

Abstract
A key question in pandemic influenza is the relative roles of innate immunity and target cell depletion in limiting primary infection and modulating pathology. Here, we model these interactions using detailed data from equine influenza virus infection, combining viral and immune (type I interferon) kinetics with estimates of cell depletion. The resulting dynamics indicate a powerful role for innate immunity in controlling the rapid peak in virus shedding. As a corollary, cells are much less depleted than suggested by a model of human influenza based only on virus-shedding data. We then explore how differences in the influence of viral proteins on interferon kinetics can account for the observed spectrum of virus shedding, immune response, and influenza pathology. In particular, induction of high levels of interferon (“cytokine storms”), coupled with evasion of its effects, could lead to severe pathology, as hypothesized for some fatal cases of influenza.
Influenza A virus causes an acute respiratory disease in humans and other mammals; in humans, it is particularly important because of the rapidity with which epidemics develop, its widespread morbidity, and the seriousness of complications. Every year, an estimated 500,000 deaths worldwide, primarily of young children and the elderly, are attributed to seasonal influenza virus infections (49). Influenza pandemics may occur when an influenza virus with new surface proteins emerges, against which the majority of the population has no preexisting immunity. Both the emergence of H5N1 virus (34) and the current H1N1 virus pandemic (43) underline the importance of understanding the dynamics of infection and disease. A key question is, what regulates virus abundance in an individual host, causing the characteristic rapid decline in virus shedding following its initial peak? The main contenders in primary influenza virus infection are depletion of susceptible target cells and the impact of the host's innate immune response (2, 20).
On infection, the influenza virus elicits an immune response, including a rapid innate response that is correlated with the observed decline in the virus load after the first 2 days of infection (1). The slower adaptive response, including both humoral and cell-mediated components, takes several days to consolidate but is important for complete virus clearance and establishment of protective immunity. During infection of an immunologically naïve host, the innate immune response is particularly important as the first line of defense against infection. The innate immune response is regulated by chemokines and cytokines, chemical messengers produced by virus-infected epithelial cells and leukocytes (23), and natural interferon-producing cells, such as plasmacytoid dendritic cells (13). Among the key cytokines induced by epithelial cells infected with influenza A virus are type I interferons (IFNs) (IFN-α/β) (23), which directly contribute to the antiviral effect on infected and neighboring cells (38).
Like other viruses, influenza A viruses have evolved strategies to limit the induction of innate immune responses (38). The NS1 protein plays a dominant role, and without it, the virus is unable to grow well or to cause pathology in an immunocompetent host (14). NS1 is multifunctional and counteracts both the induction of IFN expression and the function of IFN-activated antiviral effectors via multiple mechanisms (12, 17). Individual strains of influenza A virus possess these activities to various degrees (15, 21, 22, 26), and accordingly, NS1 has been implicated as a virulence factor (3, 17). A striking effect of the failure to control the innate response to virus infection is seen as a “cytokine storm,” which causes severe pathology (8).
While there is an extensive literature on modeling influenza virus spread at the population level, the individual-host scale has received much less attention (2, 4, 5, 18, 19, 20, 27, 28). In a recent important paper, Baccam et al. modeled the kinetics of influenza A virus (2). The innate dynamics were included in the form of an IFN response that delayed and reduced virus production but did not prevent it; thus, the infection was resolved primarily through near-total depletion of epithelial cells. Their model was fitted to virus titers from human volunteers exposed to H1N1 influenza virus, but no data were available on the innate immune response or epithelial cell pathology. This has been a general difficulty in developing and validating more refined within-host models; there is a lack of detailed biological data from natural host systems, in particular, measures of immune kinetics and patterns of cellular depletion.
The model presented here explicitly includes the ability of IFN to induce a fully antiviral state in order to explore the relative regulatory role of innate immunity and target cell depletion. Data from experimental infections of immunologically naïve horses with an equine influenza virus (36) allowed us to calibrate our model, not only to viral kinetics, but also to IFN dynamics and cell depletion in the context of infection of a naïve natural mammalian host. With our fitted model, we then investigate modulation of the immune response.
MATERIALS AND METHODS
Description of data sets.
The study used data from experiments that were previously undertaken for other purposes and that had gone through full institutional ethical review at the time, and most of the data have been published (36). Virus shedding data (daily measures of RNA copies per milliliter of nasal secretion up to 10 days) and cytokine data (IFN fold change daily from days 1 to 5 postinfection) were obtained as part of an experiment to study the effect of vaccination on proinflammatory cytokine expression in horses after exposure to influenza virus (36). Data for individual horses from the control group, immunologically naïve horses, are presented here (only grouped data were given in reference 36).
Archived samples of lung tissues were available from four immunologically naïve yearling Welsh mountain ponies that were experimentally infected in 1988 by exposure to an aerosol of 106.6 50% egg infectious doses of A/equine/Newmarket/79 (H3N8) influenza virus (31) and that were killed at days 2.5, 3.5, 4.5, and 5.5 after infection. Sections of left cranial lung samples were stained with horseradish peroxidase-linked polyclonal antibody to influenza A virus nucleoprotein (as described previously [9]), and the number of infected cells was determined. The numbers of uninfected and infected epithelial cells per bronchiole in the sample from the left cranial lung were then scored visually. The cumulative percentage of infected cells over time was estimated by interpolation (of a cubic spline) to the total percentage of infected cells at each time point. The area under the curve was calculated (by numerical integration) and normalized by the life span of an infected cell. This gave an estimated total cell death, as it was assumed that all infected cells were killed by infection.
Fitting procedure.
A weighted nonlinear least-squares procedure was used to fit the model. The sum of the squared errors (SSE), given below, between model values and data points was minimized.
