

Abstract
The role of p53 in primary effusion lymphoma (PEL) is complicated. The latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus (KSHV) binds p53. Despite this interaction, we had found that p53 was functional in PEL, i.e., able to induce apoptosis in response to DNA damage (C. E. Petre, S. H. Sin, and D. P. Dittmer, J. Virol. 81:1912-1922, 2007), and that hdm2 was overexpressed. To further elucidate the relationship between LANA, p53, and hdm2, we purified LANA complexes from PEL by column chromatography. This confirmed that LANA bound p53. However, the LANA:p53 complexes were a minority compared to hdm2:p53 and p53:p53 complexes. The half-life of p53 was not extended, which is in contrast to the half-life of simian virus 40 T antigen-transformed cells. p53:p53, LANA:p53, and LANA:LANA complexes coexisted in PEL, and each protein was able to bind to its cognate DNA element. These data suggest that under normal conditions, p53 is inactive in PEL, thus allowing for exponential growth, but that this inactivation is driven by the relative stoichiometries of LANA, hdm2, and p53. If p53 is activated by DNA damage or nutlin-3a, the complex falls apart easily, and p53 exercises its role as guardian of the genome.
Kaposi's sarcoma-associated herpesvirus (KSHV) is found in Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and tumors from patients with the plasmablastic variant of multicentric Castleman disease (MCD) (reviewed in reference 15). The latency-associated nuclear antigen (LANA) is encoded by open reading frame 73 (ORF 73) of the viral genome. It is expressed in every KSHV-infected cell during the latent as well as lytic phase of the viral life cycle. LANA is required for replication and maintenance of the viral DNA during latent infection (2, 13). Experimental abrogation of LANA expression through small interfering RNA (siRNA) or genomic knockout leads to loss of KSHV from latently infected cells, genetically demonstrating that LANA is necessary for maintenance of latency (28, 103). LANA is a large protein composed of 1,162 amino acids (KSHV M type, reference sequence NC_003409). It has many known biochemical activities, and many more have yet to be determined. One way to think about LANA is as a nuclear scaffolding protein for viral DNA replication and transcription, analogous to the large T antigen of the polyomaviruses.
We hypothesized that there exist multiple biochemically distinct LANA complexes in KSHV-infected PEL, as both LANA and its many potential interaction partners are present at physiological molar ratios to each other. Biochemical analysis of purified native complexes in PEL allowed us to establish that not all of LANA was part of a LANA:p53 complex and that not all of p53 was bound to LANA.
The C terminus of the LANA protein has sequence-specific DNA binding activity. LANA can bind to a 20-bp repeated cis element in the terminal repeats (TRs) of the viral genome (3, 26, 27, 45, 50, 78, 85, 101). We previously identified a similar cis element that was present as a single copy within the LANA core promoter (39). Purified LANA C-terminal peptide bound to this element in vitro, and LANA positively transactivated the LANA promoter, albeit modestly, in transient-transfection experiments. The N terminus of LANA tethers the viral genome to host cell chromosomes through protein-protein interaction (2, 13, 41, 42, 44, 74, 90). To date, chromatin-associated structural proteins, including the linker histone H1 and the core histones H2A and H2B, have been shown to bind LANA in latently infected cells (4, 13, 81). By immunofluorescence, the LANA protein appears with a characteristic punctate pattern in interphase nuclei. It also decorates mitotic chromosomes. These phenotypes are consistent with a model that places the majority of LANA within high-molecular-weight, chromosome-associated structural complexes.
In addition, LANA can bind many cellular proteins directly. The list of LANA interaction partners is ever increasing and to date includes cellular replication and replication licensing factors (62, 87, 96), cellular methyltransferases (80), and the cellular transcription factor Sp1 (62, 95). The list also includes RBP-jkappa (also known as CSL) (55), p53 (23), GSK-3beta (24), CBP (58), ATF4/CREB2 (59), Ring3 (63, 68, 72, 98), Rta (56), SAP30, mSin3A, and CIR (52), meCP2, and DEK (51). LANA binds to and inhibits Rb (73), and LANA binds to and inhibits p53 function (23, 100). As suggested above, it is unlikely that a single, giant LANA complex contains to all of these proteins at the same time.
p53 is the pivotal protein that coordinates the cellular response to molecular insults and stress. The primary function of p53 is to sense DNA damage. DNA damage induction is the molecular mechanism of many chemotherapeutic drugs, for instance as induced by the anthracycline drug doxorubicin. Upon activation, p53 is stabilized and induces cell cycle arrest or apoptosis through transcriptional activation of downstream targets (reviewed in reference 30). p53 is among the most frequently mutated genes in human cancer (67). Many believe that mutation of p53 is mutagenic and allows rapid accumulation of subsequent genomic abnormalities. Typically, p53 sustains missense amino acid point mutations, accompanied by reduction to homozygocity (33). Unlike with the prototypical tumor suppressor gene Rb, gene deletions are rare. p53 missense mutations cluster in a tumor lineage-specific pattern (67). They almost invariably affect p53 DNA binding and transcriptional capability. Since p53 acts as a tetramer, many tumor-derived p53 mutations exhibit a dominant negative phenotype, while others show evidence for gain of function (17).
p53 is rarely mutated in the KSHV-associated malignancies KS or PEL (40, 66, 71). KS and PEL respond clinically to doxorubicin (reviewed in reference 19), a phenotype consistent with wild-type p53 status. Treatment of PEL with p53-activating drugs, such as doxorubicin, induces serine 15 (S15) phosphorylation on p53 followed by a robust transcriptional induction of p53 target genes, including p21 (71, 77). This suggests that both the upstream p53-activating signaling pathway and the downstream p53 transcriptional activation-dependent signaling pathways are operational in PEL. Exceptions are two PEL cell lines which contain mutant p53 and which are resistant to doxorubicin (71). Evaluation of their clinical history revealed that these two cell lines (BCP-1 and BCBL-1) were isolated from patients who were heavily treated with and failed DNA-damaging-agent-based chemotherapy.
hdm2 (mdm2 in mice) is the most important cellular binding partner of p53 (reviewed in reference 30). Under normal growth conditions, p53 protein levels are low. The half-life (t1/2) of p53 protein is short (5 to 20 min) because hdm2 targets p53 for degradation. hdm2 is an E3 ligase (32, 53). hdm2 itself is a p53 target gene whose transcription is induced upon p53 activation and thus generates a feedback mechanism for regulating p53 activity (102). hdm2 also targets itself for ubiquitination and subsequent degradation (22, 34).
hdmX/hdm4 (mdmX in mouse) is an hdm2 homologue that also binds to p53. It inhibits p53-dependent transcription (82). hdmX does not have E3 ligase activity. It is thought to inhibit hdm2 function by heterodimer formation (37, 86, 91). However, there is also evidence suggesting that hdmX can act as a positive regulator of hdm2. The relative ratios of p53, hdm2, and hdmX determine the sensitivity of wild-type p53 tumor cells to the hdm2:p53-intercalating agent nutlin-3a and to DNA damage (70, 99).
Our biochemical approach confirmed that LANA bound p53. We found that this interaction was nutlin-3a sensitive, even though only a minority of LANA bound hdm2. The LANA:p53 complexes were a minority compared to hdm2:p53 and p53:p53 complexes.
MATERIALS AND METHODS
Cell culture and antibodies.
PEL cell lines BC-3, and BCP-1 and the virus-negative lymphoma cell lines BJAB and DG-75 were cultured in RPMI 1640 medium (Cellgro) containing 2 mM l-glutamine, 10% fetal bovine serum, penicillin G (100 U/ml), and streptomycin sulfate (100 μg/ml). The medium was supplemented with 0.05 mM 2-mercaptoethanol (Sigma), 0.075% sodium bicarbonate (Life Technologies), and 1 U/ml human interleukin 6 (IL-6) (Roche). Cells were grown at 37°C in a humidified environment supplemented with 5% carbon dioxide (CO2).
