phenotypes are caused by lack of N-terminal acetylation of specific substrates, misfolding/aggregation of endogenous proteins, or both. If indeed HYPK is absolutely required for hNatA-mediated acetylation in general, it should be defined as a subunit of the NatA complex per se. However, a large-scale N-terminal-acetylation analysis of HYPK knockdown cells or something similar should be performed before such conclusions can be drawn. The lack of HYPK in yeast suggests the presence of efficient NatA activity independent of HYPK, and also, HYPK expression in yeast was not required for effective in vivo acetylation of yeast NatA substrates by hNaa10p and hNaa15p (12). However, it should be noted that the degree of NatA-mediated N-terminal acetylation is significantly higher in human cells than in yeast cells and that the underlying cause(s) for this difference has not yet been revealed (12). A human-specific factor like HYPK, a factor increasing the efficiency of NatA-mediated N-terminal acetylation, might contribute to this phenomenon.
The S. cerevisiae protein most similar to HYPK is NACA. It is interesting that the heterodimeric NAC, consisting of NACA and NACB, is the first to contact nascent polypeptides as they emerge from the ribosome (44), thus potentially placing the NAC and NatA complexes in close proximity. A phylogenetic analysis of HYPK and NACA is presented in Fig. S3 in the supplemental material. Given the similarity between NACA and HYPK and the association of HYPK with ribosomes and hNaa15p-hNaa10p, it is tempting to speculate that HYPK also contacts nascent polypeptides. HYPK then may mediate effective contact between the peptide and hNatA and/or act as a chaperone preventing nascent polypeptide misfolding in order for effective acetylation to occur. In human cells, HYPK and NACA coexist, and it would be interesting to define whether NACA and/or the NAC also directly contacts the NatA complex at the ribosome and might be involved functionally in N-terminal acetylation of certain substrates.
An interesting aspect relates to whether the effect of HYPK and hNatA on Htt polyQ aggregation is mediated via a cotranslational process. There are indications that this might be the case. First, both HYPK and hNatA are associated with ribosomes. Second, an N-terminal EGFP tag (EGFP-HttQ74) masking the N terminus of Htt polyQ from HYPK and hNatA on the ribosome abrogated the effects of siHYPK and sihNAA10 treatment on aggregation (Fig. 9). This suggests that hNatA and HYPK must be in contact with the natural N terminus of Htt at the ribosome in order to prevent Htt aggregation. Third, we did not detect any significant colocalization between Htt aggregates and HYPK/hNaa15p (Fig. 10), which might be expected if hNatA-HYPK acted posttranslationally on the aggregates. Thus, the reported interaction between HYPK and Htt (19, 36) is probably dynamic and perhaps confined to Htt during translation and shortly after, before aggregation occurs in the case of long stretches of polyQ. Finally, it should be mentioned that the N terminus of Htt is (Met)-Ala-Thr-Leu, making this a likely hNatA substrate (12). Very recently, it was indeed demonstrated that Htt is N-terminally acetylated, and given the fact that the N-terminal 17 amino acid residues are critical for Htt aggregation and subcellular localization (1), it is possible that this acetylation may directly affect Htt behavior. hNatA is the natural candidate enzyme for the acetylation of the Ala N terminus of Htt, and future experiments might reveal whether there is a direct link between hNatA-mediated N-terminal acetylation of Htt and Htt aggregation.
In conclusion, HYPK is a novel potential RPB directly and functionally coupled to hNatA-mediated N-terminal acetylation and also with an important role in the prevention of protein aggregation.
Supplementary Material
Acknowledgments
We thank N. Glomnes, K. Jacobsen, C. Hoff, and E. Skjelvik for technical assistance.
This work was supported by The Meltzer Foundation (grant to T.A.), the Western Norway Regional Health Authority (grant to T.A.), and the University of Bergen (research grant to T.A.). P.V.D. is a postdoctoral fellow at the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen. The Department of Medical Protein Research further acknowledges support by research grants from the Fund for Scientific Research-Flanders (Belgium), the Concerted Research Actions from Ghent University, the Interuniversity Attraction Poles, and the European Union Interaction Proteome (6th Framework Program).
