

Abstract
We showed previously that loss of the integrin β8 subunit, which forms αvβ8 heterodimers, results in abnormal vascular development in the yolk sac, placenta, and brain. Animals lacking the integrin β8 (itgβ8) gene die either at midgestation, because of insufficient vascularization of the placenta and yolk sac, or shortly after birth with severe intracerebral hemorrhage. To specifically focus on the role of integrins containing the β8 subunit in the brain, and to avoid early lethalities, we used a targeted deletion strategy to delete itgβ8 only from cell types within the brain. Ablating itgβ8 from vascular endothelial cells or from migrating neurons did not result in cerebral hemorrhage. Targeted deletion of itgβ8 from the neuroepithelium, however, resulted in bilateral hemorrhage at postnatal day 0, although the phenotype was less severe than in itgβ8-null animals. Newborn mice lacking itgβ8 from the neuroepithelium had hemorrhages in the cortex, ganglionic eminence, and thalamus, as well as abnormal vascular morphogenesis, and disorganized glia. Interestingly, adult mice lacking itgβ8 from cells derived from the neuroepithelium did not show signs of hemorrhage. We propose that defective association between vascular endothelial cells and glia lacking itgβ8 is responsible for the leaky vasculature seen during development but that an unidentified compensatory mechanism repairs the vasculature after birth.
Keywords: brain, cortex, hemorrhage, integrin, neuroepithelium, vasculature
Introduction
Vascular development of the CNS depends on cells within the brain that secrete factors, such as vascular endothelial growth factor (VEGF), which promote and guide capillary growth (Ruhrberg et al., 2002; Gerhardt et al., 2003). To fully appreciate the complexity of vascular development within the CNS, however, requires a better understanding of the molecules used for endothelial cell and neuroepithelial cell communication. These complex interactions between endothelial cells of the vasculature and neuroepithelial cells are also essential for formation of the blood-brain barrier (BBB), which is important for homeostatic regulation of the brain microenvironment and is necessary for development and healthy function of the CNS (Abbott and Romero, 1996; Engelhardt, 2003).
Vascular development within the CNS is regulated by cell-matrix and cell-cell interactions. Integrins are important heterodimeric extracellular matrix (ECM) receptors that mediate cell adhesion, cell migration, and tissue organization (Calderwood, 2004). Several molecules have been identified that function during vasculogenesis and development of the BBB (Engelhardt, 2003; Park et al., 2003), but only integrins containing either the αv or β8 subunits have been shown to be required for proper capillary development within the CNS (Bader et al., 1998; McCarty et al., 2002, 2005; Zhu et al., 2002). The αv integrin subunit is the only known partner for β8, and the integrin αvβ8 has been shown to bind the latency-associated peptide of TGF-β1 and vitronectin (Moyle et al., 1991; Nishimura et al., 1994; Mu et al., 2002). Other evidence suggests αvβ8 may also bind laminin and collagen IV (Venstrom and Reichardt, 1995).
Deletion of the integrin β8 (itgβ8) gene during development results in severe cerebral hemorrhage, with death of null mice occurring during embryogenesis or shortly after birth (Zhu et al., 2002). Similarly, deletion of integrin αv (itgαv) also results in severe cerebral hemorrhage and neonatal death (Bader et al., 1998; McCarty et al., 2002). In addition to the β8 subunit, αv can associate with several other β subunits including β1, β3, β5, and β6. These subunits, however, are not likely to be essential in vascular development of the neuroepithelium, because no vascular defects were observed in itgβ3/itgβ5 double knock-out mice or itgβ6 mutants (Huang et al., 1996; McCarty et al., 2002). Furthermore, no cerebral hemorrhage has been observed in mice lacking itgβ1 in neuroepithelial cells (Graus-Porta et al., 2001). This evidence strongly suggests that αv and β8 function together as a heterodimer in the CNS during vasculogenesis.
To determine the cellular basis for the cerebral hemorrhage observed in itgβ8-null neonates, we generated a conditional itgβ8 knock-out mouse (itgβ8 flox). Using these mice, we selectively ablated itgβ8 from neuroepithelial cells, endothelial cells, and migrating neurons. Whereas deletion of itgβ8 from the neuroepithelium results in intracerebral hemorrhage, deletion from endothelial cells or from neurons does not. Tissue-specific deletion of itgβ8 from the neuroepithelium also resulted in morphologically abnormal capillaries and disorganized astroglial cells. Therefore, the presence of integrins containing the β8 subunit on neuroepithelial-derived glial cells is essential for proper regulation of vascular morphogenesis in the developing CNS.