Anti-LANA polyclonal antibody YT041 came from a rabbit immunized with the LANA peptide coupled to keyhole limpet hemocyanin (KLH), H2N-LEEQEQELEEQEQELEEQEQEC-COOH. The underlined sequence indicates the epitope of the commercially available monoclonal rat anti-LANA antibody (Advanced Biotechnology Inc.). This antibody recognizes the LANA H2N-EQEQE-COOH repeat (43). Additional antibodies were anti-mdm2 mouse monoclonal antibody (MAb) (Calbiochem), anti-hdm2 (SMP-14) mouse MAb (Santa Cruz), anti-hdmX affinity-purified polyclonal rabbit antibodies (Bethyl), anti-53 mouse MAb (PAb421) (Calbiochem), anti-53 mouse MAb (DO-1), anti-cdk6 (Santa Cruz), anti-β-actin mouse MAb (Sigma), and anti-SP1 mouse MAb (Chemicon).
Purification of LANA.
For the isolation of nuclear LANA complexes, nuclear extraction was performed upon BC-3 and BJAB cells (all steps were performed at 4°C) by following the method of Dignam et al. (16). Approximately 5 × 109 to approximately 8 × 109 BC-3 cells were harvested and washed with cold phosphate-buffered saline (PBS) twice. First, the cells were suspended in buffer A (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT], 0.5 mM phenylmethylsulfonyl fluoride [PMSF], and 0.5% cocktail protein inhibitor) for 30 min and then lysed by stroking them in a glass Dounce homogenizer. The crude nuclei were then collected by centrifugation at 10,000 rpm for 20 min and suspended in buffer C (20 mM HEPES-KOH, pH 7.9, 10% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, and 0.5% cocktail protein inhibitor). After several strokes of gentle Dounce homogenization, the resulting suspension was shaken gently with rotation for 45 min and then centrifuged at 10,000 rpm for 30 min. The supernatant was collected and used for subsequent procedures. The nuclear extract was subjected to chromatography. After optimization, we arrived at the following conditions. First, the extract was loaded onto a Sepharose 6B column (Sigma) equilibrated with Dignam buffer D (20 mM HEPES-KOH, pH 7.9, 10% glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM PMSF, 0.5 mM DTT, and 0.5% cocktail protein inhibitor). At a flow rate of 0.5 ml/min, each of the column fractions (1.5-ml fraction size) was collected and analyzed for the presence of LANA by Western blotting with corresponding antibodies. Continuous fractions containing LANA were pooled and loaded onto a Hiprep 16/10 Heparin FF column. After a thorough washing, the fractions were eluted with 0.5 M KCl containing buffer D (1-ml/min flow rate, 1.5-ml/min fraction size). The Heparin FF fractions that contained LANA were diluted into 0.1 M KCl and loaded onto a MonoQ column (Sigma). This was followed by two-step elution using 0.4 M KCl and 0.7 M KCl in buffer D. Finally, the corresponding fractions from different peaks were collected (0.5-ml/min flow rate, 1.5-ml/min fraction size) and analyzed.
Western blotting and immunoprecipitation.
The column fractions indicated in the figures were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Hybond P membranes (Amersham), nonspecific binding was blocked with 5% nonfat milk overnight at 4°C, and the membranes were incubated with the corresponding primary antibody for 2 h at room temperature at the concentration indicated below and washed three times with TBST (10 mM Tris-HCl, 100 mM NaCl, pH 8.0, 0.1% Tween 20). After the membranes were incubated with a horseradish peroxidase-linked secondary antibody (VWR International) at 1:3,000, bands were visualized by enhanced chemiluminescence (Thermo Scientific) and exposed to X-ray film (Genesee Scientific). For immunoprecipitation assays, fractions were precleared with protein A/G agarose beads (Sigma) for 1 h and then incubated with the appropriate antibodies overnight and then with 40 μl protein A/G agarose beads, with a gentle rotation for 2 h at 4°C. The resulting immunoprecipitates were collected by centrifugation at 10,000 × g for 1 min. The pellets were washed four times with ice-cold RIPA buffer (150 mM NaCl, 1% NP-40, 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, and 0.5% cocktail protein inhibitor). The samples were then loaded onto an SDS-PAGE gel.
Subcellular fractionation.
BC-3 cells (5 × 106) were collected by centrifugation at 250 × g for 5 min at 4°C, followed by washing with ice-cold PBS twice. Then extraction buffers I, II, III, and IV were added separately according to the manufacturer's instructions using the subcellular proteasome extraction kit (Calbiochem).
Protein half-life determination.
BC-3, BJAB, BCP-1, and DG-75 cells were grown with RPMI 1640 medium in six-well plates. Cells were treated with 50 μg/ml of the protein translation inhibitor cycloheximide (Sigma). Samples were collected from individual wells at 0, 10, 30, 60, and 120 min after cycloheximide treatment (50 μg/ml). The steady-state levels of LANA complexes (LANA, p53, and hdm2) were determined with Western blotting as mentioned above, and β-actin or cdk6 was used as a control.
p53 DNA binding assay.
The TransAm p53 DNA binding kit (Active Motif) was used according to the manufacturer's instructions to assess p53 DNA binding activity. The enzyme-linked immunosorbent assay (ELISA) plate was precoated with p53 consensus binding oligonucleotide sequence (5′-GGACATGCCCGGGCATGTCC-3′). The series of samples from the chromatographic purifications were loaded onto the ELISA plate. The specific activity of each protein preparation was determined by measuring the absorbance at a wavelength of 450 nm. Specificity of p53 DNA binding was determined using competitive wild-type and mutant oligonucleotides in the DNA binding assay.
Nutlin treatment.
BC-3 cells were cultured in T-75 cm2 flasks, treated with nutlin-3a at 0, 6, 12, 24, and 48 h separately, and then harvested for immunoprecipitation assay and Western blotting. The final concentration of nutlin-3a was 10 μM, and the control was treated with dimethyl sulfoxide (DMSO). For in vitro treatment, samples were derived from isolated products of Heparin FF column purification. For immunoprecipitation assays, aliquot samples were precleared with protein A/G agarose beads and then nutlin-3a was added to a final concentration of 0, 10, 30, 90, 270, and 810 μM; p53 antibody (DO-1) was also added for overnight incubation at 4°C. Then 40 μl of protein A/G agarose beads was added, and the mixture was incubated with rotation for 2 h and washed for Western blotting. Mouse IgG was used as a control.
Deconvolution immunofluorescence.
BC-3 cells were grown in six-well plates, fixed with 3% paraformaldehyde for 20 min at room temperature, and then permeabilized with 0.2% Triton X-100 in PBS for 15 min; they were then incubated in blocking buffer (10% horse serum in PBS) for 120 min. Samples were then processed for immunofluorescence by using a rabbit polyclonal anti-LANA YT041 antibody, a mouse monoclonal anti-p53 (DO-1) antibody, and mouse hdm2 MAb (SMP14) as primary antibodies. The secondary antibodies used were horse anti-rabbit immunoglobulin (fluorescein isothiocyanate [FITC]) (Vector) and anti-mouse immunoglobulin (TRITC) (Invitrogen). Images were obtained by using a Leica deconvolution microscopy system (Leica model DM4000B, oil lens with100-fold magnification; software, SimplePCI version 6.2]), where the fluorescent signals were mapped to green and red.
RESULTS
Biochemical separation of LANA:p53, LANA:LANA, and p53:p53 complexes.
A key advantage of biochemical purifications of interacting proteins from naturally KSHV-infected tumor cell lines is that all partners are present at their physiological levels and stoichiometries. This is in contrast to yeast two-hybrid screens or tap-tag-based purification, where one binding partner is expressed at superphysiological levels and thus drives low-affinity interactions. The disadvantage of this method is that some interactions may not survive the purification procedure and that transient interactions cannot be captured. Biochemical purification of LANA complexes from PEL thus defines the minimal complex of LANA-interacting proteins.
We employed the traditional Dignam et al. method as the initial purification step (16). This method has been used to purify functional RNA polymerase II complexes, to maintain many transcription factor interactions, and to yield properly folded proteins as judged by DNA binding, including Sp1 (20), p53:hdm2 (65), and p53:simian virus 40 (SV40) (25, 57, 60), which are relevant to our investigation. We showed earlier that this purification yields DNA binding-competent LANA complexes (reference 39 and data not shown).