Footnotes
Published ahead of print on 12 February 2010.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Aiken, C. T., J. S. Steffan, C. M. Guerrero, H. Khashwji, T. Lukacsovich, D. Simmons, J. M. Purcell, K. Menhaji, Y. Z. Zhu, K. Green, F. Laferla, L. Huang, L. M. Thompson, and J. L. Marsh. 2009. Phosphorylation of threonine-3: implications for huntingtin aggregation and neurotoxicity. J. Biol. Chem. 284:29427-29436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ametzazurra, A., E. Larrea, M. P. Civeira, J. Prieto, and R. Aldabe. 2008. Implication of human N-alpha-acetyltransferase 5 in cellular proliferation and carcinogenesis. Oncogene 27:7296-7306. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, D. C., W. Li, D. G. Payan, and W. S. Noble. 2003. A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores. J. Proteome Res. 2:137-146. [DOI] [PubMed] [Google Scholar]
- 4.Arnesen, T., D. Anderson, C. Baldersheim, M. Lanotte, J. E. Varhaug, and J. R. Lillehaug. 2005. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 386:433-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnesen, T., D. Anderson, J. Torsvik, H. B. Halseth, J. E. Varhaug, and J. R. Lillehaug. 2006. Cloning and characterization of hNAT5/hSAN: an evolutionarily conserved component of the NatA protein N-alpha-acetyltransferase complex. Gene 371:291-295. [DOI] [PubMed] [Google Scholar]
- 6.Arnesen, T., M. J. Betts, F. Pendino, D. A. Liberles, D. Anderson, J. Caro, X. Kong, J. E. Varhaug, and J. R. Lillehaug. 2006. Characterization of hARD2, a processed hARD1 gene duplicate, encoding a human protein N-alpha-acetyltransferase. BMC Biochem. 7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnesen, T., D. Gromyko, O. Horvli, O. Fluge, J. Lillehaug, and J. E. Varhaug. 2005. Expression of N-acetyl transferase human and human Arrest defective 1 proteins in thyroid neoplasms. Thyroid 15:1131-1136. [DOI] [PubMed] [Google Scholar]
- 8.Arnesen, T., D. Gromyko, D. Kagabo, M. J. Betts, K. K. Starheim, J. E. Varhaug, D. Anderson, and J. R. Lillehaug. 2009. A novel human NatA Nalpha-terminal acetyltransferase complex: hNaa16p-hNaa10p (hNat2-hArd1). BMC Biochem. 10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnesen, T., D. Gromyko, F. Pendino, A. Ryningen, J. E. Varhaug, and J. R. Lillehaug. 2006. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-alpha-acetyltransferase complex. Oncogene 25:4350-4360. [DOI] [PubMed] [Google Scholar]
- 10.Arnesen, T., X. Kong, R. Evjenth, D. Gromyko, J. E. Varhaug, Z. Lin, N. Sang, J. Caro, and J. R. Lillehaug. 2005. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett. 579:6428-6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnesen, T., P. R. Thompson, J. E. Varhaug, and J. R. Lillehaug. 2008. The protein acetyltransferase ARD1: a novel cancer drug target? Curr. Cancer Drug Targets 8:545-553. [DOI] [PubMed] [Google Scholar]
- 12.Arnesen, T., P. Van Damme, B. Polevoda, K. Helsens, R. Evjenth, N. Colaert, J. E. Varhaug, J. Vandekerckhove, J. R. Lillehaug, F. Sherman, and K. Gevaert. 2009. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 106:8157-8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birney, E., D. Andrews, M. Caccamo, Y. Chen, L. Clarke, G. Coates, T. Cox, F. Cunningham, V. Curwen, T. Cutts, T. Down, R. Durbin, X. M. Fernandez-Suarez, P. Flicek, S. Graf, M. Hammond, J. Herrero, K. Howe, V. Iyer, K. Jekosch, A. Kahari, A. Kasprzyk, D. Keefe, F. Kokocinski, E. Kulesha, D. London, I. Longden, C. Melsopp, P. Meidl, B. Overduin, A. Parker, G. Proctor, A. Prlic, M. Rae, D. Rios, S. Redmond, M. Schuster, I. Sealy, S. Searle, J. Severin, G. Slater, D. Smedley, J. Smith, A. Stabenau, J. Stalker, S. Trevanion, A. Ureta-Vidal, J. Vogel, S. White, C. Woodwark, and T. J. Hubbard. 2006. Ensembl 2006. Nucleic Acids Res. 34:D556-D561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reference deleted.