Materials and Methods
Generation of “floxed” itgβ8 mice. Using standard procedures, the targeting construct was generated by inserting a phosphoglycerate kinase-neomycin (PGK-neo) cassette, flanked by frt sites and containing one loxP site at its 3′ terminus, into the EcoRV restriction site between exons 4 and 5 in a genomic fragment of itgβ8 containing exons 4, 5, and 6. A second loxP site was inserted into the intron between exons 3 and 4. The targeted allele was subsequently generated via homologous recombination by introducing the linearized targeting construct into undifferentiated SVJ129 mouse embryonic stem (ES) cells using standard methods. A 5′ 670 bp and a 3′ 700 bp genomic DNA fragment of itgβ8 were used as probes for Southern blot identification of ES cells containing the targeted allele. Four independently targeted ES cell clones were identified. Two clones were injected into C57BL/6J blastocysts and then placed in pseudopregnant C57BL/6J females. One gave germline transmission, which was confirmed by Southern blot. Chimeric mice carrying the floxed itgβ8 allele were crossed to C57BL/6J mice. The progeny of that cross was crossed to mice carrying the enhanced flp recombinase gene (flpE) under the control of the ubiquitous β-actin promoter (Rodriguez et al., 2000) to remove the PGK-neo cassette. These mice were then interbred to obtain itgβ8flox/flox mice. Total RNA for Northern blots was obtained from control and mutant β-actin-cre itgβ8 postnatal day 0 (P0) brains using the Qiagen (Valencia, CA) RNeasy Mini kit. A 700 bp fragment from the 3′ untranslated region of itgβ8 was used as a probe. A 500 bp fragment of the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene was used as a probe as a control for RNA load.
PCR genotyping of mice. Female itgβ8flox/flox mice were crossed to male itgβ8null/+;cre/+ mice to generate itgβ8flox/null;cre/+ mutant progeny, as well as heterozygous and wild-type littermate progeny. All progeny were genotyped by standard PCR analysis using DNA from tail tissue. Primers specific for each allele were used to identify progeny and are as follows: itgβ8 wild-type (250 bp) and floxed allele (370 bp) (5′-GAGATGCAAGAGTGTTTACC-3′) and (5′-CACTTTAGTATGCTAATGATGG-3′); itgβ8-null allele (450 bp) (5′-AGAGGCCACTTGTGTAGCGCCAAG-3′) and (5′-GGAGGCATACAGTCTAAATTGT-3′); cre (400 bp) (5′-CTGGCAATTTCGGCTATACGTAACAGGGTG-3′) and (5′-GCCTGCATTACCGGTCGATGCAAC-3′).
Mouse lines. The β-actin-flpE mice (Rodriguez et al., 2000), the nestincre mice (Tronche et al., 1999), and the nescre8 mice (Petersen et al., 2002) have been described previously. The tie2-cre mice were generously provided by R. Wang [University of California, San Francisco (UCSF), San Francisco, CA] (R. Braren and R. Wang, unpublished observations). The nex-cre mice have been described previously (Beggs et al., 2003; Brockschnieder et al., 2004) but were kindly provided before publication by K. A. Nave (Max-Planck Institute, Göttingen, Germany). The β-actin-cre mice were generously provided by G. Martin (UCSF) and have been described previously (Lewandoski et al., 1997). Mice were cared for according to animal protocols approved by the UCSF Committee on Animal Research.
Morphological and histological analysis. Whole brains, dissected from P0 mice, were photographed using a CCD camera mounted on a dissecting microscope. Brains were then submerged in 4% paraformaldehyde in PBS overnight at 4°C, followed by submersion in 30% sucrose at 4°C until saturated, and then frozen in Tissue-Tek OCT (Miles, Elkhart, IN) for cutting 20 μm sections using a cryostat. Embryos used for immunohistochemistry were decapitated, and the heads were then fixed in 4% paraformaldehyde in PBS for 1-2 h, cryoprotected in 30% sucrose, embedded in OCT, and cut on the cryostat (20 μm sections). For LacZ staining, whole embryos or P0 brains were fixed for 2 h in 0.2% glutaraldehyde at 4°C, washed in PBS containing 0.02% NP-40 for 15 min, and stained with a freshly made X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) solution according to the standard protocol. These embryos were subsequently fixed in 4% paraformaldehyde in PBS overnight, cryoprotected in 30% sucrose, embedded in OCT, and cut on the cryostat (15 μm sections). All sections were counterstained with Nuclear Fast Red according to a standard protocol. Adult mice were anesthetized with 2.5% avertin in 0.9% NaCl, using 15 μl per gram of mouse weight and perfused with 4% paraformaldehyde in PBS. This tissue was frozen directly in 30% sucrose for cutting 40 μm sections using a sliding microtome. Nissl stain was used according to standard procedures.
