

Abstract
The rabies virus Ni-CE strain causes nonlethal infection in adult mice after intracerebral inoculation, whereas the parental Nishigahara (Ni) strain kills mice. We previously reported that the chimeric CE(NiN) strain with the N gene from the Ni strain in the genetic background of the Ni-CE strain kills adult mice, indicating that the N gene is related to the different pathogenicities of Ni and Ni-CE strains. In the present study, to obtain an insight into the mechanism by which the N gene determines viral pathogenicity, we compared the effects of Ni, Ni-CE, and CE(NiN) infections on host gene expressions using a human neuroblastoma cell line. Microarray analysis of these infected cells revealed that the expression levels of particular genes in Ni- and CE(NiN)-infected cells, including beta interferon (IFN-β) and chemokine genes (i.e., CXCL10 and CCL5) were lower than those in Ni-CE-infected cells. We also demonstrated that Ni-CE infection activated the interferon regulatory factor 3 (IRF-3)-dependent IFN-β promoter and induced IRF-3 nuclear translocation more efficiently than did Ni or CE(NiN) infection. Furthermore, we showed that Ni-CE infection, but not Ni or CE(NiN) infection, strongly activates the IRF-3 pathway through activation of RIG-I, which is known as a cellular sensor of virus infection. These findings indicate that the N protein of rabies virus (Ni strain) has a function to evade the activation of RIG-I. To our knowledge, this is the first report that the Mononegavirales N protein functions to evade induction of host IFN and chemokines.
Rabies virus, which belongs to Lyssavirus of the family Rhabdoviridae, which belongs to the order Mononegavirales, is known as a highly neurotropic virus and causes fatal encephalitis accompanied by severe neurological symptoms in almost all mammals, including humans. The genome is an unsegmented negative sense RNA and contains five genes (N, P, M, G, and L genes) encoding nucleoprotein (N protein), phosphoprotein (P protein), matrix (M) protein, glycoprotein (G protein), and large (L) protein, respectively (12). The N, P, and L proteins form helical ribonucleoprotein (RNP), together with the viral genomic RNA. The N protein participates in encapsidation of the genomic RNA. Only the encapsidated genomic RNA can be a template for replication of the viral genome and transcription of the viral mRNAs by the RNA-dependent RNA polymerase, L protein. The P protein binds to both N and L proteins and functions as a cofactor of the viral RNA polymerase. During virus assembly, the RNP is wrapped into an envelope containing an inner layer of the M protein and the transmembrane spike protein, G protein.
In response to viral infection (e.g., picornavirus, bunyavirus, and flavivirus infections), neurons in the brain produce type I interferon (IFN) comprised of the IFN-α family and IFN-β, which induces an antiviral status of a cell and functions as a main player for the host innate immunity (8, 9). The brain neurons are also capable of responding to the produced type I IFN. The fact that rabies virus can efficiently replicate in brain neurons strongly suggests that the virus has a certain mechanism to circumvent the host innate immunity. Interestingly, it has recently been reported that the P protein counteracts the innate immunity by inhibiting the cellular IFN system (6, 7, 38, 39).
Recently, it has been reported that rabies virus infection is recognized by a cellular sensor protein, retinoic acid-inducible gene I (RIG-I), and then induces type I IFN (16). RIG-I contains two N-terminal caspase recruitment domains (CARDs) and a DExD/H-box helicase domain (43). The helicase domain of RIG-I recognizes viral RNAs, and their CARDs are responsible for signaling through interaction with IFN-β promoter stimulator 1 (IPS-1) (also known as MAVS, VISA, or CARDIF) (21). It has been proposed that RIG-I adopts a “closed (inactivated)” conformation in the absence of viral RNAs but changes to an “opened (activated)” structure upon binding to viral RNAs, exposing the CARDs (37). Interaction of RIG-I and IPS-1 results in activation of TANK-binding kinase 1 (TBK-1). Activated TBK-1 catalyzes phosphorylation and dimerization of interferon regulatory factor 3 (IRF-3). Dimerized IRF-3 is translocated into the nucleus, where, together with nuclear factor (NF)-κB, it activates the transcription of type I IFN (1, 14, 20). Brzozka et al. (6) reported that rabies virus P protein interferes with the phosphorylation of IRF-3 by TBK-1 and consequently inhibits type I IFN induction.
Type I IFN that is produced and secreted by infected cells interacts with its receptor on the cell surface and then activates JAK/STAT-medicated signal pathways that result in expression of antiviral proteins (1, 33). It has been reported that rabies virus P protein binds to STAT1 and STAT2, which are components of the transcription factor ISGF-3 for the type I IFN signaling pathway, and that the P protein inhibits the translocation of STAT1 and STAT2 to the nucleus and consequently suppresses cellular antiviral responses (7, 38, 39). As mentioned above, rabies virus P protein counteracts host innate immunity by inhibiting both type I IFN induction and cellular antiviral responses induced by IFN. On the other hand, the other protein of rabies virus that is responsible for circumvention of host innate immunity has not been reported yet.
The fixed rabies virus Nishigahara (Ni) strain kills adult mice after intracerebral (i.c.) inoculation, whereas the chicken embryo fibroblast cell-adapted strain Ni-CE causes nonlethal infection in adult mice (35). We have also reported that a chimeric virus, CE(NiN) strain, which has the N gene from Ni strain in the genetic background of Ni-CE strain, kills adult mice after i.c. inoculation. This indicated that the N gene is related to the different pathogenicities of Ni and Ni-CE strains. However, the mechanism by which the N gene from Ni strain determines the pathogenicity has not been clarified yet. In the present study, in order to obtain an insight into the mechanism, we comprehensively examined and compared the effects of Ni, Ni-CE, and CE(NiN) infections on host gene expressions of a human neuroblastoma cell line. DNA microarray analysis of these infected cells revealed that expression levels of particular genes in Ni and CE(NiN)-infected cells such as the IFN-β and chemokine genes, which are known to be regulated by IRF-3, were lower than the levels in Ni-CE-infected cells. Further analyses demonstrated that N protein of rabies virus (Ni strain) has a function to evade activation of the RIG-I-mediated antiviral response. To our knowledge, this is the first report that the Mononegavirales N protein functions to evade induction of host IFN and chemokines.
MATERIALS AND METHODS
Cells and viruses.
Human neuroblastoma SYM-I cells (kindly provided by A. Kawai) (15) and mouse neuroblastoma NA cells were maintained in Eagle minimal essential medium supplemented with 10% fetal calf serum. 293T cells were maintained in Dulbecco minimal essential medium (high glucose) supplemented with 10% fetal calf serum. Recombinant Ni and Ni-CE strains were recovered from the cloned cDNA of the respective strains, as reported previously (35, 42). The chimeric CE(NiN) strain was previously generated by using the reverse genetic system of Ni-CE strain (35). The genomic organizations of Ni, Ni-CE, and CE(NiN) strains and their pathogenicities for adult mice are shown in Fig. 1A. Stocks of all rabies virus strains were prepared in NA cells. The B-1 vaccine strain of Newcastle disease virus (NDV) was kindly provided by H. Fukushi. NDV was grown in 10-day-old embryonated chicken eggs.
FIG. 1.

Schematic diagrams of genome organizations and replication efficiency of Ni, Ni-CE, and CE(NiN) strains. (A) Schematic diagrams of genome organizations of Ni, Ni-CE, and chimeric CE(NiN) strains. Shaded and open boxes represent open reading frames derived from Ni and Ni-CE strains, respectively. The pathogenicity of each strain for adult mice determined by our previous study (35) is also indicated. The pathogenicity was previously evaluated by i.c. inoculation with 1,000 FFU of each virus. ++, Lethal (all mice died within 7 days); +, lethal (all mice died within 10 days); -, nonlethal. (B) SYM-I cells were infected with Ni, Ni-CE, and CE(NiN) strains at an MOI of 2. Total cellular RNA was extracted at 6, 12, and 24 hpi and analyzed for viral genomic and antigenomic RNA levels by real-time PCR. Expression levels of genes were normalized to mRNA levels of GAPDH. Each point represents the mean (± the SD) of three independent replicates. ns, no significant difference.
Propagation of Ni, Ni-CE, and CE(NiN) strains in SYM-I cells.