We used Epstein-Barr virus (EBV)-negative, p53 wild-type BC-3 cells as starting material. These cells are unique, since most other PEL cell lines carry either EBV or a mutation in p53 (reference 71 and data not shown). The experimental approach is outlined in Fig. 1A. First, we prepared nuclear extracts (NE). This step excluded any complexes that are localized in the cytoplasm, such as hdm2:p53 (64). The NE were loaded onto a Sepharose 6B sizing column, which also served as a desalting step, as used by Sarek et al. (77). The rationale for this step was to isolate intact high-molecular-weight LANA complexes, consisting at least of DNA binding-competent dimers or tetramers (4 at 220 kDa). LANA eluted as a high-molecular-weight complex ahead of monomeric actin (Fig. 1B). p53 coeluted with LANA, though p53 was also found in fractions that contained lower-molecular-weight complexes. This suggests that a fraction of p53 is not associated with LANA or associates at a stoichiometry that differs from that of the tetrameric LANA:p53 complexes that eluted prior.
FIG. 1.

Analysis of LANA complexes after purification with a sizing column (Sepharose 6B) and Heparin FF column. (A) Outline of the experimental design and summary of LANA complex partition. CYT, cytosol. (B) The purification nuclear extract from BC-3 cells was loaded onto a Sepharose 6B column with a flow rate of 0.5 ml/min and then washed with buffer D (1.5 ml elution per collective tube). Traces show total protein based on UV absorption (absorbance units [A.U.]) and absorbance in mS/cm. The results of Western blotting are shown on the right. For these results, we used the indicated fractions (denoted by arrows and numbers beneath the x axis) from the Sepharose 6B purification. The following primary antibodies were used: anti-β-actin, anti-LANA (YT041), and anti-p53 (DO-1). The bar with the arrow and letter “C” indicates the fractions that were pooled and put on a subsequent Heparin column. (C) The profile of the eluted peak (eluted with wash buffer with 0.2 M KCl [pH 7.9] and with step elution buffer with 0.5 M KCl and 1 M KCl) is shown. The results of Western blotting are shown on the right. For these results, we used the indicated fractions (denoted by arrows and numbers beneath the x axis) from the heparin column. We used the same antibodies as indicated above.
Independent repetition of this purification step showed that the high-molecular-weight LANA complex after Sepharose 6B was able to bind its cognate site within the LANA promoter (data not shown).
Following size exclusion chromatography, the LANA-positive fractions were pooled and loaded onto a Heparin FF column, which enriches for DNA binding proteins (Fig. 1C). NE was bound to heparin at 0.1 M salt and was washed extensively with 0.2 M KCl until no further protein eluted. As expected, nuclear actin did not bind to the column at all. Next we applied 0.5 M KCl. At this salt concentration, LANA and p53 coeluted from the column. Finally, we applied 1 M KCl in a step gradient. A second fraction of proteins eluted, but this fraction contained neither LANA nor p53.
Independent repetition of the heparin purification step showed that p53 and hdm2 coeluted with LANA (Fig. 2). This was expected since hdm2 is localized to the nucleus as well as to the cytoplasm. It is important to keep in mind that we used conventional chromatography with 0.5-ml fraction volumes. Of those fractions, we used 10 μl in a Western blot, which somewhat limited our sensitivity. Sp1 coeluted with LANA, consistent with an earlier report by Verma et al. (95). Hence, Sp1 was used as a positive control for all subsequent experiments.
FIG. 2.

Analysis of LANA binding complexes after heparin affinity chromatography. (A) Profiles of the eluted peaks, using wash buffer with 0.2 M KCl (pH 7.9) and a step gradient up to 0.5 M KCl. Traces show total protein based on UV absorption (absorbance units [A.U.]) and absorbance in mS/cm. The bar with the arrow and letter “Q” indicates the fractions that were pooled and subsequently loaded onto a Mono Q column. (B) Components (1 to 9 tubes of continuous fractions) of the elution peak (0.2 M to 0.5 M) were analyzed with Western blotting (WB) and by immunoprecipitation (IP) followed by Western blotting. (C) Immunoprecipitation followed by Western blotting of nuclear extract prior to chromatography. We used rat monoclonal anti-LANA antibody for immunoprecipitation and anti-p53, anti-hdm2, and anti-LANA antibodies as indicated. Components (1 to 9 tubes of continuous fractions) of the elution peak (0.2 M to 0.5 M) were analyzed with Western blotting and by immunoprecipitation assay.
We confirmed interactions of LANA with p53 and of LANA with hdm2 by coimmunoprecipitation (co-IP) from heparin column fractions (Fig. 2). When we immunoprecipitated p53 (DO-1), we could detect LANA by Western blotting. When we immunoprecipitated hdm2, we could detect LANA by Western blotting. Notably, only a single band of LANA was precipitated with hdm2, whereas after precipitation with either p53 or Sp1 antibodies, the typical LANA triplet of bands could be observed. This suggested that only a fraction of LANA, perhaps with a particular phosphorylation pattern, is able to bind to hdm2.
These results showed further that a portion of LANA existed in complex with p53 and hdm2. Since we found p53 present in the high-molecular-weight LANA-enriched fractions, as well as in the lower-molecular weight LANA-depleted fractions, during size exclusion chromatography, this suggested that not all p53 was complexed with LANA. As expected for a DNA binding protein, heparin chromatography significantly enriched for LANA. More importantly, LANA heparin binding could be reversed and LANA eluted under moderate salt conditions, which maintained active LANA and active p53 (see below).
The heparin fractions containing LANA complexes were pooled and diluted to 0.1 M KCl in buffer D. They were then loaded onto an anion ion-exchange column (Fig. 3A). After optimization of elution condition, we identified two peaks: one within the 0.1 M-to-0.4 M KCl range and the second one within the 0.4 M-to-0.7 M range. LANA was not detectable by Western blotting in the first, low-salt elution peak (Fig. 3B). p53 was not detectable by Western blotting alone either, since we followed standard procedures and used only 10 μl of the 1-ml fractions. However, we were able to detect p53 after immunoprecipitation. Neither LANA nor hdm2 was in complex with p53 under these conditions (Fig. 3B). These data are consistent with the existence of a p53:p53 homodimer or tetramer complex.
FIG. 3.

Analysis of LANA complexes isolated with an ion-exchange column. (A) Profiles of the eluted peaks when 0.4 M KCl (pH 7.0) and 0.7 M KCl (pH 7.0) were used separately as elution buffers. (B) The collected small peak (1 to 9 tubes of step gradient), eluted with 0.4 M KCl, was assayed by Western blotting and IP. (C) Detection of LANA complexes in the main sharp peak (0.4 M to 0.7 M KCl). Traces show total protein based on upon UV absorption (absorbance units [A.U.]) and absorbance in mS/cm. The analysis of 1 to 9 tubes of continuous fractions assayed by Western blotting and immunoprecipitation with different monoclonal antibodies of anti-p53 (DO-1 and pab421), anti-hdm2, and anti-hdmX.
LANA eluted with 0.7 M KCl. Thus, this purification step separated p53 and LANA and enriched for LANA and other tightly LANA-associated proteins. We analyzed the components of the LANA complexes by immunoprecipitation assays (Fig. 3C). We used two different mouse monoclonal p53 antibodies (DO-1 and PAb421) to show that this p53:LANA complex was resistant to 0.7 M KCl. The Sp1:LANA complex was also resistant to 0.7 M KCl. The results of this purification are summarized in Fig. 1A. We corroborated the results of earlier experiments using a different, unbiased approach. There exists a p53:LANA complex in PEL. However, only a fraction of cellular p53 and only a fraction of LANA take part in this complex.
Specific DNA binding activity of p53 in PEL despite LANA.