- 15.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eng. J. K., A. L. McCormack, and J. R. Yates. 1994. An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976-989. [DOI] [PubMed] [Google Scholar]
- 17.Ennis, H. L., and M. Lubin. 1964. Cycloheximide: aspects of inhibition of protein synthesis in mammalian cells. Science 146:1474-1476. [DOI] [PubMed] [Google Scholar]
- 18.Evjenth, R., K. Hole, O. A. Karlsen, M. Ziegler, T. Arnesen, and J. R. Lillehaug. 2009. Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J. Biol. Chem. 284:31122-31129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faber, P. W., G. T. Barnes, J. Srinidhi, J. Chen, J. F. Gusella, and M. E. MacDonald. 1998. Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 7:1463-1474. [DOI] [PubMed] [Google Scholar]
- 20.Fisher, T. S., S. D. Etages, L. Hayes, K. Crimin, and B. Li. 2005. Analysis of ARD1 function in hypoxia response using retroviral RNA interference. J. Biol. Chem. 280:17749-17757. [DOI] [PubMed] [Google Scholar]
- 21.Gautschi, M., S. Just, A. Mun, S. Ross, P. Rucknagel, Y. Dubaquie, A. Ehrenhofer-Murray, and S. Rospert. 2003. The yeast N(alpha)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell. Biol. 23:7403-7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gururaja, T., W. Li, J. Bernstein, D. G. Payan, and D. C. Anderson. 2002. Use of MEDUSA-based data analysis and capillary HPLC-ion-trap mass spectrometry to examine complex immunoaffinity extracts of RBAp48. J. Proteome Res. 1:253-261. [DOI] [PubMed] [Google Scholar]
- 23.Ji, J., A. Chakraborty, M. Geng, X. Zhang, A. Amini, M. Bina, and F. Regnier. 2000. Strategy for qualitative and quantitative analysis in proteomics based on signature peptides. J. Chromatogr. B 745:197-210. [DOI] [PubMed] [Google Scholar]
- 24.Lee, K. J., A. Panzera, D. Rogawski, L. E. Greene, and E. Eisenberg. 2007. Cellular prion protein (PrPC) protects neuronal cells from the effect of huntingtin aggregation. J. Cell Sci. 120:2663-2671. [DOI] [PubMed] [Google Scholar]
- 25.Lim, J. H., J. W. Park, and Y. S. Chun. 2006. Human arrest defective 1 acetylates and activates beta-catenin, promoting lung cancer cell proliferation. Cancer Res. 66:10677-10682. [DOI] [PubMed] [Google Scholar]
- 26.Mullen, J. R., P. S. Kayne, R. P. Moerschell, S. Tsunasawa, M. Gribskov, M. Colavito-Shepanski, M. Grunstein, F. Sherman, and R. Sternglanz. 1989. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 8:2067-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto, H., C. Conz, P. Maier, T. Wolfle, C. K. Suzuki, P. Jeno, P. Rucknagel, J. Stahl, and S. Rospert. 2005. The chaperones MPP11 and Hsp70L1 form the mammalian ribosome-associated complex. Proc. Natl. Acad. Sci. USA 102:10064-10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park, E. C., and J. W. Szostak. 1992. ARD1 and NAT1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO J. 11:2087-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pestana, A., and H. C. Pitot. 1974. N-terminal acetylation of histone-like nascent peptides on rat liver polyribosomes in vitro. Nature 247:200-202. [DOI] [PubMed] [Google Scholar]
- 30.Pestana, A., and H. C. Pitot. 1975. Acetylation of nascent polypeptide chains on rat liver polyribosomes in vivo and in vitro. Biochemistry 14:1404-1412. [DOI] [PubMed] [Google Scholar]