Immunohistochemistry. The following primary antibodies were used: GFAP polyclonal antibody (pAb) (1:250; Dako, High Wycombe, UK), platelet-endothelial cell adhesion molecule (PECAM) (CD31) monoclonal antibody (mAb) (1:150; PharMingen, San Diego, CA), RC2 mAb (1:4; Hybridoma Bank, Iowa City, IA), collagen IV pAb (1:1000; Cosmo Bio, Tokyo, Japan), Englebreth-Holm-Swarm laminin pAb (1:3000; Sigma, St. Louis, MO), α-smooth muscle actin mAb (1:500; Sigma), and β-galactosidase (1:5000; ICN Biochemicals, Costa Mesa, CA). All sections stained with the RC2 monoclonal antibody were subjected to heat-based antigen retrieval in 10 mm sodium citrate buffer, pH 6.0, using the 34700 BioWave Microwave (Ted Pella, Redding, CA). OCT embedded frozen sections were placed in 5% goat serum, 5% BSA, and 0.3% Triton X-100 for 2 h at room temperature. Sections were incubated with primary antibodies overnight at 4°C, followed by fluorescent labeling with mouse or rabbit Alexa 488 (1:250; Invitrogen, Eugene, OR) or Texas Red (1:500; Invitrogen) in blocking buffer. For the isolectin B4 staining, paraformaldehyde-fixed sections were blocked as above for primary antibodies, followed by permeabilization in PBS containing 1% Triton X-100, 1 mm CaCl2, 1 mm MgCl2, and 0.1 mm MnCl2. These sections were then incubated with biotin-conjugated isolectin B4 (20 ng/μl; L-2140; Sigma) overnight at 4°C in permeabilization solution. After five washes in PBS, sections were incubated with streptavidin-conjugated Alexa 594 (1:500; Invitrogen) in blocking buffer. Some sections were counter-stained with the nuclear marker TO-PRO-3 (1:4000; Invitrogen). All sections were analyzed using a Zeiss LSM 5 Pascal confocal microscope. Radial glia morphology near the pial surface was analyzed by first collecting a z-series of images and then, using the Pascal program, collapsing the images into one projection image.
Results
Targeting strategy for conditional inactivation of the itgβ8 gene
In a previous study, we observed that two-thirds of itgβ8-null mutant homozygotes die during midgestation because of defects in vascularization of the yolk sac and placenta. In the remaining one-third, itgβ8-null mice were born with severe cerebral hemorrhages and died shortly after birth (Zhu et al., 2002). To gain a better understanding of the cell types responsible for the cerebral hemorrhage in the itgβ8-null mutant, we generated a conditional floxed allele of itgβ8 using cre/loxP technology. The itgβ8 locus was targeted in ES cells using a construct containing loxP sites flanking exon 4 and a PGK-neo selection cassette inserted into the intron between exons 4 and 5 (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Exon 4 encodes the integrin I-like domain, which has been shown to contribute to ligand binding in other integrin heterodimers (Tuckwell and Humphries, 1997; Green et al., 1998; Xiong et al., 2001). Cre-mediated deletion of exon 4 results in a translational frameshift that generates a premature stop codon. As a result, itgβ8 mRNA is destabilized by the mRNA surveillance mechanism that degrades mRNA containing untranslated exons (Mendell et al., 2004). Thus, there is no detectable mRNA expressed in recombined cells, as assessed by Northern blot (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). This is consistent with our previous observation that disruption of exon 4, in itgβ8-null animals, leads to a lack of itgβ8 mRNA expression (Zhu et al., 2002). Therefore, we predict that there are no functional αvβ8 heterodimers expressed by mutant cells in itgβ8 conditional mutants. Homozygous itgβ8flox/flox mice were viable, fertile, and showed no obvious phenotype.
Loss of itgβ8 from neuroepithelial cells results in cerebral hemorrhage
Integrin αvβ8 is expressed in neurons and glia in the mouse CNS (Milner et al., 1997; Nishimura et al., 1998). Although itgαv and itgβ8 mRNA are both strongly expressed in neuroepithelial cells, neither has been detected on the endothelial cells of the vasculature (Pinkstaff et al., 1999; Zhu et al., 2002). Nevertheless, it remained possible that undetectable amounts of itgβ8 expression in the CNS vasculature could contribute to the hemorrhagic phenotype in mice resulting from complete deletion of itgβ8. To determine which cell types were responsible for the hemorrhage observed in itgβ8-null animals, itgβ8 was separately ablated from neuroepithelial cells, endothelial cells, and cortical neurons. Deletion of itgβ8 was accomplished by crossing female itgβ8flox/flox mice with male itgβ8null/+ mice expressing cre-recombinase in the specified cell type (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material), resulting in mutant animals hemizygous for the cre transgene and carrying one itgβ8-floxed allele and one itgβ8-null allele.
