

Abstract
Today, global attention is focused on two influenza virus strains: the current pandemic strain, swine origin influenza virus (H1N1-2009), and the highly pathogenic avian influenza virus, H5N1. At present, the infection caused by the H1N1-2009 is moderate, with mortality rates of less <1%. In contrast, infection with the H5N1 virus resulted in high mortality rates, and ca. 60% of the infected patients succumb to the infection. Thus, one of the world greatest concerns is that the H5N1 virus will evolve to allow an efficient human infection and human-to-human transmission. Natural killer (NK) cells are one of the innate immune components playing an important role in fighting against influenza viruses. One of the major NK activating receptors involved in NK cell cytotoxicity is NKp46. We previously demonstrated that NKp46 recognizes the hemagglutinin proteins of B and A influenza virus strains. Whether NKp46 could also interact with H1N1-2009 virus or with the avian influenza virus is still unknown. We analyzed the immunological properties of both the avian and the H1N1-2009 influenza viruses. We show that NKp46 recognizes the hemagglutinins of H1N1-2009 and H5 and that this recognition leads to virus killing both in vitro and in vivo. However, importantly, while the swine H1-NKp46 interactions lead to the direct killing of the infected cells, the H5-NKp46 interactions were unable to elicit direct killing, probably because the NKp46 binding sites for these two viruses are different.
Natural killer (NK) cells, which comprise 5 to 15% of peripheral blood lymphocytes, are a key frontline defense against a number of pathogens, including intracellular bacteria, parasites, and most importantly with respect to the present study, viruses (6, 40). The antiviral mechanisms by which NK cells operate include both cytotoxic activity and cytokine/chemokine secretion (21). The NK killing activity is executed by numerous receptors, including NKG2D, NKp80, CD16, and the natural cytotoxic receptors (NCRs): NKp30, NKp44, and NKp46 (7, 10, 25).
Although the cellular ligands for NKG2D were identified (31, 38), the identity of several of the cellular ligands for the human NCRs is still unknown, except for BAT3 and B7-H6, which are ligands for NKp30 (8, 30). In contrast, viral ligands were identified for the NCRs, and we demonstrated that pp65 of HCMV interacts with NKp30 (3) and that various influenza virus hemagglutinins (HAs) are ligands for the NKp44 and NKp46 receptors (5, 22). Supporting these observations, it was recently shown that the HA-neuraminidase of Newcastle disease virus could also interact with NKp46 and NKp44 but not with NKp30 (17). Furthermore, we have shown in vivo that in the absence of NCR1 (the mouse homologue of NKp46), A/PR8 influenza virus infection is lethal (14).
Human influenza virus (H1 and H3 subtype) infections pose a major threat to the entire population, as exemplified by the three major influenza pandemics that occurred during the 20th century. The Asian (A/H2N2) in 1957 to 1958 and the Hong Kong (A/H3N2) pandemics in 1968 to 1969 resulted in the deaths of 1 to 2 million people and the 1918 “Spanish flu” (A/H1N1) pandemic killed around 50 million people (18). At present, the worldwide concern regarding influenza pandemics concentrates mainly on two viruses: the A/H1N1 swine origin influenza virus (H1N1-2009), which currently causes only a moderate pandemic (the mortality rates are ca. 1%) but is more pathogenic than a regular seasonal influenza virus (19, 26, 27), and the avian influenza virus carrying the unique H5 HA (20). The avian influenza virus is quite deadly and, although it remains a zoonotic infection, ca. 60% of infected humans died due to the infection (28).
The unique properties of the H5 protein of the avian influenza virus are one of the main reasons for the virulence of the virus. The H5 of the avian influenza virus binds to cell surface glycoproteins or glycolipids containing terminal sialyl-galactosyl residues linked by 2-3-linkage [Neu5Ac(α2-3)Gal] that are found in the human conjunctiva and ciliated portion of the respiratory columnar epithelium (33). In contrast, human viruses (including all three strains that caused the pandemics described above and the H1N1-2009) bind to receptors that mostly contain terminal 2-6-linked sialyl-galactosyl moieties [Neu5Ac(α2-6)Gal]. Such glycosylations are predominant on epithelial cells in the nasal mucosa, paranasal sinuses, pharynx, trachea, and bronchi (33, 37). It has been suggested that the lack of human-to-human transmission of avian influenza viruses is due to their α2,3-SA receptor binding preference, and the concern is that genetic changes in H5 might alter its preference from α2,3-SA to α2,6-SA, allowing human-to-human transmission.
In our previous studies (4, 22) we showed that the interaction between NKp46 and influenza virus HAs depends on the sialylation of the NKp46 receptor. We further demonstrated that the sialic acid residues, which are linked via α2,6 to the threonine 225 residue of NKp46, are crucial for the NKp46 interactions with the various influenza virus HAs (4).
We show that, both in vitro and in vivo, the killing of H1N1-2009-infected cells is correlated with the degree of NKp46 binding. Surprisingly, we observed that although NKp46 efficiently recognized the avian H5 HA, such interactions were unable to elicit the direct killing of the infected cells. By using mutagenesis analysis experiments and killing assays we demonstrate that NKp46 interacts with H1 and H5 at distinct sites, since we show that the sugar carrying residue at position 225 is crucial for the NKp46-H1N1-2009 interactions, whereas the interaction of H5 with NKp46 depends on both residues 216 and 225.
MATERIALS AND METHODS
Cells.