SYM-I cells grown in a 24-well tissue culture plate (Greiner Bio-One Co., Ltd) were inoculated with Ni, Ni-CE, and CE(NiN) strains at a multiplicity of infection (MOI) of 2. At 24 h postinfection (hpi), viruses in the culture supernatants were harvested and titrated in NA cells by indirect focus assay using monoclonal antibody 13-27 specific for N protein (27).
Total RNA preparation.
A monolayer culture of SYM-I cells was infected with each rabies virus at an MOI of 2. Total cellular RNA was extracted at 6, 12, and 24 hpi using an RNeasy Mini Total RNA extraction kit (Qiagen). The extracted RNA was treated with an RNase-free DNase kit (Qiagen) and suspended in nuclease-free water. RNA preparations used for DNA microarray analysis were analyzed with a lab-on-a-chip Agilent Bioanalyzer (RNA 6000 LabChip kit; Agilent) to confirm the concentration, integrity, and purity.
DNA microarray hybridization and analysis.
cRNA used for DNA microarray hybridization was prepared according to the One-Color microarray-based gene expression analysis protocol (Agilent). Probes were synthesized from 600 ng of total RNA isolated from two independent biological replicates in two steps according to the manufacturer's instructions. In the first step, double-stranded cDNA was synthesized with mouse Moloney murine leukemia virus reverse transcriptase (Agilent) and an oligo(dT)-T7 RNA polymerase promoter (Agilent). In the second step, we synthesized antisense cRNAs that were labeled by the incorporation of Cy3-CTP during in vitro transcription. All reagents were from Agilent's fluorescent linear amplification kit adapted for use with small amounts of total RNA. Labeled cRNAs were fragmented to an average size of 50 to 100 nucleotides by heating the samples at 60°C in a fragmentation buffer provided by Agilent. Hybridization was performed on whole-human-genome 4 × 44K oligonucleotide microarrays (G4112F; Agilent) with reagents and protocols provided by the manufacturer. After hybridization, the arrays were washed and scanned using a DNA microarray scanner (Agilent). Feature extraction software provided by Agilent (version 9.1) was used to quantify the intensity of fluorescent images and to normalize results by subtracting local background fluorescence, according to the manufacturer's instructions. The expression level of each gene was analyzed by GeneSpring GX software (version 7.3.1; Agilent). Briefly, after importing the processed data into the software, they were normalized based on the default normalizing settings for one-color experiments (GeneSpring 7.3 user's guide; Agilent). Cluster analysis was performed by using Cluster 3.0 and Java TreeView.
Real-time reverse transcription-PCR (RT-PCR).
To measure levels of virus genomic and antigenomic RNAs in infected cells, total RNA was reverse transcribed into cDNAs by using SuperScript III reverse transcriptase (Invitrogen) with reverse transcriptase primers specific to rabies virus genomic and antigenomic RNAs (Table 1) or oligo(dT)20 [for detection of human housekeeping glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA, Invitrogen]. Real-time PCR was performed by using an ABI 7300 real-time PCR system (Applied Biosystems) and TaqMan 2×PCR Universal Master Mix (for detection of virus genomic and antigenomic RNA; Applied Biosystems) or SYBR Premix Ex Taq II (for detection of human GAPDH mRNA; TaKaRa Bio). PCR conditions were as follows: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min (TaqMan assay) or 95°C for 10 s and 40 cycles of 95°C for 5 s and 60°C for 31 s (SYBR green assay). To detect virus genomic and antigenomic RNAs, we used primers and a TaqMan probe set that corresponded to the trailer sequence of rabies virus genomic RNA, which is conserved between Ni and Ni-CE strains. Sequences of the primers and a TaqMan probe are shown in Table 1.
TABLE 1.
Sequences of the primers and TaqMan probe
| Analysis | Primer or probe | Sequence (5′→3′) |
|---|---|---|
| RT | Rabies RT for genome | CTGCTTGTAAACCAGGCATTCCCGGATGTCTG |
| Rabies RT for anti-genome | AAACAATCAAACAGCCAGAGGTCCAGATTC | |
| SYBR green assay | Human GAPDH F | CCTCCTGTTCGACAGTCAGC |
| Human GAPDH R | CGCCCAATACGACCAAATC | |
| TaqMan assay | Rabies TaqMan probe | TGATGTGTCTCGAAAA |
| Rabies genome F | GTCTGCACATGCTGAGACTCTTG | |
| Rabies genome R | ACAGCCAGAGGTCCAGATTCA |
For validation of microarray data, total RNA was reverse transcribed into cDNAs using SuperScript III reverse transcriptase and random hexamer (TaKaRa Bio). Primer and probe sets for relative quantification of human genes were selected from the product list of TaqMan gene expression assays (Applied Biosystems). A TaqMan assay was performed as described above.
Data are expressed as number of copies of specific mRNA per copy of human GAPDH mRNA. All assays were carried out in triplicate and the results are expressed as means ± the standard deviation (SD).
Plasmids.
Using conventional cloning techniques, we subcloned a PCR-amplified cDNA fragment of the full-length N gene from Ni or Ni-CE strain into a polymerase II-based expression plasmid, pCAGGS/MCS (kindly provided by Y. Kawaoka) and named the respective resulting plasmids pCAGGS-NiN and -CEN. Similarly, we constructed pCAGGS-NiP and -CEP plasmids expressing Ni and Ni-CE P protein, respectively. Details of the construction of these plasmids and sequences of primers are available from the authors on request. 4×IRF-3-Luc (kindly provided by S. Ludwig) (11) contains four copies of the IRF-3-binding positive regulatory domain (PRD) I/III motif of the IFN-β promoter upstream of the luciferase reporter gene. pNF-κB-Luc (Stratagene) contains five copies of the NF-κB-binding motif of the IFN-β promoter upstream of the luciferase reporter gene. pRL-TK (Promega), used as an internal control for the reporter assay, contains the Renilla luciferase gene downstream of the herpes simplex virus thymidine kinase promoter that is activated in mammalian cells. pEGFP-C1-hIRF-3 (kindly provided by C. F. Basler) (5) express the human IRF-3 protein fused to green fluorescent protein (GFP-IRF-3). Expression plasmids for wild-type RIG-I (pEF-Flag-RIG-I), constitutively active mutant (pEF-Flag-RIG-IN), dominant-negative mutant (pEF-Flag-RIG-IC), wild-type IPS-1 (pEF-Flag-IPS-1), and empty plasmid [pEF-BOS(+)] were kindly provided by T. Fujita (24, 43).
Transfection and reporter assay.
Transfection was performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. SYM-I cells were transfected with 1 μg of viral N or P protein-expressing plasmids or pEF-Flag-RIG-I, 0.25 μg of 4×IRF-3-Luc or pNF-κB-Luc, and 0.04 μg of pRL-TK. At 24 hpi, cells were infected with Ni, Ni-CE, or CE(NiN) strains (MOI of 2) or NDV (MOI of 1). In another series of experiments, SYM-I cells were inoculated, in suspension, with Ni, Ni-CE, or CE(NiN) strain at an MOI of 2 and seeded in a 24-well tissue culture plate at 2 × 105 cells per well. At 24 hpi, cells were transfected with 0.5, 1, or 2 μg of pEF-Flag-RIG-IC or 1 μg of pEF-Flag-RIG-IN in addition to 0.25 μg of 4×IRF-3-Luc and 0.04 μg of pRL-TK.
At the completion of the experiments, cells were lysed, and the activities of firefly and Renilla luciferases were determined by a dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions. The data represent firefly luciferase activity normalized to Renilla luciferase activity. All assays were carried out in triplicate, and the results expressed as means ± the SD.
IRF-3 nuclear translocation assay.
SYM-I cells were inoculated, in suspension, with Ni, Ni-CE, or CE(NiN) strain at an MOI of 2 and seeded in a 24-well tissue culture plate at 2 × 105 cells per well. At 24 hpi, cells were transfected with 1 μg of pEGFP-C1-hIRF-3 using Lipofectamine 2000. At 24 h posttransfection, cells were fixed in 4% paraformaldehyde for 60 min and 100% methanol for 1 min. Then, infected cells were stained with anti-N protein mouse monoclonal antibody 13-27 and then TRITC-goat anti-mouse IgG (H+L) conjugate (Zymed). The localization of GFP-IRF-3 was examined with a Biozero fluorescence microscope (BZ-8000 series; Keyence). The percentage of virus-infected cells with nuclear GFP-IRF-3 localization was then determined by counting 100 infected cells. The results are expressed as means ± the SD of three independent wells.