To address the hypothesis that itgβ8 expression in neuroepithelial cells is necessary for proper vascular morphogenesis in the mouse brain, we first ablated itgβ8 specifically from the neuroepithelium using a nestin-cre transgenic mouse line, which expresses cre under the control of the neural enhancer element of the nestin promoter (Tronche et al., 1999). Because only the neural enhancer element, and not the entirety of the nestin promoter-enhancer region, is used to drive cre expression, not all cells that express nestin endogenously express cre. It has been reported that neural precursor cells that give rise to both neurons and glia are recombined as early as embryonic day 10.5 (E10.5) using this nestin-cre transgene and that endothelial cells remain unrecombined (Graus-Porta et al., 2001). In contrast to a control animal (Fig. 1A), examination of whole brains from P0 mice demonstrated that excision of itgβ8 from the neuroepithelium using nestin-cre results in cerebral hemorrhage (n = 6) (Fig. 1D). All mutant animals observed had bilateral hemorrhages distributed across their cortices. Brain morphology in coronal sections was examined using both Nissl (Fig. 1) and hematoxylin and eosin staining (data not shown). We found that 100% of nestin-cre mutant brains had visible hemorrhages throughout the dorsal cortex (Fig. 1E), as well as large hemorrhages present within the thalamus (Fig. 1F) and in the ganglionic eminence near the deep mesencephalic nucleus (data not shown). This phenotype is less severe and less widespread across the cortex than that observed in the itgβ8-null animals (Fig. 1G). The reduced severity of the hemorrhagic phenotype using this nestin-cre line may be attributable to incomplete recombination of the neuroepithelium. To address this possibility, we crossed the nestin-cre line with the R26R reporter strain and collected embryos at E15, just one-half day after the onset of hemorrhage in this mutant (Fig. 1H). Recombination appeared complete throughout the cortex and ganglionic eminence (Fig. 1I), and no β-galactosidase was detected in endothelial cells (Fig. 1J-L′) or vascular smooth-muscle cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 1.
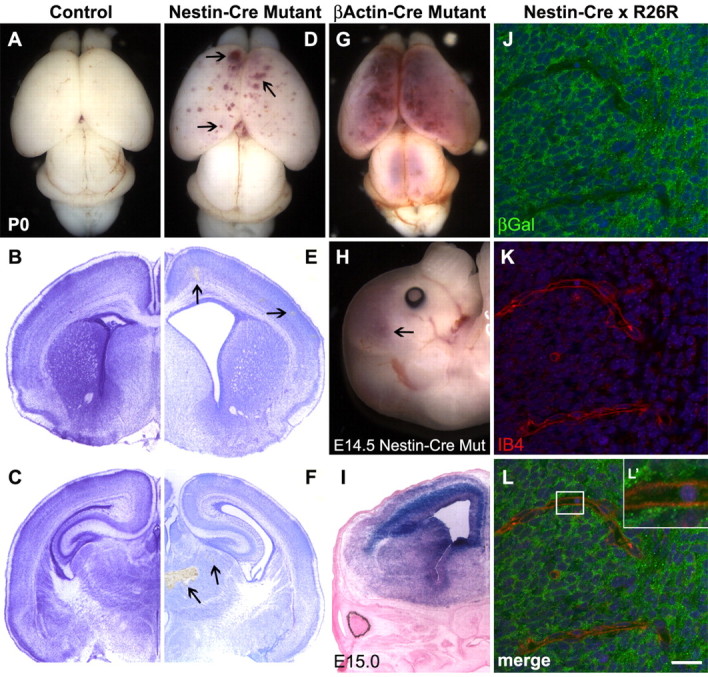
Phenotypic defects observed after conditional deletion of itgβ8 from the neuroepithelium using nestin-cre. A, Wild-type brain from P0 neonate. B, C, Nissl-stained sections of a control brain. D, P0 brain lacking itgβ8 because of conditional deletion mediated by neuroepithelial-specific nestin-cre. Note the blood produced from hemorrhages throughout the dorsal and rostral cortices (arrowheads). E, F, Nissl-stained sections of a nestin-cre mutant brain. Notice the hemorrhages (arrowheads) throughout the cortex (E) in addition to the massive hemorrhage in the thalamus (F). G, P0 mutant animal generated using the β-actin-cre line. Mutants generated with this cre line display phenotypes not obviously different from the null animal. An avas-cularized yolk sac and placenta is seen in the majority of embryos that die by E11.5, and severe hemorrhage is seen in the brain of embryos that survive to birth. H, E14.5 brain lacking itgβ8 from the neuroepithelium because of conditional deletion mediated by neuroepithelial-specific nestin-cre. Note the blood produced from hemorrhages throughout the dorsal and rostral cortices (arrowhead). I, LacZ-stained section from an E15 nestin-cre-positive animal crossed to the R26R reporter strain. Note that the neuroepithelium is almost entirely recombined. J-L′, β-galactosidase expression analysis of E15 embryo cortices (one-half day after hemorrhage was observed in nestin-cre mutants) obtained from a cross of a nestin-cre-positive animal and the R26R reporter line. Note the strong expression of β-galactosidase (green) in cells of the neuroepithelium but the lack of β-galactosidase immunolabeling of the endothelial cells labeled with isolectin B4 (red) (L′). Cell nuclei were counterstained with TO-PRO-3 (blue). βGal, β-Galactosidase; Mut, mutant; IB4, isolectin B4. Scale bar: L, 20 μm.