The cell lines used in the present study were the human hepatoma cell line Hep3b, the human choriocarcinoma cell line Jeg3, and the mouse lymphoblastlike mastocytoma cell line P815. For the generation of Jeg3 cells expressing MICB (Jeg3/MICB), we inserted the cDNA sequence of human MICB instead of the green fluorescent protein (GFP) of the lentiviral vector SIN18-pRLL-hEFIap-EGFP-WRPE (35) and stably infected Jeg3 cells. As a control, we infected Jeg3 cells with the intact vector SIN18-pRLL-hEFIap-EGFP-WRPE, generating Jeg3/GFP cells. Primary NK cells were isolated from PBLs using a human NK cell isolation kit and the autoMACS instrument (Miltenyi Biotec, Auburn, CA). NK cells were kept in culture as described previously (24).
MAbs and fusion proteins.
The monoclonal antibodies (MAbs) used in the present study included anti-NKp46 MAbs, 461-G1 (IgG1) (4), and clone 9E2 (IgG1) (11), as well as anti-influenza virus type A (H1), H17-L2, and H28-E23 anti-H1 (kindly provided by Jonathan W. Yewdell). As negative controls, we used anti-CD3 MAb T3D and the anti-CD99 antibody, 12E7 (IgG1). The anti-MICA, MICB, ULBP1-3, and NKp30 antibodies were all purchased from R&D Systems. Anti-NKG2D hybridoma C7 used for the in vivo NKG2D blocking was kindly provided by W. M. Yokoyama.
The fusion proteins used in the present study were generated by fusion of the extracellular of the various human receptors to a human IgG1, as described previously (23). The point mutations in the NKp46 protein—T125V, N216V, and T225V—were generated by using a PCR-based, site-directed mutagenesis approach, as described previously (4).
Viruses and viral infection.
vnh5n1-pr8/cdc-rg H5N1 (abbreviated as H5 [avian]) (16) and the human flu viruses A/Puerto Rico/8/34 H1N1 (abbreviated as A/PR8), A/Texas H3N2 (abbreviated as H3N2), and A/Swine/Israel/2009 H1N1 (abbreviated as H1N1-2009) were generated, and the cells were infected as described previously (1).
Cytotoxicity assay.
The cytotoxic activity of primary bulk NK cells against the various target cells was assessed in 5-h 35S release assays, as previously described (24). In experiments in which MAbs were included, the final MAb concentration was 5 μg/ml. In all assays, the spontaneous release was <25% of the maximal release.
Mice experiments.
All experiments were performed using 12- to 16-week-old C57BL/6 mice. The generation of NKp46/NCR1 knockout mice was previously described (14). For influenza virus infection, mice were anesthetized and inoculated intranasally with 1.6 50% tissue culture infective dose(s) (TCID50) of the H5N1 virus/mouse or with 16 TCID50 of the H1N1-2009 virus/mouse. For NKG2D blocking, 6 h prior to infection 300 μg of C7 purified MAb/mouse was injected intraperitoneally. To measure the infection efficiency, mice were sacrificed 5 or 6 days after virus inoculation; the lungs were then removed, homogenized in Dulbecco modified Eagle medium, and stored at −70°c. To determine the virus titer, the lungs were thawed and homogenized with an OMNI homogenizer, RNA was extracted from the lungs homogenate, and the virus titer was measured by a real-time reverse transcription-PCR (RT-PCR) assay. The virus titer was determined based on the viral genome copy numbers (15).
RESULTS
NKp46 recognizes avian influenza virus-infected cells.
We have previously shown that NKp46, but not NKp30, recognizes cells infected with human influenza viruses (5, 22) and that this interaction is dependent on the sialic acid residues of NKp46 (4). To test whether NKp46 will also recognize avian virus-infected cells, we infected the choriocarcinoma cell line with A/PR8 H1N1 and with another virus containing the core of the A/PR8 virus and the envelope of the H5N1 virus (H5 avian virus) (16). We used this particular H5N1 virus because it differs from A/PR8 H1N1 only in its HA and NA proteins. As shown in Fig. 1A, the H1 and H5 proteins are expressed on the cell surface after the infection. Importantly, as we previously reported earlier (4), increased binding of NKp46-Ig was observed both to the H1 and, as seen here, also to the H5-infected cells (Fig. 1B). In agreement with our previous results, no binding of NKp30-Ig was observed to any of the cells irrespective of whether they were infected or not (Fig. 1B).
FIG. 1.
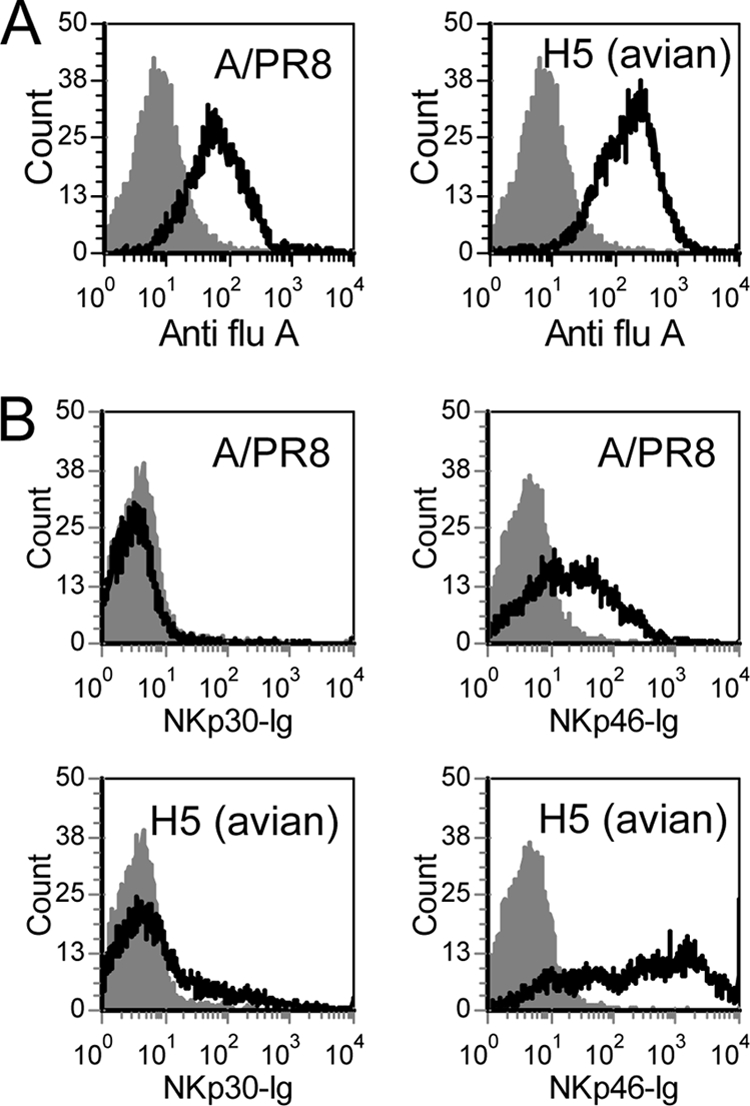
NKp46 recognition of influenza virus-infected Jeg3 cells. fluorescence-activated cell sorting (FACS) staining of uninfected Jeg3 cells (gray filled histogram) or Jeg3 cells infected with A/PR8 or H5 (avian) viruses (black line). Staining was performed with anti-influenza virus A (Anti-flu A), NKp30-Ig, and NKp46-Ig fusion proteins (B) (indicated in the x axis of the figure). The figure shows the results for one representative experiment out of four performed.