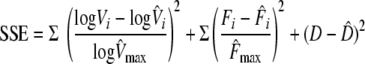 |
Weights are used to make errors dimensionless. For the virus shedding, the difference between model, Vi, and observed, V̂i, shedding is divided by the maximum observed shedding of an individual horse, V̂max, each of them in the log scale. An analogous definition is used for the errors corresponding to IFN secretion between the model, Fi, and the data, F̂i, with maximum IFN secretion, ^Fmax. The squared difference between predicted, D, and estimated, D̂, proportions of total cell death is also added to the SSE. Minimum detection levels for virus shedding are included in the fitting procedure to take into account negative experimental results. These detection levels are implemented by assuming a zero difference between the model and the data whenever both quantities are below these levels. Similarly, if the model is above the minimum detection level but the data are not, then only the difference between the model and the minimum detection level is considered. The numerical procedure was implemented using the function fminsearch in Matlab R2008b (The Mathworks).
RESULTS
Estimates of lung pathology in influenza virus-infected ponies.
To examine the levels of cell depletion during an influenza virus infection, we estimated the numbers of viral-antigen-positive cells in the lungs of ponies at days 2.5, 4.5, and 5.5 after challenge with influenza A/eq/Newmarket/79 (H3N8) virus (Fig. 1a to c). Immunohistochemistry indicated only a low percentage of infected cells, corresponding to an estimated total cell death of about 27% by the end of the infection (Fig. 1d). This estimate was dependent on the estimated life span of an infected cell (Table 1). However, the majority of bronchioles with infected cells were not seriously disrupted, indicating that most cells were not destroyed. This is consistent with observations in other hosts, where only limited foci of infection are seen, even in fatal cases (16, 47). These levels of infection suggest a mechanism for infection control other than substantial cell depletion (2), prompting us to develop a model for infection that included a strong innate immune response of the host to infection.
FIG. 1.

Experimental data. (a to c) The numbers of uninfected (blue) and infected (red) respiratory epithelial cells were determined for each bronchiole in a left cranial lung sample from archived samples from an experimental infection of three immunologically naïve horses. The distribution of infection is shown for 2.5 (a), 4.5 (b), and 5.5 (c) days postinfection. The bronchioles were sorted in ascending order with respect to the number of epithelial cells they contained. (d) Total percentages of infection at 2.5, 4.5, and 5.5 days postinfection were determined to be 1.93%, 4.73%, and 1.87%, respectively (circles). A simple function (solid curve) was interpolated to these data points to estimate the cumulative percentage of cells infected over the course of infection (27%).
TABLE 1.
Parameters of the innate-response model
| Parameter | Symbol | Unit | Value | Reference |
|---|---|---|---|---|
| Epithelial cell population | T0 | Cells | 3.5 × 1011 | 46 |
| Initial virus load | V0 | RNA copies (ml NS)−1 | Fitted | |
| Infectivity rate | β | (RNA copies)−1 ml NS day−1 | Fitted | |
| IFN-reduced infectivity | m | 1 | ||
| Virus production | p | RNA copies (ml NS)−1 day−1 cell−1 | Fitted | |
| Free-virus clearance | c | Rate of virus clearance day−1 | 5.2 | 2 |
| IFN efficiency | φ | (IFN fold change)−1 day−1 | Fitted | |
| IFN production | q | IFN fold change day−1 cell−1 | Fitted | |
| IFN-reduced production | n | 1 | ||
| IFN clearance | d | Rate of IFN clearance day−1 | Fitted | |
| Eclipse phase period | 1/ki | Days | 1/2 | 23 |
| Infectious period | 1/δ | Days | 1/2 | 23 |
| Prerefractory state | 1/a | Days | 1/4 | 11 |
Mathematical model of influenza virus infection dynamics.
In this model formulation, epithelial cells of the respiratory tract (the target cells of the virus) are classified according to their infection state. Uninfected epithelial cells (T) are infected at a rate proportional to the virus load (V), with the constant of proportionality β (to directly link the model to the data, we defined the virus load, V, as the number of RNA copies per milliliter of nasal secretion). These newly infected cells spend an average time of 1/k1 days in an eclipse phase (E1) before they start releasing progeny virus particles. The infectious cells (I) release viruses at a rate p per day and have an average life span of 1/δ days. As an influenza virus infection is likely to be cleared in about 7 days (32) and it takes around 5 days for the epithelium to start to regenerate (47), epithelial cell regeneration is not considered in the model. Indeed, even if logistic growth of epithelial cells is added to the model, there is no effect on the disease dynamics: infection rates predominate over cell regeneration rates (data not shown). Thus far, the model constructed in this way is the same as the target cell-limited model with delayed virus production of Baccam et al. (2). With this model, and in agreement with the results of Baccam et al., the infection is controlled by the exhaustion of epithelial cells, which is inconsistent with the proportion of dead cells shown in Fig. 1.
The host innate immune response was added to the model to explore its relative contribution as a mechanism for infection control, reflecting the crucial and well-described role of IFN in limiting virus spread (1). We assumed that infectious cells release IFN (F) during their life span at a rate q per day. IFN induces an antiviral state (R) at a rate φF in susceptible cells (T). Refractory cells (R) first go through a prerefractory state (W) for an average time of 1/a, during which they can still be infected at a rate mβV (lower than or equal to that of fully susceptible cells, 0 < m ≤ 1) (11, 40). If infected, these cells move to an eclipse phase (E2) and secrete IFN at a rate nq per day, because they have been exposed to IFN and primed into an antiviral state (39). After an average time of 1/k2 days spent in this eclipse stage, the cells become infectious. It was assumed that the virus and IFN load decay at rates c and d, respectively. Parameter definitions and corresponding units are given in Table 1. The complete dynamics are illustrated in Fig. 2, and the corresponding equations are given in system 1.