A second explanation for the reduction in severity of hemorrhage seen in the nestin-cre mutants may be the perdurance of a small amount of itgβ8 mRNA produced before deletion of itgβ8 by nestin-cre. Because expression of this transgene begins at E10.5 (Graus-Porta et al., 2001), we used the nescre8 transgenic mouse line, which expresses cre under control of the entire nestin promoter/enhancer beginning at E8.5 (Petersen et al., 2002). Hemorrhages in these mutants appeared by day E12.5 (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), the same time at which itgβ8-null animals develop intracerebral hemorrhage, but 2 d earlier than observed using the nestin-cre line. However, all nescre8 mutants observed at P0 had hemorrhages more similar to those obtained using nestin-cre than those seen in the itgβ8-null animal (n = 5) (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material), suggesting that factors in addition to loss of itgβ8 in the neuroepithelium contribute to the more severe hemorrhage observed in itgβ8-null animals.
To determine whether integrin β8 expressed in endothelial cells is also necessary for normal development of brain vasculature, we ablated itgβ8 specifically from endothelial cells using cre driven by the tie2 promoter (Fig. 2). This transgene drives cre expression in most endothelial cells by E7.5 (R. Braren and R. Wang, unpublished observations). By E9, tie2-driven cre expression is very strong in the head vasculature before invasion of the neuroepithelium (Fig. 2E-F). Mutants generated using the tie2-cre transgene showed no sign of hemorrhage in the cortex or thalamus, nor were other brain defects observed (n = 7) (Fig. 2G-I). These data indicate that loss of αvβ8 protein from endothelial cells does not account for the hemorrhagic phenotype observed in itgβ8 nestin-cre mutants or itgβ8-null animals.
Figure 2.
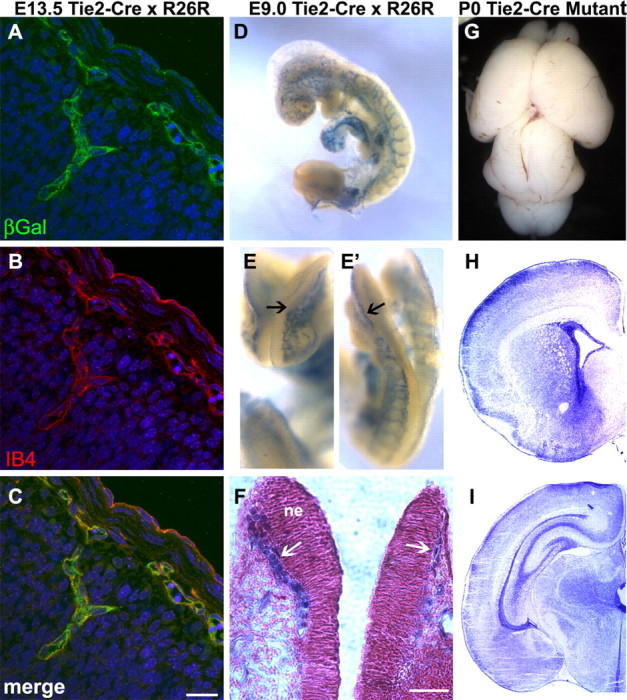
Conditional deletion of itgβ8 from vascular endothelial cells using tie2-cre does not result in cerebral hemorrhage. A-C, β-Galactosidase (βGal) expression analysis of E13.5 embryo cortices obtained from a cross between a tie2-cre-positive animal and the R26R reporter line. Note that β-galactosidase (green) expression is very strong and perfectly overlaps with the isolectinB4 (IB4) (red) (B, C) immunolabeling of endothelial cells. Cell nuclei were counterstained with TO-PRO-3 (blue). D-F, LacZ reporter analysis of E9 embryos obtained from a cross between a tie2-cre-positive animal and the R26R reporter line. Notice that cre-mediated recombination is very strong in the head vasculature before invasion of the neuroepithelium (E, E′, arrows). F, Coronal slice of embryo from E, counterstained with Nuclear Fast Red. Notice that LacZ-positive vessels (blue) have not yet invaded the neuroepithelium (ne) at 9.0 d postconception. G, P0 brain lacking itgβ8 in vascular endothelial cells because of conditional deletion mediated by tie2-cre. H, I, Nissl-stained sections of a tie2-cre mutant P0 brain. Note the absence of hemorrhage. Scale bars: C, 20 μm; F, 40 μm.