Having identified distinct biochemical complexes of p53:p53 and LANA:p53, we wondered whether we could detect p53 DNA binding activity in the PEL nuclear extracts. We hypothesized that if we could detect p53 binding activity in nuclear extracts that also contained LANA at physiological levels, this would biochemically demonstrate that LANA did not inhibit the DNA binding activity of p53. It would unify our functional results of demonstrating a p53-dependent DNA damage response in PEL (71) with other observations that LANA could interfere with p53's transactivation functions in cotransfection assays. We used ELISA to interrogate p53 binding abilities. The Heparin FF column fractions retained the ability to bind a p53 response element (RE). The histogram of the RE binding activity of p53 shows a peak after elution with 0.5 M KCl (Fig. 4A). Based upon Western blotting, these fractions contained large amounts of both LANA and p53. To verify the specificity of the ELISA kit, we conducted competition experiments. A wild-type, but not a mutant, p53 binding site oligonucleotide competed the signal (Fig. 4C). This suggests that the presence of LANA protein does not preclude p53 protein from binding to its cognate DNA element.
FIG. 4.

Analysis of the binding activity of p53 upon p53 response elements (RE). Samples were incubated with an immobilized RE oligonucleotide, and washed and bound p53 was detected by ELISA. Shown is the absorption (optical density at 450 nm [OD450]) on the vertical axis, and column fractions are shown on the horizontal axis. A higher OD indicates a larger amount of p53 binding to the immobilized RE. (A) Analysis of the binding activity of p53 after purification with the Heparin FF column. (B) Analysis of the binding activity of p53 after purification with an ion-exchange column. A1 to A9 are fractions that eluted at 0.4 M, and I1 to I9 are fractions that eluted at 0.4 to 0.7 M KCl. (C) Competition of p53 binding activity with soluble wild-type or mutant p53 oligonucleotide. Shown are the results for nuclear extracts and fractions from Heparin FF and ion-exchange columns at 0.1 to 0.4 M and 0.4 to 0.7 M KCl. Note that fractions were diluted 1:8 in binding buffer.
We could recover p53 binding activity even after purification over an anion-exchange column. p53 binding activity corresponded to the p53-containing fractions that eluted with 0.4 M KCl. In contrast, complexes of LANA and p53 found in the second peak of the anion-exchange column lost the ability to bind a p53 consensus element (Fig. 4B).
The half-life of p53 in PEL is determined by p53's mutational status.
Rapid protein turnover is a key characteristic of wild-type p53. The half-life of p53 in epithelial cells and fibroblasts is <20 min. It can be as low as 5 min in murine thymocytes (61). Mutation and binding to SV40 large T antigen or to the adenovirus E1B 55-kDa protein extend the half-life of wild-type p53 to many hours (reviewed in reference 15). Half-life is thus an indicator of p53 function (31). We hypothesized that if LANA complexed with p53, this would lead to p53 stabilization analogous to that of the SV40 T antigen:p53 complex or, as previously reported (10), accelerated degradation of p53. To determine the half-life of p53, LANA, and hdm2, representative PEL cell lines (BC-3, BCP-1) and two control KSHV-negative Burkitt lymphoma cell lines (BJAB, DG-75) were treated with 50 μg/ml of the protein translation inhibitor cycloheximide for 0, 10, 30, 60, and 120 min. The total amount of protein was detected by Western blotting and quantified by image analysis (Fig. 5A). The half-life of p53 correlated with p53 mutational status and was not extended by the presence of LANA.
FIG. 5.

Analysis of the half-lives of LANA complexes. The cell lines BC-3, BJAB, BCP-1, and DG-75 were treated separately with cycloheximide (Calbiochem) (50 μg/ml) for 0, 10, 30, 60, and 120 min. (A) Total protein was analyzed by Western blotting using anti-LANA, anti-p53, and anti-β-actin (control) antibodies. (B) Band intensities were quantified and the relative intensities plotted over time (dots). Also shown are the regression lines, which fit the data except in the case of DG-75, which exhibits a biphasic decay curve. (C) hdmX levels in PEL cell lines. Shown are Western blots for hdmX and cdk6 (as a loading control) for a panel of seven PEL cell lines. (D) Relative levels of hdmX based on densitometry. hdmX levels were normalized to cdk6 levels. wt, wild type; mt, mutant.
p53 was short-lived in BC-3 cells (t1/2 = 1.7 h) (Fig. 5B), which contain wild-type p53 (71). The half-life of p53 was extended in the other three cell lines, which carry mutant p53. The half-life was 23 h in the BJAB cell line, which has a His193Arg mutation in p53 (6). The half-life was 6 h in another KSHV-infected cell line, BCP-1, that also harbors a mutant p53 allele (insertion S262/S262) (71). The DG-75 cell line, which is heterozygous for p53 (14), carrying both a wild-type and a mutant (Arg283His) copy of p53, evidenced a biphasic curve consistent with the heterozygous phenotype.
The half-life of LANA in KSHV-infected BC-3 and BCP-1 cells was consistent with earlier reports that found the LANA half-life to exceed 24 h. We did not detect LANA protein in the KSHV-negative BJAB and DG-75 cell lines. We could not detect hdm2 in either the BJAB or DG-75 Burkitt lymphoma cell line, consistent with the absence of hdm2 RNA as previously reported (Gene Expression Omnibus data set GDS989). In contrast, hdm2 was easily detectable in both PEL cell lines, which was expected since we previously showed that hdm2 protein and RNA levels were upregulated in all PEL (71). The half-life of hdm2 was short (≤30 min). Actin was used as a control.
For the first time, we investigated the status of hdmX in PEL (Fig. 5C and D). Unlike hdm2, we could not discern a consistent expression pattern for hdmX. hdmX was detectable in JSC-1, VG-1, and BC-3 but not the other PEL lines in our experiment. hdmX levels did not correlate with either p53 status or EBV coinfection status. In sum, we failed to demonstrate any effect of LANA on the half-life of p53, as would be expected if most p53 were sequestered by LANA.
Nutlin-3a disrupts purified LANA:p53:hdm2 complex in vitro.
Nutlin-3a disrupts purified hdm2:p53 complex binding by intercalating at the p53:hdm2 surface (54), and nutlin-3a significantly reduced cell proliferation in PEL harboring wild-type p53 (71, 77). We hypothesized that nutlin-3a would also disrupt the p53:LANA complex. If so, this would demonstrate that hdm2 participates in p53:LANA interaction either directly or by changing the conformation of p53 to allow it to bind LANA. We therefore treated BC-3 cells with nutlin-3a and performed co-IP and Western blotting at various times thereafter. Nutlin-3a disrupted the p53:hdm2 complex within 6 h after addition as measured by co-IP followed by Western blotting for associated p53 (Fig. 6A). This coincided with the induction of p53, hdm2, and p21 protein as determined by Western blotting. These are direct transcriptional targets of p53. Hence, this demonstrates that nutlin-3a activated p53 in vivo. Importantly, the LANA:p53 complex was disrupted immediately after addition of nutlin-3a to BC-3 cells. Immunoprecipitation of p53 followed by Western blotting for LANA demonstrated a complex at 0 h but not at any time point thereafter. At 48 h, the cells started to die, and consequently, protein levels were reduced for all proteins assayed.
FIG. 6.
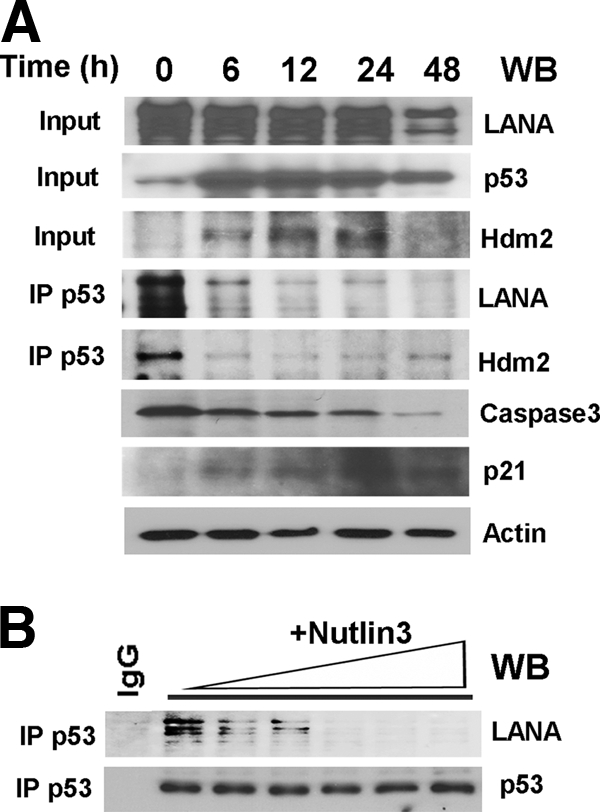
Nutlin-3a disrupts LANA:hdm2:p53 complexes in vivo and in vitro. (A) BC-3 cells were treated with nutlin-3. p53, hdm2, and LANA levels and complexes were determined by Western blotting (input) and immunoprecipitation followed by Western blotting. Also shown are results of Western blotting for p21 levels to document p53 and caspase-3 activation, with actin as a loading control. (B) The disruption of LANA:p53:hdm2 interaction by nutlin-3 is a direct effect. Purified LANA:p53:hdm2 complexes from heparin fractions 3 to 7 were pooled and incubated with increasing amounts of nutlin-3 in vitro; p53 complexes were immunoprecipitated (IP) and blotted (WB) with LANA or p53 antibodies.