 |
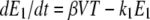 |
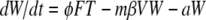 |
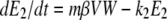 |
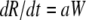 |
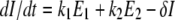 |
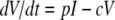 |
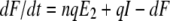 |
We estimated the parameters for viral kinetics and innate immune response (p, q, V0, d, β, and φ) by fitting the model, by nonlinear least squares, to the virus shedding and IFN profiles of six immunologically naïve horses (data from reference 36) and to the estimated proportion of total cell death (parameter estimates are shown in Table 2; average estimates were obtained by fitting the model to the pooled data). This experimental data set consisted of longitudinal measurements of the virus load estimated from nasal swabs and of interferon (relative quantification in blood) in experimentally infected naïve ponies. The remaining parameters were selected as follows. Sedmak and Grossberg (40) reported a minimum of about 6 h to be necessary for the development of maximum resistance to challenge with influenza virus in primary chicken embryo cell cultures—this value was therefore taken as the time spent by a cell in a prerefractory state (i.e., 1/a = 1/4 days). The times spent by an infected cell in the incubation and infectious classes were each taken as 12 h (i.e., ki = δ = 2). This gave 24 h for the life span of an infected cell, which is consistent with published data (23). The average rate of virus clearance was chosen as 5.2 per day (i.e., c = 5.2), as reported previously (2). Stone et al. (46) estimated the epithelial cell population size of the equine respiratory tract (T0) as 3.5 × 1011 cells. The value for the parameters m and n were chosen as 1 as a first approximation (different value choices are discussed in the Appendix). Table 1 contains a summary of all of the parameters in the model.
FIG. 2.

Diagram of the infection dynamics. Epithelial cells were classified into one of the following classes: susceptible (T), eclipse phase (E1 and E2), infectious (I), prerefractory (W), and refractory (R). Virus particles (V) are released by infectious cells, while interferon is secreted by infectious and eclipse phase (I and E2) cells.
TABLE 2.
Parameter estimates for the innate-response modela
| Pony no. | V0 | β | p | q | φ | d | SSE |
|---|---|---|---|---|---|---|---|
| 1 | 4.3 × 101 | 1.6 × 10−4 | 2.2 × 10−6 | 2.6 × 10−10 | 3.7 × 100 | 1.8 × 100 | 0.23 |
| 2 | 1.4 × 10−4 | 3.8 × 10−5 | 2.4 × 10−4 | 1.5 × 10−9 | 7.3 × 102 | 2.1 × 101 | 0.16 |
| 3 | 1.8 × 10−1 | 4.0 × 10−4 | 3.5 × 10−6 | 4.6 × 10−10 | 8.8 × 101 | 2.7 × 101 | 0.46 |
| 4 | 1.9 × 10−3 | 4.2 × 10−5 | 1.3 × 10−4 | 3.5 × 10−10 | 6.0 × 102 | 4.0 × 100 | 0.17 |
| 5 | 7.3 × 10−2 | 8.5 × 10−4 | 4.9 × 10−6 | 7.9 × 10−10 | 1.5 × 102 | 8.1 × 100 | 0.20 |
| 6 | 1.0 × 10−1 | 1.4 × 10−3 | 1.4 × 10−6 | 1.9 × 10−8 | 1.2 × 101 | 1.0 × 102 | 0.16 |
| Avgb | 3.2 × 10−1 | 1.4 × 10−4 | 1.4 × 10−5 | 5.0 × 10−10 | 5.6 × 101 | 6.8 × 100 | 1.8 |
Estimated parameters based on experimental data from Quinlivan et al. (36): V0, initial virus load; β, virus infectivity; p, virus production per cell; d, IFN clearance rate; φ, IFN efficiency; and q, IFN induction per cell. Parameters units are shown in Table 1.
For the “average horse,” all data were pooled for model fitting.
Figures 3 and 4 show the fitted model, which captures the experimental data well. In particular, the correct timings are identified for both peak virus loads and IFN activity, and the sharp peak of IFN response is remarkably well described by the model (Fig. 3, first and third rows). The model predicts that the majority of epithelial cells become refractory during infection (Fig. 3, second and fourth rows). This model does not require total cell death to control infection, consistent with observed data (Fig. 1a to c). Using the Akaike information criterion (AIC) (6), the model is very clearly supported for this data set over the IFN response model of Baccam et al. (for further details, see the Appendix).
FIG. 3.

Model dynamics. The innate-response model was fitted to individual virus shedding (RNA copies per milliliter of nasal secretion [NS]) and IFN (fold change) profiles of six immunologically naïve horses infected with A/equine/Kildare/89 (H3N8) virus (36) and to the estimated total cell death of 27%. (First and third rows) The virus shedding (red circles), IFN (blue triangles), and model output (solid and dashed curves, respectively) for each horse are shown. The minimum detection level for the virus load is drawn as a red horizontal line (negative results are plotted as zeros). (Second and fourth rows) Cell dynamics as predicted by the model (susceptible cells [T], dotted blue lines; infectious cells [I], dashed red lines; total numbers of epithelial cells, solid black lines). The purple triangles on the top right show the total percentages of cell death estimated in Fig. 1d.
FIG. 4.

Model dynamics; pooled data. The innate-response model was fitted to virus shedding and IFN responses of all six horses and to the estimated total cell death of 27%. (a) The virus shedding of each horse is drawn as a cross and the model output as a solid curve. The minimum detection level for the virus load is drawn as a horizontal line (negative results are plotted as zeros). (b) The IFN response (fold change) for each horse is given at each cross and the model output as a solid curve. (c) Cell dynamics as predicted by the model. Uninfected cells, T (dotted curve); infected cells, I (dashed line); and the total number of epithelial cells (solid curve) are shown. The triangle on the top right shows the total percentage of cell death estimated in Fig. 1d.