Immunofluorescence staining.
Confluent SYM-I cells were grown in a 24-well plate and inoculated with Ni, Ni-CE, or CE(NiN) strain at an MOI of 2. At 24 hpi, cells were fixed with 4% paraformaldehyde for 60 min and 100% methanol for 1 min. The fixed cells were double stained by using anti-N protein mouse monoclonal antibody 13-27 and anti-human IRF-3 rabbit polyclonal antibody FL-425 (Santa Cruz) as primary antibodies and using TRITC (tetramethyl rhodamine isothiocyanate)-goat anti-mouse IgG (H+L) Conjugate and fluorescein isothiocyanate (FITC)-goat anti-rabbit IgG (Cappel) as secondary antibodies. Fluorescence was visualized by using a Biozero fluorescence microscope.
Western blotting.
Cells were lysed in lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40) supplemented with 0.02 mM p-amidinophenylmethanesulfonyl fluoride at 24 hpi, and the samples were incubated on ice for 15 min. After centrifugation (15,000 × g, 10 min, 4°C), the soluble fractions were separated electrophoretically on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked with phosphate-buffered saline (PBS) containing 0.1% Tween 20 and 5% nonfat dry milk. The following antibodies were used to probe the blots: anti-N protein mouse monoclonal antibody 13-27, anti-P protein rabbit polyclonal antibody (kindly provided by A. Kawai), anti-Flag rabbit polyclonal antibody (Sigma), and anti-α-tubulin monoclonal antibody (Sigma). Antibody signals were detected by chemiluminescence using horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (H+L; Seikagaku Corp.) or HRP-conjugated anti-mouse IgG (Fab2; Cappel) and a Western Lightning Plus ECL kit (Perkin-Elmer). Chemiluminescent signals were detected and visualized by using a LAS-1000 Lumino image analyzer (Fuji Film). Densitometry analysis was carried out by using ImageJ software. Briefly, the intensity of images of scanned Western blots was determined, and the ratio of each band to its tubulin control was calculated.
IRF-3 dimerization analysis.
SYM-I cells grown in a six-well tissue culture plate were inoculated with Ni, Ni-CE, or CE(NiN) strain at an MOI of 10. Cells were lysed in the lysis buffer described above, which was supplemented with Complete mini-protease inhibitor cocktail (Roche) at 24 hpi, and the samples were incubated on ice for 15 min. After centrifugation (15,000 × g, 10 min, 4°C), the soluble fractions were separated electrophoretically on a 7.5% Ready Gels J (Bio-Rad), with 1% deoxycholate in the cathode buffer (25 mM Tris-HCl [pH 8.4], 192 mM glycine). IRF-3 was detected by using Western blotting with polyclonal anti-IRF-3 (Santa Cruz) and HRP-conjugated anti-rabbit IgG (H+L; Seikagaku Corp.).
Coimmunoprecipitation analysis.
SYM-I cells in a six-well plate were cotransfected with 4 μg of pCAGGS-CEP and 4 μg of pCAGGS-NiN or pCAGGS-CEN. After the cells were washed with PBS at 48 h posttransfection, cell extracts were prepared by lysing cells on ice for 15 min in TN buffer (50 mM Tris-HCl [pH 8.4], 150 mM NaCl) containing 1% NP-40 and protease inhibitor (Complete mini; Roche). Lysates were centrifuged at 15,000 × g for 10 min at 4°C to remove large debris. Protein A/G Plus-Agarose (Santa Cruz) was incubated with anti-N protein mouse monoclonal antibody 13-27 or mouse normal IgG (Sigma) for 2 h at room temperature and then washed three times with TN buffer containing 1% NP-40 and protease inhibitor. Lysates were incubated with agarose beads overnight with rotation at 4°C. The agarose beads were washed five times with TN buffer containing 1% NP-40 and protease inhibitor and boiled with SDS sample buffer for 5 min. The supernatant of the agarose was subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting.
Statistical analysis.
A Student t test was used to determine statistical significance, and P values of <0.01 were considered statistically significant.
RESULTS
Genome replication and viral growth of Ni, Ni-CE, and CE(NiN) strains in SYM-I cells.
In order to obtain insights into the mechanism by which the N gene determines viral pathogenicity, we tried to comprehensively compare the gene expressions of host cells infected with Ni, Ni-CE, and CE(NiN) strains using a DNA microarray. Human neuroblastoma SYM-I cells are known to be susceptible to rabies virus and to produce IFN-β in response to viral infection (15). Therefore, this cell line is suitable for examining the effects of rabies virus infection on the expression of host genes, especially genes related to host innate immunity.
First, we measured levels of viral genomic and antigenomic RNAs in SYM-I cells infected with Ni, Ni-CE, and CE(NiN) strains at 6, 12, and 24 hpi using quantitative real-time RT-PCR (Fig. 1B). The amount of genomic RNA of each strain increased markedly between 12 and 24 hpi (Fig. 1B top), indicating genome replication in this cell line. We found that genome replication of Ni strain was less efficient than that of Ni-CE and CE(NiN) strains, probably due to the fact that Ni strain has been maintained by rabbit brain passages (17) and is not well adapted to cultured cells. In contrast, there was no significant difference in the amount of genomic RNA between Ni-CE- and CE(NiN)-infected cells at 24 hpi. Similar kinetics was observed in antigenomic RNA in the cells infected with each strain (Fig. 1B, bottom).
Next, we examined growth of Ni, Ni-CE, and CE(NiN) strains in SYM-I cells at 24 hpi. Consistent with the viral RNA replication, virus titers of Ni-CE and CE(NiN) strains in the culture media were almost identical (9.7 × 103 and 1.6 × 104 focus-forming units [FFU]/ml, respectively), whereas the titer of Ni strain (4.4 × 103 FFU/ml) was ∼2-fold lower than that of Ni-CE and CE(NiN) strains. Hence, we chose 24 hpi as the condition for DNA microarray analysis, in order to minimize the influence of replication efficiencies of Ni-CE and CE(NiN) strains.
DNA microarray analysis of SYM-I cells infected with Ni, Ni-CE, and CE(NiN) strains.
We successfully collected normalized data for 40,613 human genes by the DNA microarray analysis. We considered a gene to be differently expressed if the expression level was 3-fold higher or lower than the level in mock-infected cells. The total number of genes affected by Ni-CE infection (878 genes) was much larger than that affected by Ni infection (317 genes) (Table 2). This was mainly due to the difference between numbers of upregulated genes in the Ni- and Ni-CE-infected cells (241 and 765 genes, respectively). On the other hand, the number of upregulated genes in CE(NiN)-infected cells (628 genes) was very similar to that in Ni-CE-infected cells.
TABLE 2.
Number and percentage of host genes affected by infection of each virus
| Strain | No. (%) of host genes affecteda |
||
|---|---|---|---|
| Upregulation | Downregulation | Total | |
| Ni | 241 (0.59) | 76 (0.19) | 317 (0.77) |
| Ni-CE | 765 (1.86) | 113 (0.27) | 878 (2.14) |
| CE(NiN) | 628 (1.53) | 130 (0.32) | 758 (1.85) |
A total of 41,063 genes were analyzed. A gene was considered differentially expressed if the expression level was 3-fold higher or lower than the level in mock-infected cells.
To focus on the genes related to host immunity, we carried out cluster analysis using a bioset that contained a selection of genes involved in “host-pathogen interaction” (GO accession number 0030383, 1,722 genes) (Fig. 2A). The gene expression pattern of Ni-CE-infected cells was more similar to that of CE(NiN)-infected cells than to that of Ni-infected cells: changes in gene expression in Ni-CE- and CE(NiN)-infected cells were more drastic than those in Ni-infected cells. Similar results were obtained by using a bioset of “defense immunity protein activity” (GO accession number 0003793, 1,011 genes) (data not shown).
FIG. 2.