To determine whether absence of itgβ8 from cortical neurons contributed to the hemorrhage defect seen in itgβ8-null animals, nex-cre (Beggs et al., 2003; Brockschnieder et al., 2004) was used to delete itgβ8 from those cells (Fig. 3). The cre cDNA was inserted using a knock-in strategy into the nex locus, ensuring nex cell-specific expression of cre (Brockschnieder et al., 2004). This basic helix-loop-helix transcription factor drives cre expression primarily in pyramidal, postmitotic migrating neurons in the future cortical plate by E11, with robust expression throughout the forebrain by E12.5 (S. Goebbels and K. Nave, unpublished observations). When itgβ8flox/flox animals were crossed to itgβ8null/+;nex-cre/+ mice, mutant progeny showed no sign of cerebral hemorrhage or other brain defects (n = 3) (Fig. 3A-C), indicating that expression of β8 in cortical neurons is not necessary for proper vascular morphogenesis in the CNS.
Figure 3.
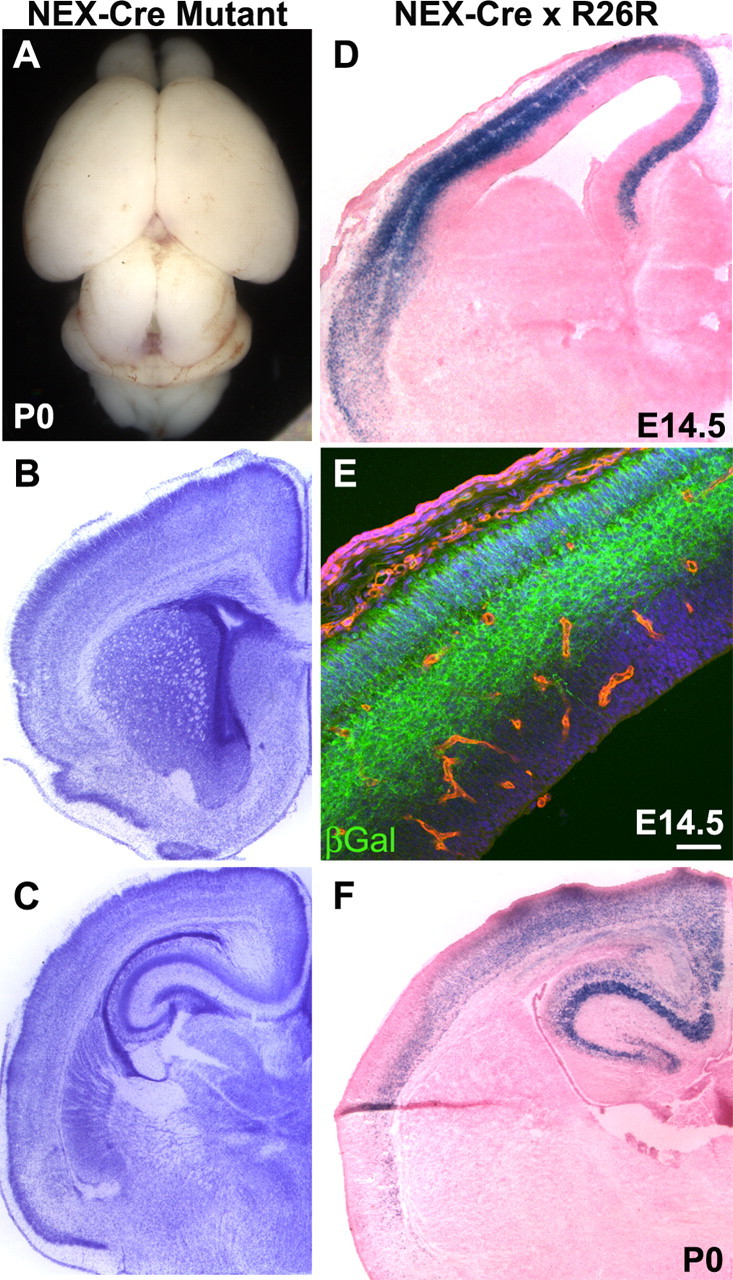
Conditional deletion of itgβ8 from postmitotic neurons using nex-cre does not result in cerebral hemorrhage. A, P0 brain lacking itgβ8 in postmitotic neurons because of conditional deletion mediated by nex-cre. B, C, Nissl-stained sections of a nex-cre mutant P0 brain. D, F, LacZ-stained sections from an E14.5 embryo cortex and P0 cortex obtained from a cross between a nex-cre-positive animal and the R26R reporter line. Cre-mediated recombination is very strong in the future cortical plate by E14.5 when hemorrhage is observed in the nestin-cre mutant and remains strong after birth. E, β-Galactosidase (βGal) expression analysis of E14.5 embryo cortex. Note that βGal (green) immunolabeling is limited to cells within the future cortical plate. Endothelial cells were immunolabeled with isolectin B4 (red) and were not recombined by this cre line. Cell nuclei were counterstained with TO-PRO-3 (blue). Scale bar: E, 50 μm.