While our model gives clear qualitative results and captures the data well, some care must be taken when interpreting individual parameter estimates. Even in this relatively simple model, there may be some redundancy among the parameters. In particular, it is hard to disentangle viral production per cell and the rate of virus transmission (see Fig. A1 and further discussion in the Appendix). Where possible from the literature, some of the parameters were preset to fixed values (i.e., δ, k1, k2, a, c, m, and n) (Table 1); the impact of varying these is discussed in the Appendix. Despite these caveats, a consistent picture emerged of strong innate immunity (and not target cell depletion) limiting typical primary influenza virus infection in horses.
FIG. A1.

Interdependence between virus-related estimates. (a) Contour plot of SSE as a function of β (virus transmission) and p (virus production per cell). The point (pmin, βmin) gives the minimum SSE (red circle). The hyperbolic curve is defined as follows: β × p = constant = βmin × pmin (red curve). (b to d) The contour plots for virus shedding (b) (in log scale), IFN production (c), and the abundance of refractory cells (d) are drawn as functions of p and time (days postinfection). β is defined as βmin × pmin/p [i.e., pairs (p, β) from the red curve in panel a]. The time scale (x axis) in panel d was cut short to 2.5 days, as the abundance of refractory cells was nearly constant beyond that point.
Immune evasion and immunopathology.
Our calibrated model of viral and immune dynamics allows exploration of how variations in IFN induction and IFN efficiency (defined here as the rate at which IFN induces a refractory state in a susceptible target cell) can affect the spectrum of virus replication, tissue damage, and subsequent pathology. Experimental data show differences between engineered influenza virus mutants (37, 44) and different virus strains (48) in how they interact with the innate immune system and cause disease; much of this variation is mediated by viral factors (including different forms of the NS1 protein [37, 44]), which can affect either the induction of IFN or its efficiency in limiting virus growth (12, 17).
Varying the rate of IFN induction per cell gives an inverse relationship (Fig. 5a) between the percentage of cell death and the total amount of IFN produced (as the rate of IFN induction per cell is increased, total cell death decreases while total IFN produced increases). This variation can account for the findings of Solórzano et al. (44) and Quinlivan et al. (37), in which genetically engineered mutants of swine and equine influenza viruses, respectively, with truncated NS1 proteins unable to inhibit IFN induction were attenuated. Moreover, Solórzano et al. (44) reported a correlation between virus titers and lung lesions and a negative correlation between virus titers and IFN induction, which are both consistent with our model's predictions (Fig. 5a). In contrast, when the IFN efficiency is varied (Fig. 5b), a large percentage of cell death can occur despite the presence of increased amounts of IFN, as the virus evades the antiviral effects of IFN.
FIG. 5.

Spectrum of the immune response. (a and b) Both the total IFN produced (blue lines) and the peak virus load (red lines) were normalized to their baseline values and plotted on log base 10; cell death (black lines) is shown as a percentage. These values are all plotted against the rates of IFN induction per cell, q (a), or IFN efficiency, φ (b), both on a log scale normalized by their baseline values (average estimates from Table 2). (c to e) Contours plotted for various rates of IFN induction per cell, q, and IFN efficiency, φ, both on a log scale normalized by their baseline values (X marks the baseline values). Cell death (c) is shown as a percentage, while both total IFN produced (d) and the peak virus load (e) were normalized by baseline values and plotted on log base 10. The color scale on the right gives the values of this measure in each contour of the graph.
The full spectrum of the immune response can be explored by varying both the IFN induction and IFN efficiency together (the effects on cell death, total IFN production, and peak virus load are shown in Fig. 5c to e). With this dynamic range, the model can describe some observed phenomena. For example, Wattrang et al. compared virus shedding and IFN levels in two groups of horses infected with different strains of equine influenza virus (48). While the titers of virus shed were similar, the total IFN produced was greater and the clinical signs were more severe following infection with one strain. This can be captured in our model by high IFN induction and a low IFN efficiency against that strain (Fig. 5d to e, lower right of each plot).
Overall pathology of influenza virus infection.
Both cell death and damage to surviving cells caused by excessive amounts of IFN must be taken into account when considering the impact of infection on host pathology. A high level of cytokines may itself be harmful to the host (7). We defined a quantitative measure for the overall damage to the host as follows: damage = death + (1 − death) × f(IFN), where death is the proportion of cell death at the end of infection and f(IFN) is an increasing function of the total amount of IFN (defined as shown in Fig. 6a). This function f is chosen so that moderate amounts of IFN have little detrimental effect while the effect of large amounts of IFN on surviving cells is no worse than cell death.
FIG. 6.

Host damage and benefit to the virus. (a) Relative damage of IFN to surviving cells (a logistic function between 0 and 1). (b) Relative increase in benefit for the virus as a function of IFN (a logistic function between 1 and 1.5). (c and d) Both host damage (c) and benefit to the virus (d), as defined in the text, are plotted as functions of IFN induction, q, and IFN efficiency, φ. Both axes are shown on a log scale normalized by baseline values. The color scales on the right give the values of these measures in each contour of the graphs.
Similarly, a measure of the benefit to the virus can be defined in terms of the total amount of virus shed and the total IFN produced. Higher levels of IFN are associated with increased nasal discharge and coughing (19, 42, 48), which could increase the chances of between-host transmission, which is of benefit to the virus. This measure is explicitly defined as follows: benefit = total virus × g(IFN), where g is taken as shown in Fig. 6b. This particular choice of g assumes that IFN could increase the transmission probability by up to 50%.
Figures 6c and d show the host damage and the benefit to the virus against both IFN induction and efficiency. The worst damage is obtained in the region where IFN efficiency is low and IFN induction is either very high or low, whereas the greatest benefit to the virus is seen only where efficiency is low and induction is high. The exact regions of critical host damage and benefit to the virus (Fig. 6c and d) depend on specific choices of the functions f and g, but we expect these results to hold qualitatively across a range of appropriate f and g functions.