We next tested the killing of the Jeg3-infected cells by NK cells. Surprisingly, although NKp46-Ig efficiently recognized the H5-infected Jeg3 cells (Fig. 1), the killing of NK cells was not enhanced (Fig. 2A), even when higher effector-to-target ratios were used (data not shown). In marked contrast, and as we previously reported (22), an increased killing was observed when the Jeg3 cells were infected with the A/PR8 influenza virus (Fig. 2A) or with other human influenza viruses (data not shown). This increased killing of A/PR8 influenza virus-infected cells was due to the interaction between NKp46 and the H1 of A/PR8 virus, as it was blocked by an anti-H1 MAb (Fig. 2B). We therefore concluded that NKp46 could interact with H5, but this recognition was insufficient to activate killing by NK cells.
FIG. 2.
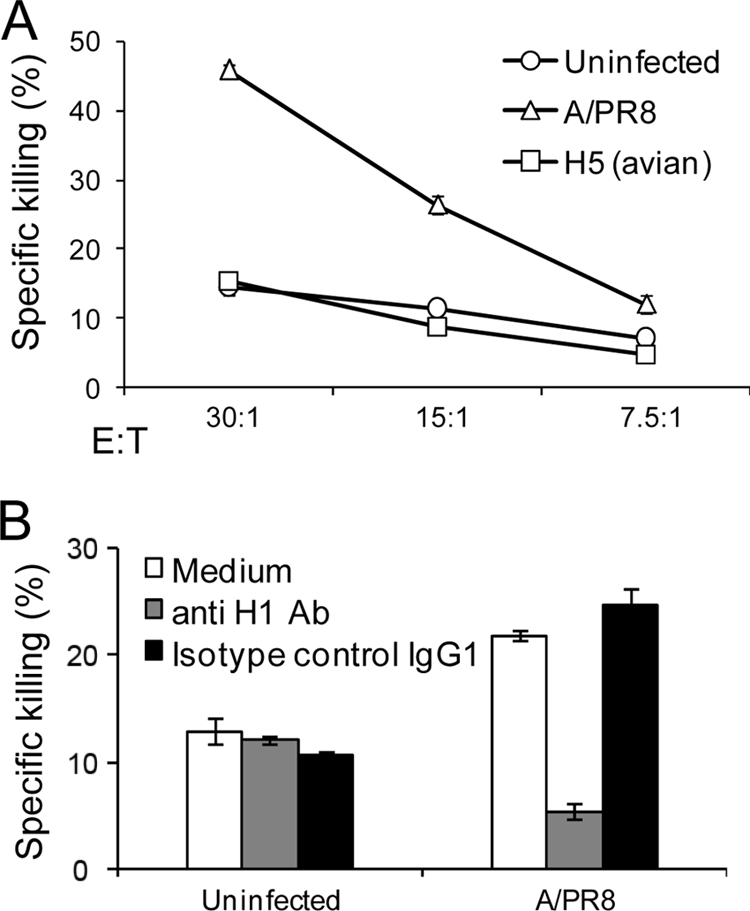
Jeg3 cells infected with avian influenza virus are not killed by NK cells. (A) Killing assay. Jeg3 cells, either uninfected or infected with A/PR8 or H5 (avian) influenza viruses, were tested in killing assays by using bulk NK cells. The effector to target ratio (E:T) is indicated in the figure. (B) The killing of Jeg3 cells is mediated by the interaction with HA. Jeg3 cells infected or not with A/PR8 influenza virus were preincubated with anti-H1 MAb, H28-E23 or with anti-CD99 MAb (negative control) and were tested in killing assays by bulk NK cells at an E:T ratio of 10:1. The figure shows the results for one representative experiment of three performed. Error bars indicate the standard deviations.
Killing of the avian virus-infected cells is possible when several NK activating receptors are engaged.
Our hypothesis to explain these results was that maybe the NKp46-H5 interactions are insufficient by themselves to induce direct killing and that maybe for this particular interaction the help of other killer receptors such as NKG2D, which was shown to participate in influenza virus eradication (12, 34), is needed. This might explain why, when we infected Jeg3 cells that do not express any of the known killer ligands (Fig. 3A) and, indeed, are almost resistant to killing (Fig. 2), the interaction of H5 with NKp46 was insufficient to induce direct killing. To test this option, we used the human hepatoma cell line, Hep3b, which expresses killer ligands such as MICA, MICB, and unknown cellular ligands for the NCRs (Fig. 3B); infected these cells with the A/PR8 or with the H5 viruses; and tested their killing by NK cells. Interestingly, a similar increase in the killing of the infected cells was observed when Hep3b cells were infected with A/PR8 or with H5N1 influenza viruses (Fig. 3C).