Comparison of the gene expressions of SYM-I cells infected with Ni, Ni-CE, and CE(NiN) strains using a DNA microarray. SYM-I cells were infected with Ni, Ni-CE, and CE(NiN) strains at an MOI of 2. After 24 h, the total cellular RNA was extracted and used for DNA microarray analysis. The data were normalized by GeneSpring GX software. (A) Cluster analysis of genes of SYM-I cells infected with each virus. The expression pattern of genes involved in “host-pathogen interaction” is represented as a hierarchical clustering, using Cluster and Java TreeView. Genes shown in red are upregulated, and those shown in green are downregulated relative to mock-infected cells. (B) Expression levels of 10 host immunity-related genes, most of which were differentially expressed in Ni-CE- and CE(NiN)-infected cells. Each bar represents the fold change in expression compared to the expression level of each gene in mock-infected cells.
Although overall gene expression patterns of Ni-CE- and CE(NiN)-infected cells were very similar as mentioned above, the expression levels of a part of the host genes were clearly different in Ni-CE- and CE(NiN)-infected cells. For example, expression level of the IFN-β gene in Ni-CE-infected cells was 2-fold higher than the level in CE(NiN)-infected cells (Fig. 2B). Similar results were obtained for the gene expression levels of type III IFN (IFN-λ1, IFN-λ2, and IFN-λ3); ISG54, which is known to be IFN-inducible (29); and chemokines (CXCL9, CXCL10, and CXCL11; CCL5; and IL-8) (Fig. 2B). Since Ni-CE and CE(NiN) strains genetically differ only in the N gene, we considered that the different gene expressions of Ni-CE- and CE(NiN)-infected cells were due to functional difference in the N gene. Notably, expression levels of these host genes in Ni-infected cells were much lower than those in Ni-CE- and CE(NiN)-infected cells (Fig. 2B).
Validation of microarray data by using quantitative real-time RT-PCR.
To confirm the different gene expression patterns of Ni-, Ni-CE-, and CE(NiN)-infected cells that were revealed by DNA microarray analysis, we quantified IFN-β, IFN-λ1, CXCL10, and CCL5 mRNAs in these infected cells by using quantitative real-time RT-PCR (Fig. 3). Corresponding to the DNA microarray data, expression level of the IFN-β gene in Ni-CE-infected cells was 40- and 4-fold higher than the levels in Ni- and CE(NiN)-infected cells, respectively (Fig. 3A). Similarly, it was shown that Ni-CE infection induced the expression of IFN-λ1, CXCL10, and CCL5 genes more efficiently than did Ni and CE(NiN) infections (Fig. 3B, C, and D, respectively). These results clearly showed that the Ni N gene functions to suppress expression of innate immunity and inflammatory genes in infected host cells.
FIG. 3.

Validation by real-time RT-PCR of DNA microarray results for IFN-β (A), IFN-λ1 (B), CXCL10 (C), and CCL5 (D). The assay was performed with the same total RNA used in the DNA microarray experiment. Expression levels of genes were normalized to mRNA levels of GAPDH. Each bar represents the mean (± the SD) of three independent replicates. *, Significant difference (P < 0.01); ND, no detection.
Identification of the signaling pathway involved in the suppressed expressions of IFN and chemokine genes by Ni N gene.
Expressions of IFN-β, IFN-λ, and chemokine genes have been shown to be regulated by the transcription factors NF-κB and IRF-3, which are activated by the respective upstream signaling pathway (13, 14, 23, 29, 30, 40). In order to identify the signaling pathway that is involved in the suppressed expressions of IFN-β gene in Ni- and CE(NiN)-infected cells, we measured NF-κB- or IRF-3-dependent IFN-β promoter activities in Ni-, Ni-CE-, and CE(NiN)-infected cells by luciferase-based reporter assays with reporter plasmids having the NF-κB- or IRF-3-binding site (PRD II or PRD I/III, respectively) of the IFN-β promoter (Fig. 4). Although NF-κB-dependent IFN-β promoter activity in Ni-infected cells was significantly lower than that in Ni-CE-infected cells (P < 0.01), there was no difference between the activities in Ni-CE- and CE(NiN)-infected cells (Fig. 4A). In contrast, IRF3-dependent IFN-β promoter activities in Ni- and CE(NiN)-infected cells (4- and 38-fold inductions, respectively) were significantly lower than that in Ni-CE-infected cells (126-fold induction) (P < 0.01) (Fig. 4B). These results indicated that the IRF-3 pathway, rather than the NF-κB pathway, is involved in suppressed expression of the IFN-β gene in Ni- and CE(NiN)-infected cells.
FIG. 4.
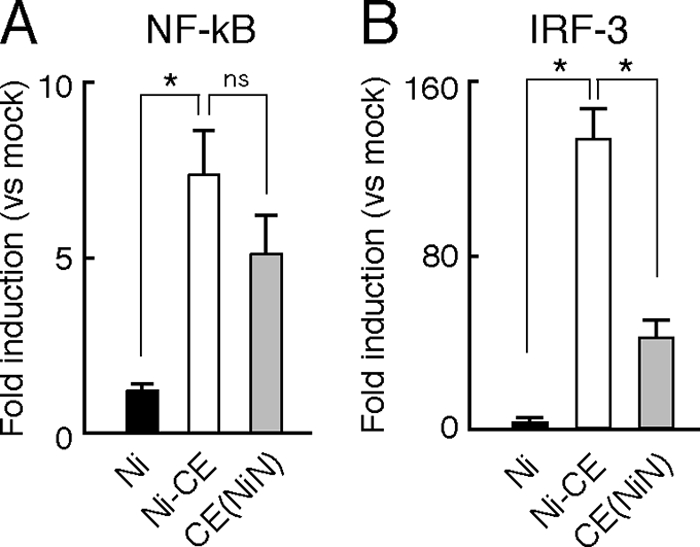
Infection of CE(NiN) strain inhibits activation of IRF-3-dependent but not NF-κB-dependent IFN-β promoter. SYM-I cells were cotransfected with pRL-TK and pNF-κB-Luc (NF-κB-responsive reporter plasmid) (A) or 4×IRF-3-Luc (IRF-3-responsive reporter plasmid) (B). After 24 h, the cells were mock infected or infected with each strain at an MOI of 2. The luciferase activities were measured 24 h after transfection. The data represent firefly luciferase activity normalized to Renilla luciferase activity and are presented as means (± the SD) of three independent replicates. *, Significant difference (P < 0.01); ns, no significant difference.
Subcellular localization and dimerization of IRF-3 in SYM-I cells infected with each virus.
In order to check subcellular localization of IRF-3 in infected cells, we transfected pEGFP-C1-hIRF-3 expressing GFP-IRF-3 into Ni-, Ni-CE-, and CE(NiN)-infected cells (Fig. 5A). We also determined the percentage of infected cells with nuclear GFP-IRF-3 in GFP-positive infected cells (Fig. 5B). In mock-infected cells, GFP-IRF-3 was localized in the cytoplasm, whereas the signals in Ni-CE-infected cells were observed mainly in the nucleus. It was shown that 79% of GFP-positive Ni-CE-infected cells had the signal in the nucleus. In contrast, in Ni- and CE(NiN)-infected cells, GFP-IRF-3 was localized mainly in the cytoplasm: only 22% of GFP-positive Ni-infected cells and 33% of CE(NiN)-infected cells had signals in the nucleus. Similar results were obtained by immunostaining of endogenous IRF-3 in Ni-, Ni-CE-, and CE(NiN)-infected cells (Fig. 5C). These results indicated that Ni and CE(NiN) infections suppress translocation of IRF-3 to the nucleus or the upstream signaling pathway. We next examined the IRF-3 homodimerization in Ni-, Ni-CE-, and CE(NiN)-infected cells by native-PAGE (Fig. 5D). In contrast to Ni-CE-infected cells, in which a prominent band of IRF-3 dimers was detectable, dimerization of IRF-3 was suppressed in Ni- and CE(NiN)-infected cells. Taken together, these results indicated that Ni and CE(NiN) infection suppress the dimerization of IRF-3 and subsequent translocation of IRF-3 to the nucleus.
FIG. 5.