Severe human cases of H5N1 infection have been associated with hypercytokinemia (7, 10, 35), and recent work suggests that the 1918 pandemic strain may also have stimulated an unusually strong innate immune response (24, 25). This is sometimes described as a cytokine storm (8) and corresponds in our model to high IFN induction (Fig. 6c and d, right edge). However, our model output suggests that high IFN induction alone is not sufficient to produce severe damage; it must be coupled with poor IFN efficiency (Fig. 6c). Interestingly, there is some evidence to suggest that the H5N1 influenza virus may evade the effect of IFN (41).
DISCUSSION
The within-host dynamics of natural virus infection and the resulting immune responses underlie both the disease signs seen in infected hosts and virus transmission, thus providing the foundation for the evolutionary dynamics and public health impact of the virus on a global scale. Our model, calibrated by experimental data from an influenza virus infection of a natural host, successfully captures several aspects of the within-host dynamics of influenza virus in immunologically naïve individuals and demonstrates the critical role of the innate immune response in controlling early infection. Resource limitation of the virus “predator,” in terms of depletion of susceptible cells, plays an important role as a regulatory mechanism in primary infection. However, innate immunity seems to be the central regulatory force, providing a protected state for target cells, rather than total cell death, as suggested from models in recently published work (2). The relevance of innate immunity was also highlighted for murine influenza virus, where it was required to describe the infection dynamics in a model that included adaptive immunity (20).
The qualitative results of viral modulation of immunity apply in a wide range of settings and can be used to explore specific scenarios. In particular, the model allows a dynamic interpretation of the spectrum of pathology, which is key for understanding the impact of an emerging virus. Interestingly, the relationship between virus shedding and pathology is far from simple. Virus abundance is predicted to be fairly stable across a wide spectrum of viral immune escape phenotypes, consistent with data from natural hosts (48); this arises from the balance of nonlinear effects on virus growth. These dynamics are even more complex when infections in primed hosts are considered; a modification to our model suggests a potentially important trade-off between innate and adaptive immunity (data not shown), a key area for future research. Overall, a modeling approach provides us with a greater understanding of the complex relationship between disease and viral dynamics for an important pathogen.
Even though this is a simple model, a number of caveats arise. The model was fitted to observed equine data, but even this simple model is somewhat overspecified, requiring richer data sets to produce precise parameter estimates for specific virus strains and host species. Ideally, parallel viral, innate and adaptive immune, and pathology data from natural hosts would be available; indirect (nondestructive) methods for measuring target cell depletion would be particularly useful. Another interesting frontier is within-host spatial variation in virus abundance, pathology, and immunity.
Infectious epithelial cells (and IFN-primed eclipse phase cells) are assumed to be the only producers of antiviral cytokines in our model. However, it is recognized that plasmacytoid dendritic cells are potent producers of IFN in the body (13). In addition, our model focuses mainly on primary infection, where infection is at its highest and only the innate immune response may play an important role (as would be the case in pandemic influenza). However, to have the complete picture, the explicit dynamics of the adaptive immune response must be included, as antibodies and cytotoxic T lymphocytes (CTLs) are recognized as playing important roles in the clearance of infection (30, 33). While it is relatively easy to extend compartmental models to include additional components of the immune system, such as these, model development must be guided by appropriate data for meaningful progress to be made.
The current paper provides a template for models based on data on influenza dynamics within hosts. Extending this approach to humans, birds, and other host species, as well as to the impact of interventions, such as drug treatment (29), would yield a rich harvest in terms of elucidating the cross-scale dynamics and evolution of this key virus.
Acknowledgments
This work was supported by BBSRC research grant BBS/B/00522. J.R.G. was supported by a Royal Society University Research Fellowship. J.L.N.W. was supported by Defra grant VT0105 and the Alborada Trust. B.T.G. was supported by NIH grant R01 GM083983-01 and NSF grant 0742373. J.R.G., B.T.G., and J.L.N.W. were supported by the RAPIDD program of the Science and Technology Directorate, Department of Homeland Security, and the Fogarty International Center, National Institutes of Health.
We thank Tovah Shaw, Jodie Miller, and Annette Flindall for performing the immunohistochemistry on archived samples; Filipe Nunes for his assistance in the counting of infected epithelial cells; and Wendy Barclay and members of her laboratory for discussions. We also thank our anonymous referees for their comments on the manuscript.
APPENDIX
Robustness of the model and fitting algorithm: cross-comparison of models and data sets.
To test the robustness of our model and to validate our fitting algorithm, we fitted our model of innate immunity to the data from Baccam et al. (2), and also vice versa (fitting the model from Baccam et al. to the data set used in our study). Our innate-immunity model was successfully fitted to the virus-shedding data from studies of human patients used by Baccam et al. (see Table 1 in reference 2). Parameter values similar to those reported by Baccam et al. were obtained, and the sum of squared errors was slightly reduced (data not shown).
Similarly, the IFN response model from Baccam et al. (2) was fitted to the equine data used in the present study (36), consisting of both virus shedding and interferon activity. The SSEs were similar to those obtained with our model (Table A1), but as previously discussed, the infection was controlled by the total depletion of target cells. Furthermore, using the AIC (6), our innate-response model had the lowest score (with a difference of 52.9 in the AIC), which suggests that our model provides a more parsimonious explanation of the data from the equine studies (Table A1). Some care needs to be taken in these AIC computations, due to the large number of parameters (36 or 42) in comparison with the size of the data set (90). We use a small-sample bias adjustment suggested by Burnham and Anderson (6), giving the AIC formula AIC = 2K + n log(SSE/n) + [2K(K + 1)]/(n − K − 1), where K is the total number of parameters, n is the total number of data points, and SSE is the cumulative sum of squared errors.
TABLE A1.