To exclude, as much as possible, indirect effects of nutlin-3 on LANA:p53 binding, we explored the effect of nutlin-3 on LANA:p53 complexes in vitro. To do so, we used purified LANA:p53 complex from our heparin fractions and added increasing amounts of nutlin-3a in vitro. Nutlin-3a was able to dissociate the LANA:p53 complex in a dose-dependent manner, as shown by co-IP (Fig. 6B). Since nutlin-3 disrupts the hdm2:p53 interaction interface, we conclude that hdm2 contributes to LANA's interaction with p53 either directly or by changing the conformation of p53 to enable it to bind LANA.
Subcellular distribution of p53, hdm2, and LANA.
p53 activities are also governed by localization. Nuclear-cytoplasmic shuttling is a defining characteristic of p53:hdm2 (8, 92). We therefore investigated the cellular localization of LANA, p53, and hdm2 using either a commercial assay (Fig. 7A) or the Dignam et al. method (Fig. 7B). The commercial assay gave better separation of the cellular fractions. The majority of LANA was present in the nucleus. p53 was also localized in the nucleus in exponentially growing BC-3 cells. Sp1 was exclusively localized to the nucleus and served as a positive control for fractionation. Actin was cytosol and cytoskeleton associated and served as a positive control for proper fractionation of these two compartments.
FIG. 7.

Cellular fractions and immunofluorescence assay of LANA, hdm2, and p53 in BC-3 cells. (A) Cellular fraction of BC-3 using a commercial assay (cytosol, membrane/organelle, nucleus, and cytoskeleton) followed by Western blotting. (B) Cellular fractionation of BC-3 cells by the Dignam et al. method. WC, whole-cell extract; B, 10 mM KCl; NE, nuclear extraction per Dignam C (0.42 M NaCl); P, pellet after Dignam et al. nuclear extraction. Antibodies used were anti-LANA, anti-p53, anti-Sp1, and anti-β-actin. (C and D) Deconvolution scanning immunofluorescence of BC-3 with antibodies of anti-LANA (green) and anti-p53 (red) (C) and with anti-LANA (green) and anti-hdm2 (red) (D).
By immunofluorescence, all LANA was located in the nuclei of BC-3 cells (Fig. 7C, green). In interphase nuclei, LANA was focally concentrated in a characteristic punctuate pattern at sites of KSHV plasmids, which have been shown to roughly correspond to the number of KSHV plasmid copies, ranging from 15 to 30 per cell (69). p53 was similarly nuclear, and some p53 colocalized with LANA (Fig. 7C, yellow). This is consistent with our biochemical purification, which found evidence for both free and LANA-bound p53. We also investigated the binding of LANA to hdm2 and found some evidence for colocalization, though most of the hdm2 (Fig. 7D, red) was not bound to LANA. This is consistent with the idea of multiple LANA complexes in which LANA that is bound to histones is not bound to hdm2 and hdm2 has other functions and binding partners other than just LANA.
There is an important caveat though: wild-type p53 is typically undetectable by Western blotting in exponentially growing cells. It accumulates only after cellular insult. Hence, the p53 fluorescence signal was 10-fold-weaker than the LANA signal. We were able to detect these proteins only because LANA, p53, and hdm2 are highly expressed in BC-3 cells.
DISCUSSION
LANA is a viral binding partner of p53. Friborg et al. (23) showed that in vitro-translated LANA protein bound to a glutathione S-transferase (GST)-p53 fusion protein but not to GST alone. They also succeeded at coimmunoprecipitating LANA and p53 from BCBL-1 cells. Of note, BCBL-1 cells contain one wild-type and one mutant (M246I) allele of p53 (49), resulting in accumulation of high levels of p53 heterodimers. These initial experiments used the monoclonal anti-p53 antibody DO-1 and relied on KSHV-reactive patient sera, since at the time, no monoclonal antibody against LANA was available. Less than 10% of p53 could be immunoprecipitated with the human anti-KS antisera and less than 10% of LANA could be immunoprecipitated with the anti-p53 monoclonal antibody. Using BC-3 cells, which contain wild-type p53, we found evidence for a fraction of p53 that was stably bound to LANA even after column purification and a 0.4 M KCl salt wash. These novel results corroborate earlier studies (10, 100), but neither ectopic overexpression nor elevation due to p53 mutation was used.
This alone is not the whole story. Our model (Fig. 8) posits that there exist multiple LANA complexes in PEL. (i) The majority of LANA is present in LANA:histone complexes. These function in KSHV genome tethering and replication. (ii) A separate population of LANA takes part in transcription regulatory protein complexes, such as LANA:p53 and LANA:Sp1. The composition and abundance of these complexes are subject to regulation: p53 activation via S15 phosphorylation will release p53 from LANA (71). hdm2:p53 disruption via nutlin-3a will also release p53 from LANA (77). Nutlin-3a disrupted partially purified LANA:p53 complexes in vitro, suggesting for the first time a direct interaction between this drug and the LANA:p53 complex. Whether nutlin-3a directly interferes with the LANA:p53 interaction interface or whether only the hdm2:p53 complex has the correct structure to bind LANA is the subject of further studies.
FIG. 8.

Model of LANA:p53:hdm2 complexes in PEL. This model is based on experiments described here as well in earlier work, which confirmed doxorubicin-induced Ser 15 phosphorylation and nutlin-3-dependent p53:mdm2 and p53:LANA dissociation, as well as induction of p53 target genes in PEL, which carry wild-type p53 (71, 77).
The existence of multiple, mutually inclusive complexes can explain why targeted approaches such as coimmunoprecipitation and overexpression-based transcriptional experiments that are conducted in cells lacking the KSHV episome consistently find evidence for p53:LANA interactions (7, 10, 23, 83, 97, 100), whereas phenotypic analyses of PEL or less sensitive, mass spectrometry (MS)-MS-based screens (4, 84) of PEL could not find evidence for a prominent p53:LANA complex. Further support for multiple independent complexes comes from our half-life analysis. The half-life of p53 in PEL correlated with p53 mutation status. It was not extended by the presence of LANA. This is in contrast to p53:SV40 T antigen complexes, in which p53's half-life is extended and which prevent p53 from binding its cognate DNA (36, 89, 93).
In the context of latently infected PEL, we found that the multiple LANA complexes coexist. Purification over Sepharose and Heparin columns facilitated the isolation of distinct LANA complexes that were comprised of LANA-p53-hdm2, p53 alone, or LANA alone. In the Heparin fractions that contained LANA and p53, LANA retained the important characteristic and functionality of binding to the KSHV terminal repeat (TR) sequences (data not shown). Likewise, p53 complexes contained in the same column fractions retained the ability to bind to p53 cognate DNA elements. This is in contrast to what occurred with the SV40 large T antigen (5), which interferes with p53's DNA binding ability. Conversely, p53 interferes with the SV40 large T antigen's ability to unwind the SV40 origin of replication, and there exists a concentration-dependent trimeric complex of p53, SV40 T antigen, and polymerase-alpha (9, 25, 57). This suggests (i) that LANA and SV40 T are not analogous with respect to their p53 interactions and (ii) that there exist separate LANA-free p53 complexes or, alternatively, that the LANA:p53 complexes in PEL retain the ability to bind specifically to p53-responsive elements in a manner that is indistinguishable from KSHV-uninfected cell extracts.