Ni and CE(NiN) infections prevent nuclear translocation and dimerization of IRF-3. (A) SYM-I cells were inoculated, in suspension, with each virus strain at an MOI of 2 FFU/cells and seeded. After 24 h, cells were transfected with the GFP-IRF-3 expression plasmid (green). At 24 h posttransfection, the cells were fixed and stained with an anti-N monoclonal antibody (red). (B) Assessment of the rate of GFP-IRF-3 nuclear translocation in GFP-IRF-3-expressing and virus-infected calls. Each value is the average (± the SD) of three independent experiments in which 100 cells were counted. *, Significant difference (P < 0.01). (C) Subcellular localization of endogenous IRF-3 in SYM-I cells infected with each strain. SYM-I cells were mock infected or infected with Ni, Ni-CE, or CE(NiN) strains at an MOI of 2. After 24 h, cells were fixed and examined by double immunofluorescence staining using anti-IRF-3 polyclonal antibody (green) and anti-N monoclonal antibody (red). (D) Extracts from SYM-I cells infected with Ni, Ni-CE, and CE(NiN) strains for 24 h were analyzed by native gel electrophoresis, followed by Western blotting, to detect IRF-3. N protein and tubulin of same samples were detected by SDS-PAGE, followed by Western blotting.
Ni N protein functions to evade activation of the IRF-3 pathway in the presence of other viral components.
To determine whether single expression of Ni N protein is sufficient to inhibit the IRF-3 pathway, we checked IRF-3-dependent IFN-β promoter activities in Ni N protein- or Ni-CE N protein-expressing SYM-I cells after inoculation of NDV, which is known as a type I IFN inducer (18). Since single expression of rabies virus P protein is known to inhibit the IRF-3 pathway (6), we also expressed Ni P protein as a positive control. In contrast to expression of Ni P protein that inhibited IRF-3-dependent IFN-β promoter activity in NDV-infected cells, neither single expression of Ni nor Ni-CE N protein inhibited the promoter activity (Fig. 6A). There was no significant difference between the activities of Ni N protein- and Ni-CE N protein-expressing cells. Similar results were obtained by using transfection of a double-stranded RNA homolog, poly(I:C), instead of NDV infection, as an IFN inducer (data not shown). These results indicated that single expression of Ni N protein does not inhibit the IRF-3 pathway and strongly suggested that other rabies viral components are required to evade activation of the IRF-3 pathway by Ni N protein.
FIG. 6.
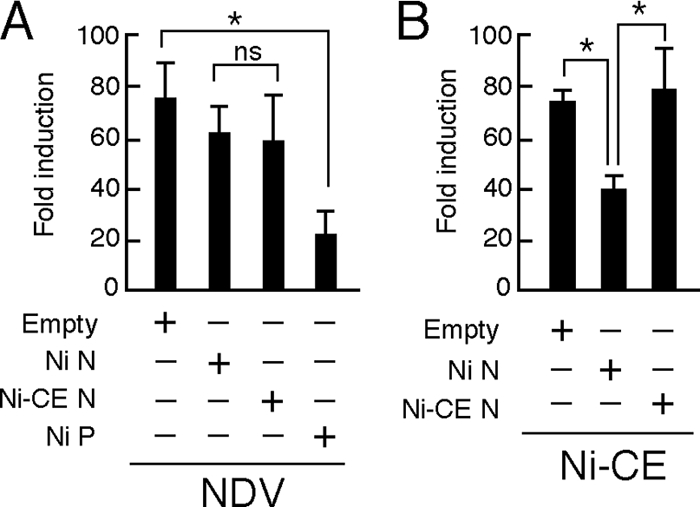
Effects of transient expression of Ni or Ni-CE N protein on IRF-3-dependent promoter activities in SYM-I cells infected with NDV or Ni-CE strain. SYM-I cells were cotransfected with pRL-TK, 4×IRF-3-Luc, and 1 μg of each plasmid driving the expression of the indicated viral protein or empty vector. At 24 h posttransfection, the cells were infected with NDV at an MOI of 1 and incubated for 12 h (A) or infected with Ni-CE strain at an MOI of 2 and incubated for 24 h (B). Then, the cells were lysed, and the luciferase activities were measured. The data represent firefly luciferase activity normalized to Renilla luciferase activity and are presented as means (± the SD) of three independent replicates. *, Significant difference (P < 0.01); ns, no significant difference.
To determine whether other rabies viral components are involved in the evasion of activation of the IRF-3 pathway by the N protein, we inoculated Ni-CE strain as an IFN inducer into Ni N protein- or Ni-CE N protein-expressing SYM-I cells and then checked the IRF-3-dependent IFN-β promoter activities (Fig. 6B). The promoter activity induced by Ni-CE infection was significantly lower in Ni N protein-expressing cells than the activity in the empty vector-transfected cells (P < 0.01). In contrast, overexpression of Ni-CE N protein did not affect the promoter activity: the activity in Ni-CE N protein-expressing cells was significantly higher than the activity in Ni N protein-expressing cells (P < 0.01). These results indicated that Ni N protein, but not Ni-CE N protein, functions to evade activation of the IRF-3 pathway in the presence of other viral components.
N protein does not affect expression level and activity of viral P protein to inhibit the IRF-3 pathway.
Rabies virus N protein is known to physically interact with the P protein (12, 26), which is known to inhibit type I IFN induction by inhibiting phosphorylation of IRF-3 by TBK-1 (6). Therefore, we hypothesized that Ni and Ni-CE N proteins would differently affect the N-P interaction and, consequently, would alter the expression levels or biological property of P protein. First, we examined expression levels of P proteins in Ni-, Ni-CE-, and CE(NiN)-infected cells by using Western blotting. We found that the expression levels of both N and P proteins in Ni-infected cells were lower than the levels of the respective protein in Ni-CE- and CE(NiN)-infected cells (Fig. 7A), reflecting the lower propagation efficiency of Ni strain described above. Also, the band mobility of Ni P protein was found to be slightly faster than that of Ni-CE P protein, probably due to the conformational differences of the Ni and Ni-CE P proteins. Importantly, we did not observe a clear difference between expression levels of P protein in Ni-CE- and CE(NiN)-infected cells. Consistent with this result, coexpression of Ni or Ni-CE N protein and P protein did not affect the expression levels of P protein from both strains (Fig. 7B). These results demonstrated that Ni and Ni-CE N proteins do not differently affect the expression level of P protein. Furthermore, to compare binding abilities of Ni and Ni-CE N proteins to Ni-CE P protein, we carried out coimmunoprecipitation analysis (Fig. 7C). Lysates of SYM-I cells coexpressing Ni-CE P protein and each of Ni and Ni-CE N proteins were subjected to IP with an anti-N protein monoclonal antibody or normal IgG. Both Ni and Ni-CE N proteins were detected in the precipitates after immunoprecipitation with an anti-N antibody (Fig. 7C, middle) but not after immunoprecipitation with normal mouse IgG (Fig. 7C, bottom), indicating that the anti-N antibody specifically binds to the N proteins. Notably, both precipitates that contained Ni and Ni-CE N proteins included comparable amounts of Ni-CE P protein (Fig. 7C, middle). The data indicated that both Ni and Ni-CE N proteins bind to Ni-CE P protein with similar efficiency.
FIG. 7.

N protein does not affect expression level of and activity of viral P protein to inhibit the IRF-3 pathway. (A) SYM-I cells were infected with the Ni, Ni-CE, or CE(NiN) strain at an MOI of 2. After 24 h, the cells were lysed and N, P, and tubulin were detected by Western blotting. (B) SYM-I cells in a 24-well plate were cotransfected with 1 μg of each plasmid driving the expression of the indicated viral protein or empty vector. At 48 h posttransfection, the cells were lysed, and N, P, and tubulin were detected by Western blotting. (C) SYM-I cells in a six-well plate were cotransfected with 4 μg of pCAGGS-CEP and 4 μg of pCAGGS-NiN or pCAGGS-CEN. Cell extracts were prepared at 48 h posttransfection and directly subjected to Western blotting with anti-N antibody, anti-P antibody, or anti-tubulin antibody (top). The same cell extracts were subjected to coimmunoprecipitation analysis with anti-N antibody (middle) or normal mouse IgG (bottom). The immunoprecipitated samples were examined by Western blotting. (D) SYM-I cells were cotransfected with pRL-TK, 4×IRF-3-Luc, and 1 μg of each plasmid driving the expression of the indicated viral protein or empty vector. At 24 h posttransfection, the cells were infected with NDV at an MOI of 1 and incubated for 12 h. Then the cells were lysed and luciferase activities were measured. The data represent firefly luciferase activity normalized to Renilla luciferase activity and are presented as means (± the SD) of three independent replicates.