FIG. 3.

The killing of the avian-infected cells is possible when several NK activating receptors are engaged. (A) FACS staining of Jeg3 cells for NKG2D ligands. Staining was performed with anti-MICA, MICB, and ULBP1-3 MAbs. (B) FACS staining of Hep3b cells for NKG2D and NCR ligands. Staining was performed with anti-MICA, MICB, ULBP1-3 MAbs, NKp30-Ig, NKp44-Ig, and NKp46-Ig fusion proteins (indicated in the x axis of the figure). (C) Killing experiment of Hep3b cells infected or not with A/PR8 or with H5N1 viruses by bulk NK cells. The various E:T ratios are indicated in the figure. (D) FACS staining of different Jeg3 cells transfected with MICB or with GFP as control. In the three left histograms, staining was performed with an anti-MICB MAb (red line) and with either a negative control MAb (blue line) or with the secondary MAb (black line). In the two right histograms the GFP intensity of the indicated cells is shown. (E) Killing experiment of H5N1-infected Jeg3/GFP and Jeg3/MICB cells by bulk NK cells. The E:T ratio is indicated in the figure. The figure shows the results for one representative experiment out of three performed. Error bars indicate standard deviations.
To directly demonstrate that H5 is able to trigger the NKp46-mediated killing only in the presence of additional ligands, we expressed the MICB protein (a ligand of the activating NKG2D receptor [38]) in Jeg3 cells (Jeg3/MICB) and also expressed GFP (Jeg3/GFP), which was used as a control (Fig. 3D). The various Jeg3 cells were infected with the H5 influenza virus and then used in killing assays (Fig. 3E). As expected, the presence of MICB in Jeg3 cells slightly enhanced the killing of the uninfected cells, probably because it is recognized by the activating receptor NKG2D. Importantly, and as we predicted, the killing of Jeg3/MICB cells infected with the H5 virus was substantially increased compared to the killing of uninfected Jeg3/MICB cells (Fig. 3E).
NKp46 interacts with H5 and H1 in distinct binding sites.
We have previously shown that the binding of NKp46 to the various HAs (excluding H5) is restricted to the membrane-proximal domain of the receptor, requires the sialylation of NKp46, and preferentially requires the Neu5Ac α(2,6)-Gal linkage (4, 22). In contrast, the avian H5 protein preferentially binds to α2,3-sialosides and only weakly binds to certain α2,6-sialosides (36).
NKp46 contains three potential glycosylation sites located at Thr125, Asn216, and Thr225 (Fig. 4A) (29), of which Thr225 is the main amino acid residue, critically involved in the HA interaction with NKp46 (4). To test whether the lack of direct killing of H5-expressing cells could be explained by a differential binding of NKp46 to H1 versus H5, we mutated the three potential glycosylation residues of NKp46 by replacing them with valine (T125V, N216V, and T225V). The mutated NKp46 proteins were appended to human Fc, and the binding of the mutated proteins was compared to the binding of the wild-type NKp46 protein. Importantly, whereas only a slight reduction in the binding of the mutated protein T125V-Ig to the H5-infected Jeg3 was observed, both N216V-Ig and T225V-Ig showed markedly reduced binding to H5-infected Jeg3 cells (Fig. 4B). Thus, whereas T225 plays a crucial role in the interaction between H1 and NKp46 (4) (see also Fig. 6 below), with regard to H5, two glycosylation sites on NKp46, located at positions N216 and T225, are involved in the binding.
FIG. 4.

Glycosylation at amino acids Asp216 and Thr225 of NKp46 are involved in its binding to H5-infected cells. (A) Amino acid sequences of human NKp46. The first Ig and the second Ig domains are indicated in green and in blue, respectively. The transmembrane domain is underlined, and the three putative glycosylation sites are indicated in bold red. (B) H5N1 (avian) influenza virus-infected or noninfected Jeg3 cells were stained with NKp46-Ig (black and blue lines, respectively) and with the indicated mutated proteins (uninfected [purple] and infected [red]). The figure shows the results of one representative experiment of three performed.
FIG. 6.

The H1N1-2009-NKp46 interaction is similar to other human origin influenza virus HAs. (A) Jeg3 cells infected or not infected with 19 different isolations of H1N1-2009 (the isolation number is indicated at the x axis) that were obtained from different Israeli patients. Jeg3 cells were stained either with NKp46-Ig or with T225V-Ig (white and gray bars, respectively). (B) The cells in panel A were tested for killing by NK cells in E:T ratio of 25:1. Error bars indicate standard deviations.
To further establish that the binding sites of H5 and H1 in NKp46 are distinct, we tested the ability of different anti-NKp46 MAbs to inhibit the NKp46-mediated killing of Jeg3/MICB cells infected with the A/PR8 or with the H5 viruses. As shown in Fig. 5, the 461-G1 MAb, which is directed against the D1 domain of NKp46 (a domain that does not contain glycosylation and is not involved in the killing of influenza virus-infected cells [4]), did not block the killing. Importantly, blocking of NKp46 with the 9E2 MAb significantly reduced the killing of the H5-infected cells but did not affect the killing of the A/PR8-infected cells (Fig. 5), suggesting that indeed NKp46 interacts with H1 and H5 at different binding sites.
FIG. 5.