The experimental details of our purification are important in order to gauge which LANA:p53 complexes we could have detected and which we would have missed based on our experimental design. Our fractionation scheme started with nuclear extract, since LANA is a nuclear protein. Any interactions involving a minor fraction of cytoplasmic LANA would have eluded our purification scheme. There is presently no indication that LANA can shuttle in and out of the nucleus. In contrast, nuclear-cytoplasmic shuttling is a defining property of p53. By implication then, any cytoplasmic p53:hdm2 complexes would not contain LANA and would be functionally wild type. This is important, since p53 can activate apoptosis via release of bax from mitochondria, followed by activation of caspase-9 (11, 12). This mechanism is independent of p53's transactivation function and has been implicated to explain the response of B-CLL to nutlin-3a (48, 79, 88). It might similarly explain the response of PEL to nutlin-3a (71, 77).
Activation of p53 by DNA damage depends on posttranslational modifications such as acetylation, phosphorylation at Ser 15, 20, 33, and 37, release of hdm2, extension of half-life, and nuclear accumulation. We and others previously showed that this pathway is intact in LANA-expressing PEL (71, 77). LANA does not block the activation of p53 by DNA damage and its subsequent transcriptional function. Hence, the nature of the LANA:p53 complex must be such as to allow access to the p53 N terminus and phosphorylation at Ser 15. hdm2 also binds to the N terminus of p53, involving amino acids (Phe 19, Trp 23, and Leu 26) (54). This interaction is sensitive to nutlin-3 (94). Nutlin-3a efficiently kills LANA-positive PEL, if they contain wild-type p53 (71). Sarek et al. showed that treatment with 7 μM nutlin-3a released p53 from both LANA and hdm2 (77) in BC-3 cells. Hence, the nature of the LANA:p53 complex must be such as to allow nutlin-3a access to the p53 N terminus and the p53:hdm2 interaction site. Alternatively, there exists a cytoplasmic hdm2:p53 complex that is not inhibited by LANA or other viral proteins.
KSHV possesses a complex series of molecular strategies to regulate cell proliferation, induce cell transformation, and prevent cell apoptosis. The detailed molecular mechanisms are the subject of ongoing investigation and may yield novel intervention targets for KSHV-associated cancers. LANA is a multifunctional protein that is consistently expressed in all KSHV-associated malignancies. The primary function of LANA is to tether the KSHV episome to cellular chromosomes and thus to ensure the proper segregation of latent episomes during mitosis. However, episome tethering is not the only function of LANA. LANA also has DNA binding and transcriptional regulatory activity on its own promoter (38, 39, 75) as well as on cellular promoters, for instance, the promoter for human telomerase (95). The transcriptional function of LANA is highlighted in experimental designs that express LANA in the absence of the viral episome (and latent origin). Here, LANA has been shown to regulate a large number of target genes and their promoters (1, 29, 35, 47, 75, 97). Likewise, B cell-specific expression of LANA in transgenic mice induces mature B cell hyperplasia and lymphoma in a fraction of animals (21). In contrast, latent infection of B cells with KSHV has consistently failed to bring about a fully transformed phenotype (18, 46, 76). These superficially discordant observations can be unified if one postulates the existence of multiple LANA complexes, one each for the replicative, transcriptional, and transforming functions.
Acknowledgments
This work was supported by the NIH (grant CA109232). C.E.B. was supported by training grant CA009156.
Footnotes
Published ahead of print on 3 February 2010.
REFERENCES
- 1.An, F. Q., N. Compitello, E. Horwitz, M. Sramkoski, E. S. Knudsen, and R. Renne. 2005. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J. Biol. Chem. 280:3862-3874. [DOI] [PubMed] [Google Scholar]
- 2.Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. [DOI] [PubMed] [Google Scholar]
- 3.Ballestas, M. E., and K. M. Kaye. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75:3250-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbera, A. J., J. V. Chodaparambil, B. Kelley-Clarke, V. Joukov, J. C. Walter, K. Luger, and K. M. Kaye. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856-861. [DOI] [PubMed] [Google Scholar]
- 5.Bargonetti, J., I. Reynisdottir, P. N. Friedman, and C. Prives. 1992. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev. 6:1886-1898. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia, K., W. Goldschmidts, M. Gutierrez, G. Gaidano, R. Dalla-Favera, and I. Magrath. 1993. Hemi- or homozygosity: a requirement for some but not other p53 mutant proteins to accumulate and exert a pathogenetic effect. FASEB J. 7:951-956. [DOI] [PubMed] [Google Scholar]
- 7.Borah, S., S. C. Verma, and E. S. Robertson. 2004. ORF73 of herpesvirus saimiri, a viral homolog of Kaposi's sarcoma-associated herpesvirus, modulates the two cellular tumor suppressor proteins p53 and pRb. J. Virol. 78:10336-10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyd, S. D., K. Y. Tsai, and T. Jacks. 2000. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol. 2:563-568. [DOI] [PubMed] [Google Scholar]
- 9.Braithwaite, A. W., H. W. Sturzbecher, C. Addison, C. Palmer, K. Rudge, and J. R. Jenkins. 1987. Mouse p53 inhibits SV40 origin-dependent DNA replication. Nature 329:458-460. [DOI] [PubMed] [Google Scholar]
- 10.Cai, Q. L., J. S. Knight, S. C. Verma, P. Zald, and E. S. Robertson. 2006. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chipuk, J. E., T. Kuwana, L. Bouchier-Hayes, N. M. Droin, D. D. Newmeyer, M. Schuler, and D. R. Green. 2004. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010-1014. [DOI] [PubMed] [Google Scholar]
- 12.Chipuk, J. E., U. Maurer, D. R. Green, and M. Schuler. 2003. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 4:371-381. [DOI] [PubMed] [Google Scholar]
- 13.Cotter, M. A., II, and E. S. Robertson. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254-264. [DOI] [PubMed] [Google Scholar]
- 14.Crook, T., G. A. Parker, M. Rozycka, S. Crossland, and M. J. Allday. 1998. A transforming p53 mutant, which binds DNA, transactivates and induces apoptosis reveals a nuclear:cytoplasmic shuttling defect. Oncogene 16:1429-1441. [DOI] [PubMed] [Google Scholar]
- 15.Damania, B., and J. B. Pipas (ed.). 2009. DNA tumor viruses. Springer, New York, NY.