Next, in order to test whether Ni N protein, but not Ni-CE N protein, enhances inhibitory activity of P protein on the IRF-3 pathway, we measured IRF-3-dependent IFN-β promoter activities in NDV-infected SYM-I cells coexpressing Ni or Ni-CE N and P proteins in different combinations (Fig. 7D). Single expression of Ni and Ni-CE P proteins equally inhibited activation of the IFN-β promoter induced by NDV infection. Importantly, we found that coexpression of Ni- or Ni-CE N and P proteins in any combinations did not affect the inhibitory activity of the respective P protein on the IRF-3 pathway.
Ni N protein, but not Ni-CE N protein, functions to evade activation of RIG-I-mediated antiviral response.
Rabies virus N protein is also known to physically interact with the viral genomic RNA (2-4). It was previously reported that genomic RNA of rabies virus is recognized by RIG-I and induces type-I IFN production (16). Hence, we hypothesized that Ni N protein, but not Ni-CE N protein, functions to inhibit recognition of viral genomic RNA by RIG-I. First, to investigate the effect of infection of each virus on the RIG-I-mediated IRF-3 pathway, we transfected a wild-type RIG-I-expressing plasmid into SYM-I cells and then checked the IRF-3-dependent IFN-β promoter activities after infection with Ni, Ni-CE, and CE(NiN) strains (Fig. 8A). Overexpression of wild-type RIG-I significantly enhanced IRF-3-dependent IFN-β promoter activity in Ni-CE-infected cells. However, we found that the overexpression did not enhance IFN-β promoter activities in Ni- and CE(NiN)-infected cells. This result indicated that Ni and CE(NiN) strains, but not Ni-CE strain, evade the activation of the RIG-I-mediated IRF-3 pathway.
FIG. 8.

Ni N protein, but not Ni-CE N protein, functions to evade activation of RIG-I-mediated antiviral response. (A) SYM-I cells were cotransfected with pRL-TK, 4×IRF-3-Luc, and 1 μg of pEF-Flag-RIG-I or empty vector. At 24 h posttransfection, the cells were infected with Ni, Ni-CE, and CE(NiN) strains at an MOI of 2 and incubated for 24 h. Then, the cells were lysed and luciferase activities were measured. *, Significant difference (P < 0.01); ns, no significant difference. (B) SYM-I cells were inoculated, in suspension, with each virus strain at an MOI of 2 FFU/cells and seeded. After 24 h, cells were transfected with pRL-TK, 4×IRF-3-Luc, and 1 μg of pEF-Flag-RIG-IC. After 24 h, the cells were lysed and luciferase activities were measured. *, Significant difference (P < 0.01); ns, no significant difference. (C) SYM-I cells were inoculated, in suspension, with each virus strain at an MOI of 2 FFU/cells and seeded. After 24 h, cells were transfected with pRL-TK, 4×IRF-3-Luc, and 0.5, 1, or 2 μg of pEF-Flag-RIG-IC. After 24 h, the cells were lysed, and the luciferase activities were measured. *, Significant difference versus mock-transfected cells (P < 0.01). (D) SYM-I cells were inoculated, in suspension, with each virus strain at an MOI of 2 FFU/cells and seeded. After 24 h, cells were transfected with pRL-TK, 4×IRF-3-Luc, and 1 μg of pEF-Flag-RIG-IN. After 24 h, the cells were lysed, and the luciferase activities were measured. The data are presented as means (± the SD) of three independent replicates. ns, No significant difference.
Next, we investigated the effect of expression of a CARD-deleted mutant RIG-I (RIG-IC), which acts as a dominant-negative mutant of RIG-I (43), on IRF-3-dependent IFN-β promoter activity in Ni-, Ni-CE-, or CE(NiN)-infected cells (Fig. 8B). Expression of RIG-IC significantly reduced IRF-3-dependent IFN-β promoter activity induced by infection of Ni-CE strain. On the other hand, RIG-IC did not change the IFN-β promoter activity induced by CE(NiN) strain. Furthermore, we found that IRF-3-dependent IFN-β promoter activity of Ni-CE-infected cells, but not that of CE(NiN)-infected cells, was reduced dose dependently by expression of RIG-IC (Fig. 8C). These findings indicated that the Ni and CE(NiN) strains, but not Ni-CE strain, evade the activation of RIG-I. To further confirm where the evasion of the RIG-I-mediated IRF-3 pathway occurs, we used a carboxy terminally truncated RIG-I (RIG-IN), which is a constitutively active mutant (43). We measured IRF-3-dependent IFN-β promoter activities in RIG-IN-transfected SYM-I cells after each virus infection (Fig. 8D). Expression of RIG-IN induced IRF-3-dependent IFN-β promoter activity without virus infection as expected. Importantly, expression of RIG-IN enhanced the IFN-β promoter activity equivalently in Ni-, Ni-CE-, and CE(NiN)-infected cells. Similar results were obtained by using an expression plasmid of IPS-1, which is an adopter molecule of RIG-I, to activate the IRF-3 pathway (data not shown). These results indicated that Ni and CE(NiN) strains do not inhibit the IRF-3-dependent IFN-β promoter activity induced by activated RIG-I. Taken together, the results showed that Ni N protein, but not Ni-CE N protein, functions to inhibit activation of RIG-I.
We then hypothesized that CE(NiN) strain, but not Ni-CE strain, can evade antiviral responses ascribed to activation of RIG-I. To confirm this, we examined the ability of Ni-CE and CE(NiN) strains to replicate in RIG-I- or RIG-IN-expressing cells. RIG-I- or RIG-IN-expressing 293T cells were infected with Ni-CE- or CE(NiN) strain, and expression levels of their N proteins were compared by Western blotting, followed by quantification using a densitometer. As the expression levels of RIG-I increased, expression level of N protein of Ni-CE strain, but not that of CE(NiN) strain, was decreased significantly (Fig. 9A and B). On the other hand, the expression levels of N protein of both strains were decreased by RIG-IN in a dose-dependent fashion (Fig. 9C and D). Taken together, these results indicate that Ni N, but not Ni-CE N, functions to evade activation of RIG-I and, subsequently, the induced antiviral responses.
FIG. 9.

CE(NiN) strain efficiently replicates in cells overexpressing RIG-I, but not RIG-IN. 293T cells were transfected with 0.05, 0.1, and 1 μg of pEF-Flag-RIG-I (A) or pEF-Flag-RIG-IN (C). After 24 h, cells were infected with Ni-CE and CE(NiN) strains at an MOI of 3. At 24 hpi, the cells were lysed and N protein, tubulin, RIG-I and RIG-IN were detected by Western blotting. (B and D) Ratio of each N protein band to its tubulin control was calculated by using ImageJ. Panels B and D show the results of experiments using pEF-Flag-RIG-I (A) and pEF-Flag-RIG-IN (C), respectively. Each bar represents the mean (± the SD) of three independent replicates. *, Significant difference versus mock-transfected cells (P < 0.01).
DISCUSSION
A DNA microarray is a strong tool for comprehensive analysis of cellular gene expression and has been widely used in many research fields, including molecular biology and medicine. It has also been used to examine the effects of rabies virus infection on host gene expression (32, 41). For example, Wang et al. (41) reported that expression levels of innate immunity-related genes, including IFN-β, CXCL10, and CCL5, in the mouse brain infected with a street rabies virus (a field isolate from a silver-haired bat) are lower than the levels in the mouse brain infected with a less-virulent fixed rabies virus. This strongly suggests that evasion of innate immunity is important for high pathogenicity of rabies virus. However, the viral gene that is related to this phenomenon has not been identified yet, mainly due to the great genetic difference between street and fixed rabies viruses. In the present study, we comprehensively compared effects of infection with avirulent Ni-CE and virulent CE(NiN) strains on host gene expressions using a DNA microarray. Since these two strains are genetically identical except for the N gene, we thought that the differences between gene expressions in Ni-CE- and CE(NiN)-infected cells would give us an insight into N gene function, which is related to the different pathogenicities of the strains. We found that Ni-CE infection induces several innate immunity-related and inflammatory genes (e.g., IFN-β, CCL5, CXCL10, and CXCL11) more efficiently than does CE(NiN) infection. This finding clearly indicated that the N gene of rabies virus (virulent Ni strain) functions to evade host innate immunity and inflammation.