Fitting results for our model and the IFN response model of Baccam et al.
| Pony no. | Our model |
Baccam et al. model |
||||
|---|---|---|---|---|---|---|
| SSE | No. of parameters | No. of data | SSE | No. of parameters | No. of dataa | |
| 1 | 0.23 | 6 | 16 | 0.14 | 7 | 15 |
| 2 | 0.16 | 6 | 16 | 0.21 | 7 | 15 |
| 3 | 0.46 | 6 | 16 | 0.17 | 7 | 15 |
| 4 | 0.17 | 6 | 16 | 0.15 | 7 | 15 |
| 5 | 0.20 | 6 | 16 | 0.22 | 7 | 15 |
| 6 | 0.16 | 6 | 16 | 0.19 | 7 | 15 |
| Cumulative | 1.38b | 36 | 96 | 1.08c | 42 | 90 |
The estimated total proportion of cell death was not used for model fitting, as total cell depletion results from this model.
AIC cumulative, −290.1.
AIC cumulative, −237.2.
We advise caution when looking at this model comparison, since not all of the parameters in both models were estimated and then used in the computation of the AIC values. However, since the numbers of neglected parameters in each model are similar and the difference in AIC values is relatively high, we still expect our model to be favored over the model of Baccam et al.
Difficulties in separating parameters.
In our innate-response model, the rate of change of virus abundance depends directly on the rate of virus production per cell, p, and the abundance of infectious cells, which in turn depends on the rate of virus transmission, β. As these parameters interact in their effects, there is a strong linkage between the fittings for the two parameters. For a given virus abundance, the greater the virus production per cell, the fewer cells that need to be infected to obtain a given virus load (Fig. A1a). While the product of these two parameters (p and β) can be reasonably estimated, it is currently very hard to give a narrow estimate of either separately. This can be seen in Fig. A1a, where the peak of best fit is stretched along a hyperbolic curve in the p-versus-β plane. This hyperbolic curve is defined by β × p = βmin × pmin, where the point (pmin, βmin) gives the minimum SSE (all other parameters are fixed to average estimates from Table 2). The dynamics of the model are obtained at each point along this hyperbolic curve (Fig. A1b to d). For each value of p, the contours of the virus load (Fig. A1b), IFN response (Fig. A1c), and number of refractory cells (Fig. A1d) are shown as a function of time (according to how the infection develops). As the value of p is varied but the product is kept constant (so that β is given by βmin × pmin/p), the contours in Fig. A1b to d are vertical for a wide range of p. This means that the dynamics of the virus load, IFN activity, and refractory cells are not sensitive to the parameters independently. To disentangle these parameters, we would need detailed data on the proportions or numbers of infected cells.
There is similar ambiguity with the immune parameters q and φ, as the overall immune response depends on both the amount of IFN available and its effect. These two parameters represent the rate of production of IFN, which scales the absolute quantity, and how effective IFN is per unit. Again, there is an inverse relationship between these two parameters: the product of the two determines the dynamics.
Sensitivity of parameter values.
We investigated the effect on the dynamics of the model when the parameter values were varied. The virus peak, the time when the peak occurred, the IFN peak, and the percentage of cell death were recorded as each of the parameter values in the model was varied (Fig. A2 and A3). The “average” estimates for the parameters in Table 2 were used as baseline values. For the rest of the parameters, the values reported in Table 1 were used as baseline values. The values for the estimated parameters (Table 2) were varied from a factor of 1/10 to 10 times their estimated values (Fig. A2). On the other hand, the values for the preset parameters (Table 1) were varied from 50% to 150% of their baseline values (Fig. A3). The uncertainty expected in the estimates in Table 2 was the main reason for having different factors for the parameters in Fig. A2 and A3. Both the transmission parameter, β, and the virus production, p, have a strong effect on all the dynamics of the model, increasing cell death and virus and IFN peaks and decreasing the time for the virus peak (Fig. A2). A similar effect was observed when the parameter m was varied (Fig. A3). The IFN efficiency, φ, and IFN production, q, have an effect on only the IFN peak and the percentage of cell death (Fig. A2). The initial virus shedding, V0, seems to have any effect on only the time when the virus peak occurs (Fig. A2). The eclipse phase period, 1/k1, determines when the virus peak occurs, while the infectious period, 1/δ, has an effect on the value of the peak (Fig. A3). Similarly, the virus clearance rate, c, has a strong effect on the virus peak, but it also affects the IFN peak and the percentage of cell death (Fig. A3). The duration of the prerefractory state, 1/a, has a negative effect on all the dynamics of the model (Fig. A3). The IFN clearance rate, d, has a strong effect on the IFN peak and a mild effect on the percentage of cell death (Fig. A2). The IFN-reduced production, n, seems to affect only the amount of IFN at its peak (Fig. A3).
FIG. A2.

Sensitivity of the model parameters. The parameters V0, β, p, φ, q, and d were varied, one at a time, and the model dynamics were calculated. The virus peak, the time when the peak occurred, the IFN peak, and the total percentage of cell death are shown (the dotted lines show the dynamics at baseline values). The parameters were varied between 10−1 (open triangles), 10−0.5 (open squares), 100.5 (filled squares), and 101 (filled triangles) of their baseline values (average estimates from Table 2).
FIG. A3.

Sensitivity of the model parameters. The parameters k1, k2, δ, c, a, m, and n were varied, one at a time, and the model dynamics were calculated. The virus peak, the time when the peak occurred, the IFN peak, and the total percentage of cell death are shown (the dotted lines show the dynamics at baseline values). The parameters were varied between 0.5 (open triangles), 0.75 (open squares), 1.25 (filled squares), and 1.5 (filled triangles) of their baseline values (shown in Table 1).
The value of the total number of epithelial cells, T0, scales the rates of virus and IFN production per cell, p and q, respectively. To show this, following a method described previously (45), let (T, E1, W, E2, R, I, V, F) be the solution for system 1 with initial condition (T0, 0, 0, 0, 0, 0, V0, 0). Then, it can be easily determined that (T̃, Ẽ1, W̃, Ẽ2, R̃, Ĩ, Ṽ, F̃) = (σT, σE1, σW, σE2, σR, σI, V, F) is the solution of system 1 with the new parameters p̃ = (1/σ)p and q̃ = (1/σ)q and for the initial condition (σT0, 0, 0, 0, 0, 0, V0, 0).