- 16.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dittmer, D., S. Pati, G. Zambetti, S. Chu, A. K. Teresky, M. Moore, C. Finlay, and A. J. Levine. 1993. Gain of function mutations in p53. Nat. Genet. 4:42-46. [DOI] [PubMed] [Google Scholar]
- 18.Dittmer, D., C. Stoddart, R. Renne, V. Linquist-Stepps, M. E. Moreno, C. Bare, J. M. McCune, and D. Ganem. 1999. Experimental transmission of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med. 190:1857-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dittmer, D. P., and S. E. Krown. 2007. Targeted therapy for Kaposi's sarcoma and Kaposi's sarcoma-associated herpesvirus. Curr. Opin. Oncol. 19:452-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dynan, W. S., and R. Tjian. 1983. The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell 35:79-87. [DOI] [PubMed] [Google Scholar]
- 21.Fakhari, F. D., J. H. Jeong, Y. Kanan, and D. P. Dittmer. 2006. The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J. Clin. Invest. 116:735-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang, S., J. P. Jensen, R. L. Ludwig, K. H. Vousden, and A. M. Weissman. 2000. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 275:8945-8951. [DOI] [PubMed] [Google Scholar]
- 23.Friborg, J., Jr., W. Kong, M. O. Hottiger, and G. J. Nabel. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889-894. [DOI] [PubMed] [Google Scholar]
- 24.Fujimuro, M., F. Y. Wu, C. ApRhys, H. Kajumbula, D. B. Young, G. S. Hayward, and S. D. Hayward. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 9:300-306. [DOI] [PubMed] [Google Scholar]
- 25.Gannon, J. V., and D. P. Lane. 1987. p53 and DNA polymerase alpha compete for binding to SV40 T antigen. Nature 329:456-458. [DOI] [PubMed] [Google Scholar]
- 26.Garber, A. C., J. Hu, and R. Renne. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277:27401-27411. [DOI] [PubMed] [Google Scholar]
- 27.Garber, A. C., M. A. Shu, J. Hu, and R. Renne. 2001. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:7882-7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godfrey, A., J. Anderson, A. Papanastasiou, Y. Takeuchi, and C. Boshoff. 2005. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105:2510-2518. [DOI] [PubMed] [Google Scholar]
- 29.Groves, A. K., M. A. Cotter, C. Subramanian, and E. S. Robertson. 2001. The latency-associated nuclear antigen encoded by Kaposi's sarcoma-associated herpesvirus activates two major essential Epstein-Barr virus latent promoters. J. Virol. 75:9446-9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris, S. L., and A. J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene 24:2899-2908. [DOI] [PubMed] [Google Scholar]
- 31.Harvey, D. M., and A. J. Levine. 1991. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 5:2375-2385. [DOI] [PubMed] [Google Scholar]
- 32.Haupt, Y., R. Maya, A. Kazaz, and M. Oren. 1997. Mdm2 promotes the rapid degradation of p53. Nature 387:296-299. [DOI] [PubMed] [Google Scholar]
- 33.Hollstein, M., D. Sidransky, B. Vogelstein, and C. C. Harris. 1991. p53 mutations in human cancers. Science 253:49-53. [DOI] [PubMed] [Google Scholar]
- 34.Honda, R., and H. Yasuda. 2000. Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19:1473-1476. [DOI] [PubMed] [Google Scholar]
- 35.Hyun, T. S., C. Subramanian, M. A. Cotter II, R. A. Thomas, and E. S. Robertson. 2001. Latency-associated nuclear antigen encoded by Kaposi's sarcoma-associated herpesvirus interacts with tat and activates the long terminal repeat of human immunodeficiency virus type 1 in human cells. J. Virol. 75:8761-8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue, H., G. Kondoh, T. M. Sudiro, and A. Hakura. 1992. Stability of p53 protein in rat cells transformed by various viral transforming genes. Virology 187:343-347. [DOI] [PubMed] [Google Scholar]
- 37.Jackson, M. W., and S. J. Berberich. 2000. MdmX protects p53 from Mdm2-mediated degradation. Mol. Cell. Biol. 20:1001-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong, J., J. Papin, and D. Dittmer. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 75:1798-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeong, J. H., J. Orvis, J. W. Kim, C. P. McMurtrey, R. Renne, and D. P. Dittmer. 2004. Regulation and autoregulation of the promoter for the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 279:16822-16831. [DOI] [PubMed] [Google Scholar]
- 40.Katano, H., Y. Sato, and T. Sata. 2001. Expression of p53 and human herpesvirus-8 (HHV-8)-encoded latency-associated nuclear antigen with inhibition of apoptosis in HHV-8-associated malignancies. Cancer 92:3076-3084. [DOI] [PubMed] [Google Scholar]
- 41.Kedes, D. H., M. Lagunoff, R. Renne, and D. Ganem. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J. Clin. Invest. 100:2606-2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kellam, P., C. Boshoff, D. Whitby, S. Matthews, R. A. Weiss, and S. J. Talbot. 1997. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J. Hum. Virol. 1:19-29. [PubMed] [Google Scholar]
- 43.Kellam, P., D. Bourboulia, N. Dupin, C. Shotton, C. Fisher, S. Talbot, C. Boshoff, and R. A. Weiss. 1999. Characterization of monoclonal antibodies raised against the latent nuclear antigen of human herpesvirus 8. J. Virol. 73:5149-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelley-Clarke, B., M. E. Ballestas, T. Komatsu, and K. M. Kaye. 2007. Kaposi's sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology 357:149-157. [DOI] [PubMed] [Google Scholar]
- 45.Kelley-Clarke, B., M. E. Ballestas, V. Srinivasan, A. J. Barbera, T. Komatsu, T. A. Harris, M. Kazanjian, and K. M. Kaye. 2007. Determination of Kaposi's sarcoma-associated herpesvirus C-terminal latency-associated nuclear antigen residues mediating chromosome association and DNA binding. J. Virol. 81:4348-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kliche, S., E. Kremmer, W. Hammerschmidt, U. Koszinowski, and J. Haas. 1998. Persistent infection of Epstein-Barr virus-positive B lymphocytes by human herpesvirus 8. J. Virol. 72:8143-8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knight, J. S., M. A. Cotter II, and E. S. Robertson. 2001. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus transactivates the telomerase reverse transcriptase promoter. J. Biol. Chem. 276:22971-22978. [DOI] [PubMed] [Google Scholar]
- 48.Kojima, K., M. Konopleva, T. Tsao, H. Nakakuma, and M. Andreeff. 2008. Concomitant inhibition of Mdm2-p53 interaction and Aurora kinases activates the p53-dependent postmitotic checkpoints and synergistically induces p53-mediated mitochondrial apoptosis along with reduced endoreduplication in acute myelogenous leukemia. Blood 112:2886-2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Komanduri, K. V., J. A. Luce, M. S. McGrath, B. G. Herndier, and V. L. Ng. 1996. The natural history and molecular heterogeneity of HIV-associated primary malignant lymphomatous effusions. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 13:215-226. [DOI] [PubMed] [Google Scholar]
- 50.Komatsu, T., M. E. Ballestas, A. J. Barbera, B. Kelley-Clarke, and K. M. Kaye. 2004. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 319:225-236. [DOI] [PubMed] [Google Scholar]
- 51.Krithivas, A., M. Fujimuro, M. Weidner, D. B. Young, and S. D. Hayward. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596-11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krithivas, A., D. B. Young, G. Liao, D. Greene, and S. D. Hayward. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 74:9637-9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kubbutat, M. H., S. N. Jones, and K. H. Vousden. 1997. Regulation of p53 stability by Mdm2. Nature 387:299-303. [DOI] [PubMed] [Google Scholar]
- 54.Kussie, P. H., S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine, and N. P. Pavletich. 1996. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274:948-953. [DOI] [PubMed] [Google Scholar]
- 55.Lan, K., D. A. Kuppers, and E. S. Robertson. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468-3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lan, K., D. A. Kuppers, S. C. Verma, N. Sharma, M. Murakami, and E. S. Robertson. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453-7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lane, D. P., and L. V. Crawford. 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261-263. [DOI] [PubMed] [Google Scholar]
- 58.Lim, C., Y. Gwack, S. Hwang, S. Kim, and J. Choe. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 276:31016-31022. [DOI] [PubMed] [Google Scholar]
- 59.Lim, C., H. Sohn, Y. Gwack, and J. Choe. 2000. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) binds ATF4/CREB2 and inhibits its transcriptional activation activity. J. Gen. Virol. 81:2645-2652. [DOI] [PubMed] [Google Scholar]
- 60.Linzer, D. I., and A. J. Levine. 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43-52. [DOI] [PubMed] [Google Scholar]
- 61.Lowe, S. W., E. M. Schmitt, S. W. Smith, B. A. Osborne, and T. Jacks. 1993. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362:847-849. [DOI] [PubMed] [Google Scholar]