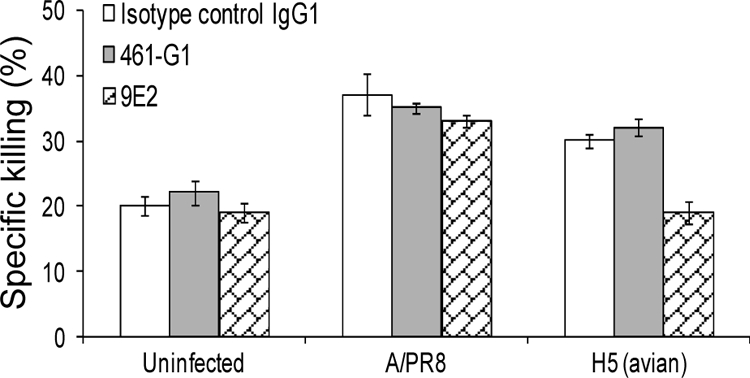
The binding sites of H1 and H5 to NKp46 are distinct. Jeg3/MICB cells were infected with A/PR8 or with H5 (avian) viruses, and their killing by bulk NK cells was tested in the presence or absence of the indicated anti-NKp46 MAbs. The E:T ratio is 60:1. The figure shows the results for one representative experiment of three performed. Error bars indicate standard deviations.
NKp46 interaction with H1 of the H1N1-2009 virus leads to direct NK cytotoxicity.
In June 2009 the World Health Organization (WHO) raised the influenza pandemic to level 6, stating that the current H1N1-2009 infection is the first pandemic of this century and the first in the last 41 years. To test whether the H1 HA of the H1N1-2009 virus would be recognized by NKp46, we infected Jeg3 cells with 19 different H1N1-2009 viruses that were isolated from Israeli patients. The infected cells were tested for binding of NKp46-Ig and NKp46 T225V-Ig proteins (Fig. 6A) and for killing by NK cells (Fig. 6B). Interestingly, we found that NKp46-Ig binds to all cells infected with the 19 different H1N1-2009 viruses (Fig. 6A) and the binding was correlated with the efficiency of Jeg3-mediated killing (Fig. 6B). Little or no binding of T225V-Ig was observed (Fig. 6A), confirming our previous observations (4) that the glycosylation at position 225 of NKp46 is important for the NKp46-H1 interactions. Thus, while the avian influenza virus recognition by NKp46 is unable to directly induce NK cytotoxicity, the recognition of the swine influenza virus by NKp46 plays a critical role in the killing of the infected cells.
Engagement of NKG2D is essential for in vitro and in vivo NKp46-mediated killing of avian influenza virus.
If indeed the NKp46-H5 interactions are insufficient to induce direct NKp46-mediated killing by themselves, then by blocking the NKG2D receptor we should be able to reduce the NKp46-mediated killing of the H5-infected cells. To test this assumption, we performed a redirected killing assay in which we infected the P815 cell line with H5N1 avian influenza virus or with the human influenza virus strain A/H3N2. We did not use the A/PR8 or the H1N1-2009 viruses since these viruses could not infect the P815 cells (data not shown). As expected, the uninfected and the H5-infected P815 cells were not killed when we used a control MAb, since uninfected P815 cells do not express lysis ligands for NK cells and, as shown above, the interaction between NKp46 and H5 is insufficient by itself to induce direct killing. In contrast, the H3N2-infected P815 cells were killed by NK cells, since the H3-NKp46 interaction leads to direct NKp46-mediated NK cell killing (4) (Fig. 7A). When we induced the redirected killing of the uninfected P815 cells with anti-NKG2D MAb, an efficient killing of the P815 cells was observed, since NKG2D is a potent killer receptor and its triggering leads to direct cytotoxicity (Fig. 7A). Importantly, when we incubated either the H3N2 or the H5N1-infected P815 cells with anti-NKG2D MAb an increased and equivalent redirected killing of both infected cells was observed (Fig. 7A). Thus, when NKp46 interacts with H5, an efficient NK killing of the H5-infected cells is observed only when the NKG2D receptor is also engaged (either by MAb as seen in Fig. 7A or through the interaction with MICB, as seen in Fig. 3E).
FIG. 7.

Blocking of NKG2D activity during infection. (A) Redirected killing assay. 35S-labeled P815 cells were infected or not with H5 (avian virus) or H3N2 (human influenza virus) and then assayed in a redirected killing assay with human NK cells at E:T of 3:1 in the presence of either anti-NKG2D MAb or control IgG1 MAb. (B) Bulk NK cells were preincubated with anti-NKG2D MAb or with anti-CD99 MAb (negative control) and were tested in killing assays at an E:T ratio of 10:1 against Jeg3/MICB cells that were infected or not infected with A/PR8 or H5N1 influenza viruses. (C) Anti-NKG2D receptor antibody was injected or not injected into C57BL/6 mice (WT) or C57BL/6 NCR1gfp/gfp mice (KO) 6 h prior to their intranasal inoculation with 16 TCID50 of the H1N1-2009 virus (isolation number 926)/mouse or with 1.6 TCID50 of the H5N1 virus/mouse. Five or six days (for avian and H1N1-2009 infections, respectively) after infection the mice were sacrificed, their lungs were homogenized, RNA was extracted, and the presence of virus in the infected lungs was detected by specific primers using real-time RT-PCR. The virus titer was determined based on the viral genome copy numbers.
Next, we tested the ability of the anti-NKG2D MAb to block the NKp46-mediated killing of the Jeg3/MICB cells. For this purpose, we infected the Jeg3/MICB cells with A/PR8 or with H5 viruses and tested their killing by NK cells that were preincubated with anti-NKG2D or with control MAb. As can be seen in Fig. 7B, the anti-NKG2D MAb did not block the killing of the H1-infected Jeg3/MICB cells since the H1-NKp46 interactions are still intact and, as shown above, such interactions lead to direct cytotoxicity. In contrast, blocking of the NKG2D interaction in the H5-infected Jeg3/MICB cells significantly reduced the killing, further supporting the observations that the killing induced by the H5-NKp46 interaction is possible only when other lysis ligands are engaged (Fig. 7B).