- 31.Pfund, C., N. Lopez-Hoyo, T. Ziegelhoffer, B. A. Schilke, P. Lopez-Buesa, W. A. Walter, M. Wiedmann, and E. A. Craig. 1998. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J. 17:3981-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polevoda, B., T. Arnesen, and F. Sherman. 2009. A synopsis of eukaryotic Nalpha-terminal acetyltransferases: nomenclature, subunits and substrates. BMC Proc. 3(Suppl. 6):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polevoda, B., J. Hoskins, and F. Sherman. 2009. Properties of Nat4, an N(alpha)-acetyltransferase of Saccharomyces cerevisiae that modifies N termini of histones H2A and H4. Mol. Cell. Biol. 29:2913-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polevoda, B., and F. Sherman. 2003. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325:595-622. [DOI] [PubMed] [Google Scholar]
- 35.Raue, U., S. Oellerer, and S. Rospert. 2007. Association of protein biogenesis factors at the yeast ribosomal tunnel exit is affected by the translational status and nascent polypeptide sequence. J. Biol. Chem. 282:7809-7816. [DOI] [PubMed] [Google Scholar]
- 36.Raychaudhuri, S., M. Sinha, D. Mukhopadhyay, and N. P. Bhattacharyya. 2008. HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity. Hum. Mol. Genet. 17:240-255. [DOI] [PubMed] [Google Scholar]
- 37.Ronquist, F., and J. P. Huelsenbeck. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572-1574. [DOI] [PubMed] [Google Scholar]
- 38.Song, O. K., X. Wang, J. H. Waterborg, and R. Sternglanz. 2003. An Nalpha-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J. Biol. Chem. 278:38109-38112. [DOI] [PubMed] [Google Scholar]
- 39.Starheim, K. K., T. Arnesen, D. Gromyko, A. Ryningen, J. E. Varhaug, and J. R. Lillehaug. 2008. Identification of the human N(alpha)-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochem. J. 415:325-331. [DOI] [PubMed] [Google Scholar]
- 40.Starheim, K. K., D. Gromyko, R. Evjenth, A. Ryningen, J. E. Varhaug, J. R. Lillehaug, and T. Arnesen. 2009. Knockdown of human N alpha-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Mol. Cell. Biol. 29:3569-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strous, G. J., A. J. Berns, and H. Bloemendal. 1974. N-terminal acetylation of the nascent chains of alpha-crystallin. Biochem. Biophys. Res. Commun. 58:876-884. [DOI] [PubMed] [Google Scholar]
- 42.Strous, G. J., H. van Westreenen, and H. Bloemendal. 1973. Synthesis of lens protein in vitro. N-terminal acetylation of alpha-crystallin. Eur. J. Biochem. 38:79-85. [DOI] [PubMed] [Google Scholar]
- 43.Vedeler, A., I. F. Pryme, and J. E. Hesketh. 1991. The characterization of free, cytoskeletal and membrane-bound polysomes in Krebs II ascites and 3T3 cells. Mol. Cell. Biochem. 100:183-193. [DOI] [PubMed] [Google Scholar]
- 44.Wang, S., H. Sakai, and M. Wiedmann. 1995. NAC covers ribosome-associated nascent chains thereby forming a protective environment for regions of nascent chains just emerging from the peptidyl transferase center. J. Cell Biol. 130:519-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wyttenbach, A., J. Carmichael, J. Swartz, R. A. Furlong, Y. Narain, J. Rankin, and D. C. Rubinsztein. 2000. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc. Natl. Acad. Sci. USA 97:2898-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng, X. C., S. Bhasin, X. Wu, J. G. Lee, S. Maffi, C. J. Nichols, K. J. Lee, J. P. Taylor, L. E. Greene, and E. Eisenberg. 2004. Hsp70 dynamics in vivo: effect of heat shock and protein aggregation. J. Cell Sci. 117:4991-5000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.