Footnotes
Published ahead of print on 3 February 2010.
REFERENCES
- 1.Ada, G. L., and P. D. Jones. 1986. The immune response to influenza infection. Curr. Top. Microbiol. Immunol. 128:1-54. [DOI] [PubMed] [Google Scholar]
- 2.Baccam, P., C. Beauchemin, C. A. Macken, F. G. Hayden, and A. S. Perelson. 2006. Kinetics of influenza A virus infection in humans. J. Virol. 80:7590-7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basler, C. F., and P. V. Aguilar. 2008. Progress in identifying virulence determinants of the 1918 H1N1 and the Southeast Asian H5N1 influenza A viruses. Antiviral Res. 79:166-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beauchemin, C., J. Samuel, and J. Tuszynski. 2005. A simple cellular automaton model for influenza A viral infections. J. Theor. Biol. 232:223-234. [DOI] [PubMed] [Google Scholar]
- 5.Bocharov, G. A., and A. A. Romanyukha. 1994. Mathematical model of antiviral immune response. III. Influenza A virus infection. J. Theor. Biol. 167:323-360. [DOI] [PubMed] [Google Scholar]
- 6.Burnham, K. P., and D. R. Anderson. 2002. Model selection and multimodel inference: a practical information-theoretic approach, 2nd ed. Springer Verlag, New York, NY.
- 7.Cheung, C. Y., L. L. M. Poon, A. S. Lau, W. Luk, Y. L. Lau, K. F. Shortridge, S. Gordon, Y. Guan, and J. S. M. Peiris. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831-1837. [DOI] [PubMed] [Google Scholar]
- 8.Clark, I. A. 2007. The advent of the cytokine storm. Immunol. Cell Biol. 85:271-273. [DOI] [PubMed] [Google Scholar]
- 9.Daly, J. M., K. E. Whitwell, J. Miller, G. Dowd, J. M. Cardwell, and K. C. Smith. 2006. Investigation of equine influenza cases exhibiting neurological disease: coincidence or association? J. Comp. Pathol. 134:231-235. [DOI] [PubMed] [Google Scholar]
- 10.de Jong, M. D., C. P. Simmons, T. T. Thanh, V. M. Hien, G. J. D. Smith, T. N. B. Chau, D. M. Hoang, N. V. V. Chau, T. H. Khanh, and V. C. Dong. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 12:1203-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dianzani, F., I. Viano, M. Santiano, M. Zucca, P. Gullino, and S. Baron. 1978. Tissue culture models of in vivo interferon production and action. Adv. Exp. Med. Biol. 110:119-131. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Sesma, A. 2007. The influenza virus NS1 protein: inhibitor of innate and adaptive immunity. Infect. Disord. Drug Targets 7:336-343. [DOI] [PubMed] [Google Scholar]
- 13.Fitzgerald-Bocarsly, P., J. Dai, and S. Singh. 2008. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 19:3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.García-Sastre, A., A. Egorov, D. Matassov, S. Brandt, D. E. Levy, J. E. Durbin, P. Palese, and T. Muster. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324-330. [DOI] [PubMed] [Google Scholar]
- 15.Geiss, G. K., M. Salvatore, T. M. Tumpey, V. S. Carter, X. Wang, C. F. Basler, J. K. Taubenberger, R. E. Bumgarner, P. Palese, and M. G. Katze. 2002. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl. Acad. Sci. U. S. A. 99:10736-10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guarner, J., C. D. Paddock, W. J. Shieh, M. M. Packard, M. Patel, J. L. Montague, T. M. Uyeki, N. Bhat, A. Balish, and S. Lindstrom. 2006. Histopathologic and immunohistochemical features of fatal influenza virus infection in children during the 2003-2004 season. Clin. Infect. Dis. 43:132-140. [DOI] [PubMed] [Google Scholar]
- 17.Hale, B. G., R. E. Randall, J. Ortin, and D. Jackson. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359-2376. [DOI] [PubMed] [Google Scholar]
- 18.Hancioglu, B., D. Swigon, and G. Clermont. 2007. A dynamical model of human immune response to influenza A virus infection. J. Theor. Biol. 246:70-86. [DOI] [PubMed] [Google Scholar]
- 19.Handel, A., I. Longini, Jr., and R. Antia. 2007. Neuraminidase inhibitor resistance in influenza: assessing the danger of its generation and spread. PLoS Comput. Biol. 3:2456-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Handel, A., I. M. Longini, Jr., and R. Antia. 2010. Towards a quantitative understanding of the within-host dynamics of influenza A infections. J. R. Soc. Interface 7:35-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayman, A., S. Comely, A. Lackenby, L. C. S. Hartgroves, S. Goodbourn, J. W. McCauley, and W. S. Barclay. 2007. NS1 proteins of avian influenza A viruses can act as antagonists of the human alpha/beta interferon response. J. Virol. 81:2318-2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayman, A., S. Comely, A. Lackenby, S. Murphy, J. McCauley, S. Goodbourn, and W. Barclay. 2006. Variation in the ability of human influenza A viruses to induce and inhibit the IFN-pathway. Virology 347:52-64. [DOI] [PubMed] [Google Scholar]
- 23.Julkunen, I., K. Melén, M. Nyqvist, J. Pirhonen, T. Sareneva, and S. Matikainen. 2001. Inflammatory responses in influenza A virus infection. Vaccine 19:S32-S37. [DOI] [PubMed] [Google Scholar]