To test whether the NKp46 interactions with H5 and the H1N1-2009 would be important under physiological conditions, we infected our NCR1gfp/gfp (the mouse homologue of NKp46 [14]) knockout mice (KO) and the corresponding C57BL/6 wild-type (WT) mice with the H5N1 and the H1N1-2009 viruses. Mice were sacrificed 5 and 6 days after infection (for H5N1 and H1N1-2009, respectively), and the amounts of viruses that were present in the lungs were measured as described in Materials and Methods. To test the in vivo involvement of NKG2D in influenza virus eradication, we also infected additional groups of mice with both viruses and injected, prior to the infection, the anti-NKG2D MAb C7 that is known to block NKG2D function in vivo. As shown in Fig. 7C, in the absence of NKp46 (KO mice) the presence of both H5N1 and H1N1-2009 viruses was observed in the lungs of the infected animals, whereas in the WT mice, few or no viruses were found (Fig. 7C). Strikingly and in agreement with our in vitro results, blocking of the NKG2D receptor in the NKp46 KO mice had no effect on the H1N1-2009 virus titers, probably because for this virus NKp46 is the major killer receptor involved in the killing of the infected cells. In contrast, when NKG2D was blocked prior to H5 infection, a 2-fold increase in the virus titer was observed, suggesting that NKG2D is involved in vivo in the killing of the H5 virus. It seems, however, that the NKG2D activity is secondary to that of NKp46 because in both viruses when the NKG2D interactions were blocked in vivo in the WT mice, viruses were still not detected in the lungs (Fig. 7C).
In conclusion, we demonstrate here both in vitro and in vivo that NKp46 directly interacts with both the H1N1-2009 and the avian viruses. However, whereas the interaction of NKp46 with H1 is sufficient to induce direct killing, the NKp46 interaction with H5 is not sufficient to induce direct killing and requires the “help” of additional receptors.
DISCUSSION
Influenza epidemics occur every year, and every several decades an influenza pandemic that claims the lives of millions arises. Although the world's greatest concern in the last few years was an avian H5N1 influenza pandemic, a novel subtype of influenza virus A (H1N1), leading to what is now considered to be the first pandemic in the last 41 years and the first in the 21st century. This virus, H1N1-2009, first appeared in Mexico in March 2009 and in 3 months spread to more than 80 countries (http://www.who.int/csr/don/2009_06_22/en/index.html). Although the WHO considers the severity of this pandemic at this time to be moderate, the severity might change over time, as occurred in the “Spanish Flu” pandemic (27).
In contrast to the limited mortality caused by the H1N1-2009, infection with the avian influenza virus (H5N1) results in a severe mortality rate of ca. 60% (http://www.who.int/csr/disease/avian_influenza/country/cases_table_2009_06_02/en/index.html). One possible explanation for the extreme virulence of the H5N1 virus might be the preferential binding of the avian H5 to α2,3-SA residues (33), which are located mainly on proteins found at the human lower respiratory tract (39).
We show here that, in contrast to H1N1-2009 virus and all other influenza viruses studied thus far (4, 17, 22), the H5-NKp46 interactions are unable to directly activate the NKp46-mediated killing by themselves. Indeed, it has been shown that under certain conditions NKp46 acts as a costimulating receptor and not as a direct cytotoxic receptor. Bryceson et al. demonstrated that in resting nonactivated NK cells, triggering of NKp46 is insufficient to activate cytotoxicity or cytokine secretion, whereas in interleukin-2-activated NK cells, NKp46 could mediate direct killing (9). Indeed, when additional ligands of NK activating receptors are present on the H5N1-infected cells, or in vivo, when other activating ligands such as the ligands for NKG2D are also upregulated due to the infection (12, 34; the present study), NKp46 interactions with H5 significantly contribute to virus killing.
We propose that the H5-NKp46 interactions are unable to induce direct cytotoxicity, probably because H5 binds NKp46 in a site that is distinct from that of H1. We have previously demonstrated that the glycosylation on the Thr225 residue is critical for the interactions of H1 with NKp46 (4); we further show here that the glycosylation at Thr225 is also important for the interaction of NKp46 with the H1-2009 protein. In contrast, the recognition of H5 by NKp46 depends on both the Asp216 and the Thr225 residues. Interestingly, both residues are located in the stalk region of NKp46, whereas the third glycosylated residue is located between domains 1 and 2 and therefore might not be accessible for binding (13).
The invading viruses and the immune system are in constant battle. The influenza viruses, similar to other viruses, have developed several ways to escape from immune system recognition. For example, a high frequency of mutations enables the virus to constantly change its surface glycoproteins and thereby escape from recognition by cytotoxic T lymphocytes and MAbs (32). We showed that the influenza virus also induces the reorganization of major histocompatibility complex (MHC) class I proteins on the cell surface. MHC class I proteins enter into rafts early after infection to enhance the recognition by NK inhibitory receptors (1, 2). On the other hand, we suggest that NKp46 has evolved to use the conserved ability of various human HAs to bind α2,6-SA and “utilize” this property to directly kill the infected cells in an α2,6-SA-NKp46-dependent manner (4). This is probably why the swine influenza virus, which interacts with α2,6-SA (19), is directly killed in an NKp46-dependent manner. Unfortunately, due to the lack of specific blocking MAbs, we could not directly demonstrate NKp46-dependent killing of the swine influenza viruses. However, the direct correlation observed between the binding of NKp46 and the killing of the H1N1-2009 influenza virus-infected cells, together with the impressive enhancement of viral load observed in the absence of NKp46, strongly suggests that similar to other human influenza viruses (4), the interaction between NKp46 and the swine H1 virus is crucial for the killing of the infected cells.