Together, these data indicate that itgβ8 in the neuroepithelium is essential for proper vascular formation during development. Deletion of itgβ8 specifically from endothelial cells or from migrating cortical neurons does not result in hemorrhage. Additionally, deletion of itgβ8 specifically from cells in the neuroepithelium using nestin-cre or nescre8 does result in cerebral hemorrhage during development that closely resembles, but is less severe than, the hemorrhage identified in the complete itgβ8-null mice (Zhu et al., 2002).
Loss of itgβ8 in neuroepithelial cells results in endothelial cell abnormalities in the developing cortex
To further characterize the nature of the defect observed in the itgβ8 nestin-cre mutants, we used confocal microscopy to visualize cortical vasculature in coronal sections of the cortex by immunostaining with an antibody recognizing PECAM, a membrane glycoprotein expressed on the surface of endothelial cells. During development, a uniformly sized, primary capillary plexus is formed in the neural tube through angiogenesis (Yancopoulos et al., 2000). Anti-PECAM immunostaining of endothelial cells in sections of the forebrain from control mice labels this uniform plexus at P0 (Fig. 4A,B). A normal plexus also developed in mice from which itgβ8 had been deleted in endothelial cells using tie2-cre (Fig. 4K,L). However, deletion of itgβ8 from the neuroepithelium using nestin-cre resulted in vessels with large irregular endothelial cell clusters (Fig. 4F,G). These clusters are similar to those observed in itgβ8-null embryonic neuroepithelium (Zhu et al., 2002). We hypothesize that these abnormal clusters of endothelial cells may reflect aberrant endothelial cell migration or hyperproliferation during development of the brain and may permit leakage and subsequent hemorrhage of the vasculature.
Figure 4.
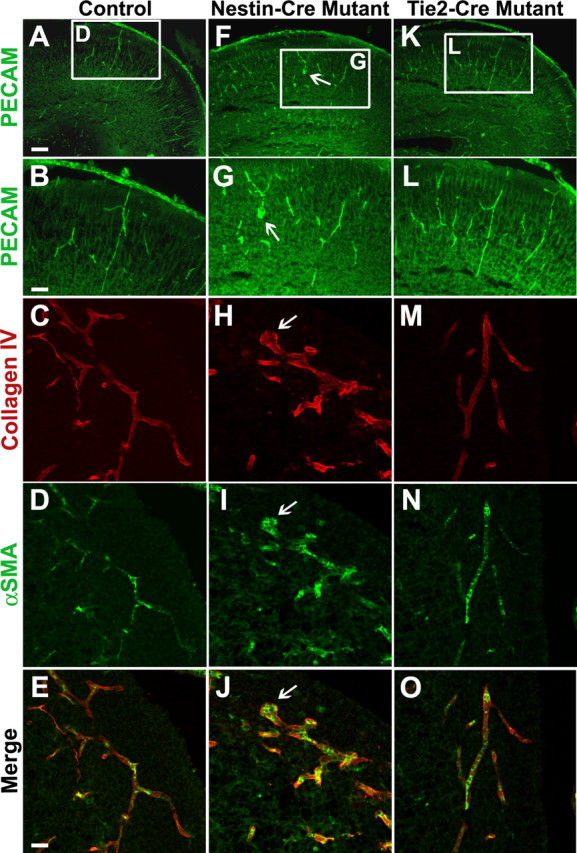
Conditional deletion of itgβ8 in the neuroepithelium leads to endothelial cell irregularities in the P0 neonatal cortex. Anti-PECAM immunolabeling of P0 coronal sections of the forebrain to visualize vascular endothelial cells is shown. A, B, Control. F, G, Nestin-cre-targeted mutant. K, L, Tie2-cre-targeted mutant. Notice the bulbous organization of endothelial cell clusters in the cortex (arrows) of the nestin-cre-targeted mutant (F, G). Anti-collagen IV immunolabeling of P0 coronal sections of the neuroepithelium to visualize the basal lamina surrounding the vasculature is shown. C, E, Control. H, J, Nestin-cre-targeted mutant. M, O, Tie2-cre-targeted mutant. Notice the intact basement membrane in the area of an endothelial cell cluster (arrowhead) in the nestin-cre-targeted mutant (H, J). Anti-α-smooth muscle actin (αSMA) immunolabeling of pericytes in P0 coronal sections of the neuroepithelium. D, E, Control. I, J, Nestin-cre mutant. N, O, Tie2-cre mutant. Note the normal recruitment of pericytes to the vasculature in both mutants. Scale bars: A, 80 μm; B, E, 40 μm.