We previously reported that Ni and CE(NiN) strains, but not Ni-CE strain, kill adult mice after i.c. inoculation (35). We found that Ni and CE(NiN) strains grow more efficiently in the mouse brain than does Ni-CE strain: titers of Ni and CE(NiN) strains reached 108 and 104 FFU/g, respectively, whereas the titer of Ni-CE strain was less than 102 FFU/g at 3 days after i.c. inoculation with 100 FFU of each virus (K. Shimizu, unpublished data). This strongly suggests that host innate immunity is involved in the different growth rates of the strains in the mouse brain at the early stage of infection. Therefore, we considered that evasion of antiviral response by Ni N protein determines the viral pathogenicity in adult mice.
Some studies have shown that N protein of measles virus, which belongs to the family Paramyxoviridae, participates in systemic immunosuppression: measles virus N protein binds to the Fc receptor on B cells and dendritic cells and consequently induces immunosuppression by inhibiting antibody production (34) and impairing dendritic cell function (25), respectively. These studies indicated that the measles virus N protein plays an important role in evasion of host acquired immunity. Furthermore, it has been suggested that nucleoprotein of Ebola virus plays a role in the evasion of the IFN-induced antiviral response (10). On the other hand, to our knowledge, the present study is the first study showing that the Mononegavirales N protein functions to evade induction of host IFN and chemokines.
In the present study, we showed that Ni N protein, but not Ni-CE N protein, functions to evade activation of the IRF-3 pathway in the presence of other viral components (Fig. 6). We previously reported that there are only three amino acid changes between the two N proteins (Phe to Leu at position 273 [indicated as a mutation from Ni strain to Ni-CE strain], Tyr to His at position 394, and Phe to Leu at position 395) (35). These mutations may change the structure of N protein and possibly affect its interaction with other viral components. It is known that N protein physically interacts with P protein and viral genomic RNA (3). Notably, a previous study indicated that rabies virus P protein functions to block the IRF-3 pathway (6). Therefore, we hypothesized that Ni N protein increases inhibitory activity of P protein on the IRF-3 pathway or the expression level. However, our data indicated that Ni N protein did not affect this activity and the expression level of P protein of each virus (Fig. 7).
Our data indicated that N protein of Ni strain functions to evade activation of RIG-I (Fig. 8 and 9). Some studies have suggested that single-stranded RNA with a 5′-triphosphate end, such as virus genomic RNA of rabies virus, activates RIG-I (16, 31). It has also been reported that nonencapsidated genomic RNA of rabies virus is recognized by RIG-I and then induces the production of type I IFN (16). Recently, Albertini et al. (4) determined the crystal structure of rabies virus N protein and showed that the N protein is composed of two domains, an N-terminal domain (NTD; amino acid residues 32 to 233) and a C-terminal domain (CTD; residues 236 to 356 and 396 to 450). It was also shown that the NTD and CTD clamp down onto the RNA strand (viral genomic RNA homolog) and enclose it completely. Based on these findings, these authors pointed out the possibility that the closed form of the N-RNA complex protects the viral genome from recognition by cellular RIG-I or Toll-like receptors, which are known to be viral RNA sensors and to play important roles in host innate immunity (3, 4). Therefore, we hypothesized that the three amino acid changes between Ni and Ni-CE N proteins, which are all located in the CTD, would affect the N-RNA interaction and, consequently, the structure of the RNP complex. It would be interesting to determine whether or not the genomic RNA encapsidated by the Ni-CE N protein is recognized more efficiently by RIG-I than the RNA with Ni N protein.
Encapsidation of genomic RNA by N protein of rabies virus plays vital roles in regulation of viral RNA replication (3). Therefore, it is also possible that Ni N protein, but not Ni-CE N protein, limits the replication efficiency of the viral genomic RNA and consequently suppresses the activation of RIG-I. However, our data indicated that the amounts of viral genomic and antigenomic RNAs in Ni-CE-infected SYM-I cells at 6, 12, and 24 hpi were comparable to those in CE(NiN)-infected cells (Fig. 1B). Therefore, we concluded that there is no clear difference between genome replications of Ni-CE strain and CE(NiN) strain. On the other hand, it has recently been shown that RIG-I is activated by short double-stranded RNA (19) and short-hairpin RNA (22). Thus, the exact structure of RNA activating RIG-I remains controversial. We are just starting to search for an actual ligand of RIG-I produced in Ni-CE-infected cells.
Although both Ni and CE(NiN) strains cause lethal infection in adult mice after i.c. inoculation, the Ni strain is much more virulent than the CE(NiN) strain: only 10 FFU of Ni strain is sufficient to kill 100% of adult mice by i.c. inoculation, whereas 1,000 FFU of CE(NiN) strain is required to do so (35). In the present study, we showed that expression levels of many host genes, including IFN-β and chemokine genes, in Ni-infected cells are much lower than those in CE(NiN)-infected cells (Fig. 2 and 3). These low expression levels of genes are attributed to low growth rate and the replication efficiency of viral genomic and antigenomic RNAs of Ni strain (Fig. 1B). In addition, we previously reported that not only the N gene but also the P and M genes are related to the difference between pathogenicities of Ni and Ni-CE strains (35). We also showed that Ni P and M genes are involved in the low sensitivity to type I IFN (36) and low cytopathogenicity (28), respectively. Therefore, P and M genes, together with the N gene, might be important for suppression of the expression of host defense-related genes.
The present study has revealed a novel function of rabies virus N protein by showing that the protein functions to evade activation of RIG-I and inhibit activation of the IRF-3 pathway. To our knowledge, this is the first report that the Mononegavirales N protein functions to evade induction of host IFN and chemokines. Further studies are needed to completely elucidate the molecular mechanism. We believe that our findings provide basic information for understanding the pathogenicity of rabies virus and also for identifying new targets for antiviral therapies.
Acknowledgments
We thank A. Kawai (Research Institute for Production and Development, Kyoto, Japan) for providing SYM-I cells and anti-P polyclonal antibody. We also thank H. Fukushi and K. Ohya (Gifu University, Gifu, Japan) for providing the NDV B-1 strain. We are grateful to Y. Kawaoka (University of Tokyo), S. Ludwig (Westfaelische-Wilhelms University, Muenster, Germany), C. F. Basler (Mount Sinai School of Medicine, New York, NY), and T. Fujita (Kyoto University, Kyoto, Japan) for providing plasmids. We thank W. Kamitani (Osaka University, Osaka, Japan) for technical advice for the IRF-3 dimerization assay.
This study was partially supported by a grant (project code I-AD14-2009-11-01) from the National Veterinary Research and Quarantine Service, Ministry for Food, Agriculture, Forestry, and Fisheries (Korea) in 2008.
Footnotes
Published ahead of print on 3 February 2010.