With regard to the avian influenza virus, we suggest that NKp46 did not evolve to interact with avian HA, and therefore the NKp46 interactions with H5 through its α2,3-SA are insufficient to activate the NKp46-mediated killing of NK cells. Our final conclusion is that it might be that the avian H5N1 virus is so deadly not only due to its ability to infect the lower respiratory track but perhaps also due to its inability to directly activate NKp46. It is therefore possible that if genetic changes in the H5N1 virus would occur to enable its transmission from human to human, the emerging virus would not be more violent but may be less dangerous, since the infected cells would be recognized directly by NKp46 and the virus might be better eliminated, in a similar manner to that of the H1N1-2009 influenza virus.
Acknowledgments
This study was supported by grants from the U.S.-Israel Binational Science Foundation, the Israeli Cancer Research Foundation, the Israeli Science Foundation, the Israel Science Foundation (Morasha), the Association for International Cancer Research, the DKFZ-MOST, and an Israel-Croatia research grant (all to O.M.). O.M. is a Crown Professor of Molecular Immunology.
Footnotes
Published ahead of print on 3 February 2010.
REFERENCES
- 1.Achdout, H., T. I. Arnon, G. Markel, T. Gonen-Gross, G. Katz, N. Lieberman, R. Gazit, A. Joseph, E. Kedar, and O. Mandelboim. 2003. Enhanced recognition of human NK receptors after influenza virus infection. J. Immunol. 171:915-923. [DOI] [PubMed] [Google Scholar]
- 2.Achdout, H., I. Manaster, and O. Mandelboim. 2008. Influenza virus infection augments NK cell inhibition through reorganization of major histocompatibility complex class I proteins. J. Virol. 82:8030-8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnon, T. I., H. Achdout, O. Levi, G. Markel, N. Saleh, G. Katz, R. Gazit, T. Gonen-Gross, J. Hanna, E. Nahari, A. Porgador, A. Honigman, B. Plachter, D. Mevorach, D. G. Wolf, and O. Mandelboim. 2005. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 6:515-523. [DOI] [PubMed] [Google Scholar]
- 4.Arnon, T. I., H. Achdout, N. Lieberman, R. Gazit, T. Gonen-Gross, G. Katz, A. Bar-Ilan, N. Bloushtain, M. Lev, A. Joseph, E. Kedar, A. Porgador, and O. Mandelboim. 2004. The mechanisms controlling the recognition of tumor- and virus-infected cells by NKp46. Blood 103:664-672. [DOI] [PubMed] [Google Scholar]
- 5.Arnon, T. I., M. Lev, G. Katz, Y. Chernobrov, A. Porgador, and O. Mandelboim. 2001. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur. J. Immunol. 31:2680-2689. [DOI] [PubMed] [Google Scholar]
- 6.Arnon, T. I., G. Markel, and O. Mandelboim. 2006. Tumor and viral recognition by natural killer cells receptors. Semin. Cancer Biol. 16:348-358. [DOI] [PubMed] [Google Scholar]
- 7.Biassoni, R. 2008. Natural killer cell receptors. Adv. Exp. Med. Biol. 640:35-52. [DOI] [PubMed] [Google Scholar]
- 8.Brandt, C. S., M. Baratin, E. C. Yi, J. Kennedy, Z. Gao, B. Fox, B. Haldeman, C. D. Ostrander, T. Kaifu, C. Chabannon, A. Moretta, R. West, W. Xu, E. Vivier, and S. D. Levin. 2009. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. doi: 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed]
- 9.Bryceson, Y. T., M. E. March, H. G. Ljunggren, and E. O. Long. 2006. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107:159-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerwenka, A., and L. L. Lanier. 2001. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 1:41-49. [DOI] [PubMed] [Google Scholar]
- 11.Chen, Y., B. Perussia, and K. S. Campbell. 2007. Prostaglandin D2 suppresses human NK cell function via signaling through D prostanoid receptor. J. Immunol. 179:2766-2773. [DOI] [PubMed] [Google Scholar]
- 12.Draghi, M., A. Pashine, B. Sanjanwala, K. Gendzekhadze, C. Cantoni, D. Cosman, A. Moretta, N. M. Valiante, and P. Parham. 2007. NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J. Immunol. 178:2688-2698. [DOI] [PubMed] [Google Scholar]
- 13.Foster, C. E., M. Colonna, and P. D. Sun. 2003. Crystal structure of the human natural killer (NK) cell activating receptor NKp46 reveals structural relationship to other leukocyte receptor complex immunoreceptors. J. Biol. Chem. 278:46081-46086. [DOI] [PubMed] [Google Scholar]
- 14.Gazit, R., R. Gruda, M. Elboim, T. I. Arnon, G. Katz, H. Achdout, J. Hanna, U. Qimron, G. Landau, E. Greenbaum, Z. Zakay-Rones, A. Porgador, and O. Mandelboim. 2006. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 7:517-523. [DOI] [PubMed] [Google Scholar]
- 15.Hindiyeh, M., V. Levy, R. Azar, N. Varsano, L. Regev, Y. Shalev, Z. Grossman, and E. Mendelson. 2005. Evaluation of a multiplex real-time reverse transcriptase PCR assay for detection and differentiation of influenza viruses A and B during the 2001-2002 influenza season in Israel. J. Clin. Microbiol. 43:589-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horimoto, T., and Y. Kawaoka. 2006. Strategies for developing vaccines against H5N1 influenza A viruses. Trends Mol. Med. 12:506-514. [DOI] [PubMed] [Google Scholar]