- 24.Kash, J. C., C. F. Basler, A. Garcia-Sastre, V. Carter, R. Billharz, D. E. Swayne, R. M. Przygodzki, J. K. Taubenberger, M. G. Katze, and T. M. Tumpey. 2004. Global host immune response: pathogenesis and transcriptional profiling of type A influenza viruses expressing the hemagglutinin and neuraminidase genes from the 1918 pandemic virus. J. Virol. 78:9499-9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobasa, D., A. Takada, K. Shinya, M. Hatta, P. Halfmann, S. Theriault, H. Suzuki, H. Nishimura, K. Mitamura, and N. Sugaya. 2004. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 431:703-707. [DOI] [PubMed] [Google Scholar]
- 26.Kochs, G., A. Garcia-Sastre, and L. Martinez-Sobrido. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81:7011-7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larson, E. W., J. W. Dominik, A. H. Rowberg, and G. A. Higbee. 1976. Influenza virus population dynamics in the respiratory tract of experimentally infected mice. Infect. Immun. 13:438-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, H. Y., D. J. Topham, S. Y. Park, J. Hollenbaugh, J. Treanor, T. R. Mosmann, X. Jin, B. M. Ward, H. Miao, J. Holden-Wiltse, A. S. Perelson, M. Zand, and H. Wu. 2009. Simulation and prediction of the adaptive immune response to influenza A virus infection. J. Virol. 83:7151-7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lipsitch, M., T. Cohen, M. Murray, and B. R. Levin. 2007. Antiviral resistance and the control of pandemic influenza. PLoS Med. 4:111-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitchell, D. M., A. J. McMichael, and J. R. Lamb. 1985. The immunology of influenza. Br. Med. Bull. 41:80-85. [DOI] [PubMed] [Google Scholar]
- 31.Mumford, J. A., D. Hannant, and D. M. Jessett. 1990. Experimental infection of ponies with equine influenza (H3N8) viruses by intranasal inoculation or exposure to aerosols. Equine Vet. J. 22:93-98. [DOI] [PubMed] [Google Scholar]
- 32.Nicholson, K., R. Webster, and A. Hay. 1998. Textbook of influenza. Blackwell Science, Cornwall, United Kingdom.
- 33.Outlaw, M. C., and N. J. Dimmock. 1991. Insights into neutralization of animal viruses gained from study of influenza virus. Epidemiol. Infect. 106:205-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peiris, J. S. M., M. D. de Jong, and Y. Guan. 2007. Avian influenza virus (H5N1): a threat to human health. Clin. Microbiol. Rev. 20:243-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peiris, J. S. M., W. C. Yu, C. W. Leung, C. Y. Cheung, W. F. Ng, J. M. Nicholls, T. K. Ng, K. H. Chan, S. T. Lai, and W. L. Lim. 2004. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 363:617-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlivan, M., M. Nelly, M. Prendergast, C. Breathnach, D. Horohov, S. Arkins, Y. W. Chiang, H. J. Chu, T. Ng, and A. Cullinane. 2007. Pro-inflammatory and antiviral cytokine expression in vaccinated and unvaccinated horses exposed to equine influenza virus. Vaccine 25:7056-7064. [DOI] [PubMed] [Google Scholar]
- 37.Quinlivan, M., D. Zamarin, A. Garcia-Sastre, A. Cullinane, T. Chambers, and P. Palese. 2005. Attenuation of equine influenza viruses through truncations of the NS1 protein. J. Virol. 79:8431-8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Randall, R. E., and S. Goodbourn. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1-47. [DOI] [PubMed] [Google Scholar]
- 39.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya, M. Katsuki, S. Noguchi, and N. Tanaka. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13:539-548. [DOI] [PubMed] [Google Scholar]
- 40.Sedmak, J. J., and S. E. Grossberg. 1973. Interferon bioassay: reduction in yield of myxovirus neuraminidases. J. Gen. Virol. 21:1-7. [DOI] [PubMed] [Google Scholar]
- 41.Seo, S. H., E. Hoffmann, and R. G. Webster. 2002. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 8:950-954. [DOI] [PubMed] [Google Scholar]
- 42.Skoner, D. P., D. A. Gentile, A. Patel, and W. J. Doyle. 1999. Evidence for cytokine mediation of disease expression in adults experimentally infected with influenza A virus. J. Infect. Dis. 180:10-14. [DOI] [PubMed] [Google Scholar]
- 43.Smith, G. J. D., D. Vijaykrishna, J. Bahl, S. J. Lycett, M. Worobey, O. G. Pybus, S. K. Ma, C. L. Cheung, J. Raghwani, and S. Bhatt. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122-1126. [DOI] [PubMed] [Google Scholar]
- 44.Solórzano, A., R. J. Webby, K. M. Lager, B. H. Janke, A. García-Sastre, and J. A. Richt. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J. Virol. 79:7535-7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stafford, M. A., L. Corey, Y. Cao, E. S. Daar, D. D. Ho, and A. S. Perelson. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203:285-301. [DOI] [PubMed] [Google Scholar]
- 46.Stone, K. C., R. R. Mercer, P. Gehr, B. Stockstill, and J. D. Crapo. 1992. Allometric relationships of cell numbers and size in the mammalian lung. Am. J. Respir. Cell Mol. Biol. 6:235-243. [DOI] [PubMed] [Google Scholar]
- 47.Taubenberger, J. K., and D. M. Morens. 2008. The pathology of influenza virus infections. Annu. Rev. Pathol. Mech. Dis. 3:499-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wattrang, E., D. M. Jessett, P. Yates, L. Fuxler, and D. Hannant. 2003. Experimental infection of ponies with equine influenza A2 (H3N8) virus strains of different pathogenicity elicits varying interferon and interleukin-6 responses. Viral Immunol. 16:57-67. [DOI] [PubMed] [Google Scholar]
- 49.World Health Organization. 2009. Influenza fact sheet no. 211. http://www.who.int/mediacentre/factsheets/fs211/en/.