- 62.Lu, F., L. Day, S. J. Gao, and P. M. Lieberman. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273-5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mattsson, K., C. Kiss, G. M. Platt, G. R. Simpson, E. Kashuba, G. Klein, T. F. Schulz, and L. Szekely. 2002. Latent nuclear antigen of Kaposi's sarcoma herpesvirus/human herpesvirus-8 induces and relocates RING3 to nuclear heterochromatin regions. J. Gen. Virol. 83:179-188. [DOI] [PubMed] [Google Scholar]
- 64.Moll, U. M., M. LaQuaglia, J. Benard, and G. Riou. 1995. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc. Natl. Acad. Sci. U. S. A. 92:4407-4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Momand, J., G. P. Zambetti, D. C. Olson, D. George, and A. J. Levine. 1992. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237-1245. [DOI] [PubMed] [Google Scholar]
- 66.Nador, R. G., E. Cesarman, A. Chadburn, D. B. Dawson, M. Q. Ansari, J. Sald, and D. M. Knowles. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88:645-656. [PubMed] [Google Scholar]
- 67.Olivier, M., R. Eeles, M. Hollstein, M. A. Khan, C. C. Harris, and P. Hainaut. 2002. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat. 19:607-614. [DOI] [PubMed] [Google Scholar]
- 68.Ottinger, M., T. Christalla, K. Nathan, M. M. Brinkmann, A. Viejo-Borbolla, and T. F. Schulz. 2006. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 80:10772-10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parsons, C. H., L. A. Adang, J. Overdevest, C. M. O'Connor, J. R. Taylor, Jr., D. Camerini, and D. H. Kedes. 2006. KSHV targets multiple leukocyte lineages during long-term productive infection in NOD/SCID mice. J. Clin. Invest. 116:1963-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patton, J. T., L. D. Mayo, A. D. Singhi, A. V. Gudkov, G. R. Stark, and M. W. Jackson. 2006. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer Res. 66:3169-3176. [DOI] [PubMed] [Google Scholar]
- 71.Petre, C. E., S. H. Sin, and D. P. Dittmer. 2007. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J. Virol. 81:1912-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Platt, G. M., G. R. Simpson, S. Mittnacht, and T. F. Schulz. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789-9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Radkov, S. A., P. Kellam, and C. Boshoff. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121-1127. [DOI] [PubMed] [Google Scholar]
- 74.Rainbow, L., G. M. Platt, G. R. Simpson, R. Sarid, S. J. Gao, H. Stoiber, C. S. Herrington, P. S. Moore, and T. F. Schulz. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 71:5915-5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Renne, R., C. Barry, D. Dittmer, N. Compitello, P. O. Brown, and D. Ganem. 2001. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:458-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Renne, R., D. Blackbourn, D. Whitby, J. Levy, and D. Ganem. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 72:5182-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sarek, G., S. Kurki, J. Enback, G. Iotzova, J. Haas, P. Laakkonen, M. Laiho, and P. M. Ojala. 2007. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J. Clin. Invest. 117:1019-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwam, D. R., R. L. Luciano, S. S. Mahajan, L. Wong, and A. C. Wilson. 2000. Carboxy terminus of human herpesvirus 8 latency-associated nuclear antigen mediates dimerization, transcriptional repression, and targeting to nuclear bodies. J. Virol. 74:8532-8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Secchiero, P., E. Barbarotto, M. Tiribelli, C. Zerbinati, M. G. di Iasio, A. Gonelli, F. Cavazzini, D. Campioni, R. Fanin, A. Cuneo, and G. Zauli. 2006. Functional integrity of the p53-mediated apoptotic pathway induced by the non-genotoxic agent nutlin-3a in B-cell chronic lymphocytic leukemia (B-CLL). Blood 107:4122-4129. [DOI] [PubMed] [Google Scholar]
- 80.Shamay, M., A. Krithivas, J. Zhang, and S. D. Hayward. 2006. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. U. S. A. 103:14554-14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shinohara, H., M. Fukushi, M. Higuchi, M. Oie, O. Hoshi, T. Ushiki, J.-I. Hayashi, and M. Fujii. 2002. Chromosome binding site of latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus is essential for persistent episome maintenance and is functionally replaced by histone H1. J. Virol. 76:12917-12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shvarts, A., W. T. Steegenga, N. Riteco, T. van Laar, P. Dekker, M. Bazuine, R. C. van Ham, W. van der Houven van Oordt, G. Hateboer, A. J. van der Eb, and A. G. Jochemsen. 1996. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 15:5349-5357. [PMC free article] [PubMed] [Google Scholar]
- 83.Si, H., and E. S. Robertson. 2006. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 80:697-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Si, H., S. C. Verma, and E. S. Robertson. 2006. Proteomic analysis of the Kaposi's sarcoma-associated herpesvirus terminal repeat element binding proteins. J. Virol. 80:9017-9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Srinivasan, V., T. Komatsu, M. E. Ballestas, and K. M. Kaye. 2004. Definition of sequence requirements for latency-associated nuclear antigen 1 binding to Kaposi's sarcoma-associated herpesvirus DNA. J. Virol. 78:14033-14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stad, R., Y. F. Ramos, N. Little, S. Grivell, J. Attema, A. J. van Der Eb, and A. G. Jochemsen. 2000. Hdmx stabilizes Mdm2 and p53. J. Biol. Chem. 275:28039-28044. [DOI] [PubMed] [Google Scholar]
- 87.Stedman, W., Z. Deng, F. Lu, and P. M. Lieberman. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566-12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steele, A. J., A. G. Prentice, A. V. Hoffbrand, B. C. Yogashangary, S. M. Hart, E. P. Nacheva, J. D. Howard-Reeves, V. M. Duke, P. D. Kottaridis, K. Cwynarski, L. T. Vassilev, and R. G. Wickremasinghe. 2008. p53-mediated apoptosis of CLL cells: evidence for a transcription-independent mechanism. Blood 112:3827-3834. [DOI] [PubMed] [Google Scholar]
- 89.Symonds, H., J. D. Chen, and T. Van Dyke. 1991. Complex formation between the lymphotropic papovavirus large tumor antigen and the tumor suppressor protein p53. J. Virol. 65:5417-5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Szekely, L., C. Kiss, K. Mattsson, E. Kashuba, K. Pokrovskaja, A. Juhasz, P. Holmvall, and G. Klein. 1999. Human herpesvirus-8-encoded LNA-1 accumulates in heterochromatin-associated nuclear bodies. J. Gen. Virol. 80:2889-9000. [DOI] [PubMed] [Google Scholar]
- 91.Tanimura, S., S. Ohtsuka, K. Mitsui, K. Shirouzu, A. Yoshimura, and M. Ohtsubo. 1999. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 447:5-9. [DOI] [PubMed] [Google Scholar]
- 92.Tao, W., and A. J. Levine. 1999. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc. Natl. Acad. Sci. U. S. A. 96:3077-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tiemann, F., and W. Deppert. 1994. Stabilization of the tumor suppressor p53 during cellular transformation by simian virus 40: influence of viral and cellular factors and biological consequences. J. Virol. 68:2869-2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vassilev, L. T., B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi, and E. A. Liu. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844-848. [DOI] [PubMed] [Google Scholar]
- 95.Verma, S. C., S. Borah, and E. S. Robertson. 2004. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 78:10348-10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Verma, S. C., T. Choudhuri, R. Kaul, and E. S. Robertson. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80:2243-2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Viejo-Borbolla, A., E. Kati, J. A. Sheldon, K. Nathan, K. Mattsson, L. Szekely, and T. F. Schulz. 2003. A domain in the C-terminal region of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus affects transcriptional activation and binding to nuclear heterochromatin. J. Virol. 77:7093-7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Viejo-Borbolla, A., M. Ottinger, E. Bruning, A. Burger, R. Konig, E. Kati, J. A. Sheldon, and T. F. Schulz. 2005. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 79:13618-13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang, Y. V., M. Wade, E. Wong, Y. C. Li, L. W. Rodewald, and G. M. Wahl. 2007. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. Proc. Natl. Acad. Sci. U. S. A. 104:12365-12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wong, L. Y., G. A. Matchett, and A. C. Wilson. 2004. Transcriptional activation by the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen is facilitated by an N-terminal chromatin-binding motif. J. Virol. 78:10074-10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wong, L. Y., and A. C. Wilson. 2005. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen induces a strong bend on binding to terminal repeat DNA. J. Virol. 79:13829-13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu, X., J. H. Bayle, D. Olson, and A. J. Levine. 1993. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7:1126-1132. [DOI] [PubMed] [Google Scholar]
- 103.Ye, F. C., F. C. Zhou, S. M. Yoo, J. P. Xie, P. J. Browning, and S. J. Gao. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 78:11121-11129. [DOI] [PMC free article] [PubMed] [Google Scholar]