REFERENCES
- 1.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124:783-801. [DOI] [PubMed] [Google Scholar]
- 2.Albertini, A. A., C. R. Clapier, A. K. Wernimont, G. Schoehn, W. Weissenhorn, and R. W. Ruigrok. 2007. Isolation and crystallization of a unique size category of recombinant rabies virus nucleoprotein-RNA rings. J. Struct. Biol. 158:129-133. [DOI] [PubMed] [Google Scholar]
- 3.Albertini, A. A., G. Schoehn, W. Weissenhorn, and R. W. Ruigrok. 2008. Structural aspects of rabies virus replication. Cell Mol. Life Sci. 65:282-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albertini, A. A., A. K. Wernimont, T. Muziol, R. B. Ravelli, C. R. Clapier, G. Schoehn, W. Weissenhorn, and R. W. Ruigrok. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360-363. [DOI] [PubMed] [Google Scholar]
- 5.Basler, C. F., A. Mikulasova, L. Martinez-Sobrido, J. Paragas, E. Muhlberger, M. Bray, H. D. Klenk, P. Palese, and A. Garcia-Sastre. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77:7945-7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brzozka, K., S. Finke, and K. K. Conzelmann. 2005. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J. Virol. 79:7673-7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brzozka, K., S. Finke, and K. K. Conzelmann. 2006. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J. Virol. 80:2675-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daffis, S., M. A. Samuel, B. C. Keller, M. Gale, Jr., and M. S. Diamond. 2007. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 3:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delhaye, S., S. Paul, G. Blakqori, M. Minet, F. Weber, P. Staeheli, and T. Michiels. 2006. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U. S. A. 103:7835-7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebihara, H., A. Takada, D. Kobasa, S. Jones, G. Neumann, S. Theriault, M. Bray, H. Feldmann, and Y. Kawaoka. 2006. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrhardt, C., C. Kardinal, W. J. Wurzer, T. Wolff, C. von Eichel-Streiber, S. Pleschka, O. Planz, and S. Ludwig. 2004. Rac1 and PAK1 are upstream of IKK-epsilon and TBK-1 in the viral activation of interferon regulatory factor-3. FEBS Lett. 567:230-238. [DOI] [PubMed] [Google Scholar]
- 12.Finke, S., and K. K. Conzelmann. 2005. Replication strategies of rabies virus. Virus Res. 111:120-131. [DOI] [PubMed] [Google Scholar]
- 13.Genin, P., M. Algarte, P. Roof, R. Lin, and J. Hiscott. 2000. Regulation of RANTES chemokine gene expression requires cooperativity between NF-κB and IFN-regulatory factor transcription factors. J. Immunol. 164:5352-5361. [DOI] [PubMed] [Google Scholar]
- 14.Honda, K., and T. Taniguchi. 2006. IRFs: master regulators of signaling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644-658. [DOI] [PubMed] [Google Scholar]
- 15.Honda, Y., A. Kawai, and S. Matsumoto. 1984. Comparative studies of rabies and Sindbis virus replication in human neuroblastoma (SYM-I) cells that can produce interferon. J. Gen. Virol. 65(Pt. 10):1645-1653. [DOI] [PubMed] [Google Scholar]
- 16.Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck, S. Akira, K. K. Conzelmann, M. Schlee, S. Endres, and G. Hartmann. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994-997. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa, Y., T. Samejima, T. Nunoya, T. Motohashi, and Y. Nomura. 1989. Biological properties of the cell culture-adapted RC-HL strain of rabies virus as a candidate strain for an inactivated vaccine. J. Jpn. Vet. Med. Assoc. 42:637-643. [Google Scholar]
- 18.Kato, H., S. Sato, M. Yoneyama, M. Yamamoto, S. Uematsu, K. Matsui, T. Tsujimura, K. Takeda, T. Fujita, O. Takeuchi, and S. Akira. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19-28. [DOI] [PubMed] [Google Scholar]
- 19.Kato, H., O. Takeuchi, E. Mikamo-Satoh, R. Hirai, T. Kawai, K. Matsushita, A. Hiiragi, T. S. Dermody, T. Fujita, and S. Akira. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katze, M. G., J. L. Fornek, R. E. Palermo, K. A. Walters, and M. J. Korth. 2008. Innate immune modulation by RNA viruses: emerging insights from functional genomics. Nat. Rev. Immunol. 8:644-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai, T., K. Takahashi, S. Sato, C. Coban, H. Kumar, H. Kato, K. J. Ishii, O. Takeuchi, and S. Akira. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981-988. [DOI] [PubMed] [Google Scholar]
- 22.Kenworthy, R., D. Lambert, F. Yang, N. Wang, Z. Chen, H. Zhu, F. Zhu, C. Liu, K. Li, and H. Tang. 2009. Short-hairpin RNAs delivered by lentiviral vector transduction trigger RIG-I-mediated IFN activation. Nucleic Acids Res. 37:6587-6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lefort, S., A. Soucy-Faulkner, N. Grandvaux, and L. Flamand. 2007. Binding of Kaposi's sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 81:10950-10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loo, Y. M., D. M. Owen, K. Li, A. K. Erickson, C. L. Johnson, P. M. Fish, D. S. Carney, T. Wang, H. Ishida, M. Yoneyama, T. Fujita, T. Saito, W. M. Lee, C. H. Hagedorn, D. T. Lau, S. A. Weinman, S. M. Lemon, and M. Gale, Jr. 2006. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 103:6001-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marie, J. C., J. Kehren, M. C. Trescol-Biemont, A. Evlashev, H. Valentin, T. Walzer, R. Tedone, B. Loveland, J. F. Nicolas, C. Rabourdin-Combe, and B. Horvat. 2001. Mechanism of measles virus-induced suppression of inflammatory immune responses. Immunity 14:69-79. [DOI] [PubMed] [Google Scholar]
- 26.Mavrakis, M., F. Iseni, C. Mazza, G. Schoehn, C. Ebel, M. Gentzel, T. Franz, and R. W. Ruigrok. 2003. Isolation and characterization of the rabies virus N degrees-P complex produced in insect cells. Virology 305:406-414. [DOI] [PubMed] [Google Scholar]
- 27.Minamoto, N., H. Tanaka, M. Hishida, H. Goto, H. Ito, S. Naruse, K. Yamamoto, M. Sugiyama, T. Kinjo, K. Mannen, and K. Mifune. 1994. Linear and conformation-dependent antigenic sites on the nucleoprotein of rabies virus. Microbiol. Immunol. 38:449-455. [DOI] [PubMed] [Google Scholar]
- 28.Mita, T., K. Shimizu, N. Ito, K. Yamada, Y. Ito, M. Sugiyama, and N. Minamoto. 2008. Amino acid at position 95 of the matrix protein is a cytopathic determinant of rabies virus. Virus Res. 137:33-39. [DOI] [PubMed] [Google Scholar]
- 29.Nakaya, T., M. Sato, N. Hata, M. Asagiri, H. Suemori, S. Noguchi, N. Tanaka, and T. Taniguchi. 2001. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem. Biophys. Res. Commun. 283:1150-1156. [DOI] [PubMed] [Google Scholar]
- 30.Onoguchi, K., M. Yoneyama, A. Takemura, S. Akira, T. Taniguchi, H. Namiki, and T. Fujita. 2007. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 282:7576-7581. [DOI] [PubMed] [Google Scholar]
- 31.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997-1001. [DOI] [PubMed] [Google Scholar]
- 32.Prehaud, C., F. Megret, M. Lafage, and M. Lafon. 2005. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 79:12893-12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Randall, R. E., and S. Goodbourn. 2008. Interferons and viruses: an interplay between induction, signaling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1-47. [DOI] [PubMed] [Google Scholar]
- 34.Ravanel, K., C. Castelle, T. Defrance, T. F. Wild, D. Charron, V. Lotteau, and C. Rabourdin-Combe. 1997. Measles virus nucleocapsid protein binds to FcγRII and inhibits human B-cell antibody production. J. Exp. Med. 186:269-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu, K., N. Ito, T. Mita, K. Yamada, J. Hosokawa-Muto, M. Sugiyama, and N. Minamoto. 2007. Involvement of nucleoprotein, phosphoprotein, and matrix protein genes of rabies virus in virulence for adult mice. Virus Res. 123:154-160. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu, K., N. Ito, M. Sugiyama, and N. Minamoto. 2006. Sensitivity of rabies virus to type I interferon is determined by the phosphoprotein gene. Microbiol. Immunol. 50:975-978. [DOI] [PubMed] [Google Scholar]
- 37.Takahasi, K., M. Yoneyama, T. Nishihori, R. Hirai, H. Kumeta, R. Narita, M. Gale, Jr., F. Inagaki, and T. Fujita. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29:428-440. [DOI] [PubMed] [Google Scholar]
- 38.Vidy, A., M. Chelbi-Alix, and D. Blondel. 2005. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J. Virol. 79:14411-14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vidy, A., J. El Bougrini, M. K. Chelbi-Alix, and D. Blondel. 2007. The nucleocytoplasmic rabies virus P protein counteracts interferon signaling by inhibiting both nuclear accumulation and DNA binding of STAT1. J. Virol. 81:4255-4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagoner, J., M. Austin, J. Green, T. Imaizumi, A. Casola, A. Brasier, K. S. Khabar, T. Wakita, M. Gale, Jr., and S. J. Polyak. 2007. Regulation of CXCL-8 (interleukin-8) induction by double-stranded RNA signaling pathways during hepatitis C virus infection. J. Virol. 81:309-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang, Z. W., L. Sarmento, Y. Wang, X. Q. Li, V. Dhingra, T. Tseggai, B. Jiang, and Z. F. Fu. 2005. Attenuated rabies virus activates, while pathogenic rabies virus evades, the host innate immune responses in the central nervous system. J. Virol. 79:12554-12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamada, K., N. Ito, M. Takayama-Ito, M. Sugiyama, and N. Minamoto. 2006. Multigenic relation to the attenuation of rabies virus. Microbiol. Immunol. 50:25-32. [DOI] [PubMed] [Google Scholar]
- 43.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730-737. [DOI] [PubMed] [Google Scholar]