- 17.Jarahian, M., C. Watzl, P. Fournier, A. Arnold, D. Djandji, S. Zahedi, A. Cerwenka, A. Paschen, V. Schirrmacher, and F. Momburg. 2009. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J. Virol. 69:8420-8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilbourne, E. D. 2006. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 12:9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maines, T. R., A. Jayaraman, J. A. Belser, D. A. Wadford, C. Pappas, H. Zeng, K. M. Gustin, M. B. Pearce, K. Viswanathan, Z. H. Shriver, R. Raman, N. J. Cox, R. Sasisekharan, J. M. Katz, and T. M. Tumpey. 2009. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 24:484-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maines, T. R., K. J. Szretter, L. Perrone, J. A. Belser, R. A. Bright, H. Zeng, T. M. Tumpey, and J. M. Katz. 2008. Pathogenesis of emerging avian influenza viruses in mammals and the host innate immune response. Immunol. Rev. 225:68-84. [DOI] [PubMed] [Google Scholar]
- 21.Manaster, I., and O. Mandelboim. 2008. The unique properties of human NK cells in the uterine mucosa. Placenta 29(Suppl. A):S60-S66. [DOI] [PubMed] [Google Scholar]
- 22.Mandelboim, O., N. Lieberman, M. Lev, L. Paul, T. I. Arnon, Y. Bushkin, D. M. Davis, J. L. Strominger, J. W. Yewdell, and A. Porgador. 2001. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409:1055-1060. [DOI] [PubMed] [Google Scholar]
- 23.Mandelboim, O., P. Malik, D. M. Davis, C. H. Jo, J. E. Boyson, and J. L. Strominger. 1999. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 96:5640-5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandelboim, O., H. T. Reyburn, M. Vales-Gomez, L. Pazmany, M. Colonna, G. Borsellino, and J. L. Strominger. 1996. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J. Exp. Med. 184:913-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moretta, A., C. Bottino, M. Vitale, D. Pende, C. Cantoni, M. C. Mingari, R. Biassoni, and L. Moretta. 2001. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19:197-223. [DOI] [PubMed] [Google Scholar]
- 26.Munster, V. J., E. de Wit, J. M. van den Brand, S. Herfst, E. J. Schrauwen, T. M. Bestebroer, D. van de Vijver, C. A. Boucher, M. Koopmans, G. F. Rimmelzwaan, T. Kuiken, A. D. Osterhaus, and R. A. Fouchier. 2009. Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets. Science 24:971-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann, G., T. Noda, and Y. Kawaoka. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pappaioanou, M. 2009. Highly pathogenic H5N1 avian influenza virus: cause of the next pandemic? Comp. Immunol. Microbiol. Infect. Dis. 32:287-300. [DOI] [PubMed] [Google Scholar]
- 29.Pessino, A., S. Sivori, C. Bottino, A. Malaspina, L. Morelli, L. Moretta, R. Biassoni, and A. Moretta. 1998. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 188:953-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pogge von Strandmann, E., V. R. Simhadri, B. von Tresckow, S. Sasse, K. S. Reiners, H. P. Hansen, A. Rothe, B. Boll, V. L. Simhadri, P. Borchmann, P. J. McKinnon, M. Hallek, and A. Engert. 2007. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 27:965-974. [DOI] [PubMed] [Google Scholar]
- 31.Raulet, D. H. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3:781-790. [DOI] [PubMed] [Google Scholar]
- 32.Scholtissek, C. 1994. Source for influenza pandemics. Eur. J. Epidemiol. 10:455-458. [DOI] [PubMed] [Google Scholar]
- 33.Shinya, K., M. Ebina, S. Yamada, M. Ono, N. Kasai, and Y. Kawaoka. 2006. Avian flu: influenza virus receptors in the human airway. Nature 440:435-436. [DOI] [PubMed] [Google Scholar]
- 34.Siren, J., T. Sareneva, J. Pirhonen, M. Strengell, V. Veckman, I. Julkunen, and S. Matikainen. 2004. Cytokine and contact-dependent activation of natural killer cells by influenza A or Sendai virus-infected macrophages. J. Gen. Virol. 85:2357-2364. [DOI] [PubMed] [Google Scholar]
- 35.Stern-Ginossar, N., N. Elefant, A. Zimmermann, D. G. Wolf, N. Saleh, M. Biton, E. Horwitz, Z. Prokocimer, M. Prichard, G. Hahn, D. Goldman-Wohl, C. Greenfield, S. Yagel, H. Hengel, Y. Altuvia, H. Margalit, and O. Mandelboim. 2007. Host immune system gene targeting by a viral miRNA. Science 317:376-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens, J., O. Blixt, L. M. Chen, R. O. Donis, J. C. Paulson, and I. A. Wilson. 2008. Recent avian H5N1 viruses exhibit increased propensity for acquiring human receptor specificity. J. Mol. Biol. 381:1382-1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens, J., O. Blixt, T. M. Tumpey, J. K. Taubenberger, J. C. Paulson, and I. A. Wilson. 2006. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 312:404-410. [DOI] [PubMed] [Google Scholar]
- 38.Sutherland, C. L., N. J. Chalupny, and D. Cosman. 2001. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 181:185-192. [DOI] [PubMed] [Google Scholar]
- 39.van Riel, D., V. J. Munster, E. de Wit, G. F. Rimmelzwaan, R. A. Fouchier, A. D. Osterhaus, and T. Kuiken. 2006. H5N1 virus attachment to lower respiratory tract. Science 312:399. [DOI] [PubMed] [Google Scholar]
- 40.Yokoyama, W. M., S. Kim, and A. R. French. 2004. The dynamic life of natural killer cells. Annu. Rev. Immunol. 22:405-429. [DOI] [PubMed] [Google Scholar]