The presence of endothelial cell clusters in nestin-cre mutant vessels suggested that improper basement membrane deposition or organization could result in the improper clustering of endothelial cells. However, anti-collagen IV immunostaining of the basement membrane of vessels in the cortex was normal in the itgβ8 nestin-cre mutants, even in areas of bulbous endothelial cell clusters (Fig. 4H,J). Anti-laminin immunostaining of the vessel basement membranes also appeared normal in the itgβ8 nestin-cre mutants compared with control animals (data not shown). This confirms our previous finding in the itgβ8-null animals that loss of itgβ8 does not cause a general defect in basement membrane assembly, although discontinuities were observed possibly because of secondary effects of hemorrhage (Zhu et al., 2002).
Pericytes also appeared to be recruited normally to endothelial cells in nestin-cre mutant brains as detected by anti-α-smooth-muscle-actin immunostaining (Fig. 4I,J). Pericytes were recruited to both stalk cells of the vessels and cells within the bulbous endothelial cell clusters. This finding is consistent with the normal recruitment of pericytes we reported previously in the itgβ8-null animals (Zhu et al., 2002).
Conditional deletion of itgβ8 from the neuroepithelium results in glial disorganization in the mouse brain
The known juxtaposition of glial cells and endothelial cells in the brain (Kacem et al., 1998; Simard et al., 2003) suggested that a primary defect in glial cells could give rise to a secondary endothelial cell phenotype in the itgβ8 nestin-cre mutant. Using confocal microscopy, we examined astroglia and radial glia in control and itgβ8 nestin-cre mutant P0 cortices. Cortical astroglial processes in wild-type P0 neonates were arrayed in a very regular pattern and were parallel to and in close association with blood vessels (Fig. 5A,A′). In a matched section from an itgβ8 nestin-cre mutant, astroglia appeared disorganized. They did not have the regular parallel organization seen in the control. Furthermore, in contrast to the wild type, blood vessels in the mutant did not run parallel to these glial processes (Fig. 5E,E′). The lack of alignment of the blood vessels and astroglial processes observed in the itgβ8 nestin-cre mutant may be explained by our previous observation that endothelial cells in the itgβ8-null mice were not well attached to the surrounding brain parenchyma, as observed by electron microscopy (Zhu et al., 2002).
Figure 5.
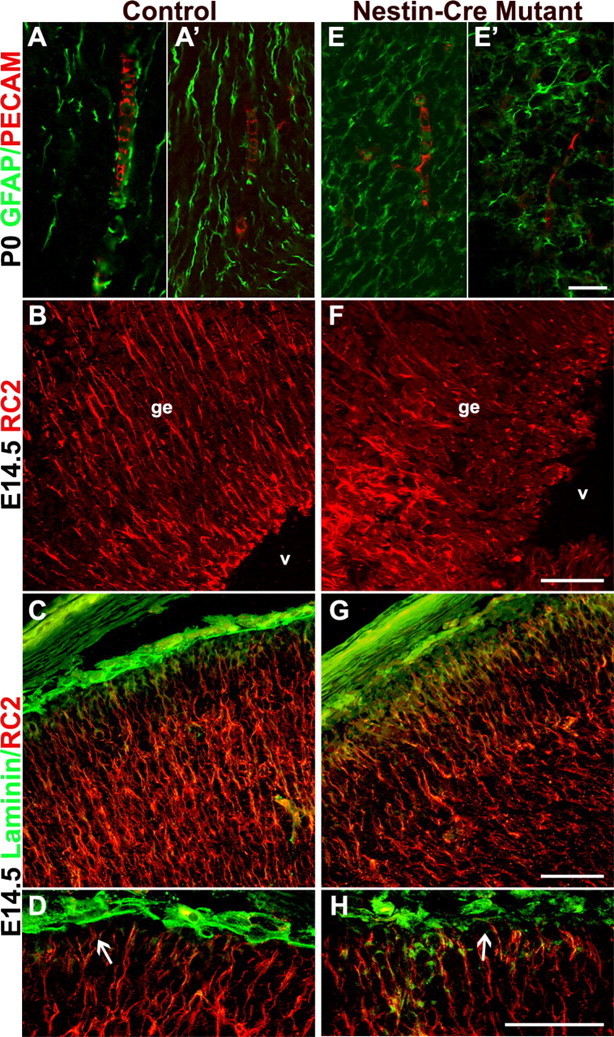
Itgβ8 nestin-cre mutants have abnormally organized cortical glia. Anti-PECAM/GFAP immunostaining of endothelial cells and glia, respectively, in P0 coronal sections of the dorsal forebrain is shown. A, A′, Control. E, E′, Nestin-cre mutant. Notice the disorganization of the astroglia and the lack of alignment between endothelial cells and the astroglial processes (E, E′). Anti-RC2 immunostaining of radial glia in E14.5 coronal sections of the ganglionic eminence is shown. B, Control. F, Nestin-cre-targeted mutant. Radial glia in F are badly disorganized, most likely because of hemorrhage within the ganglionic eminence (ge) near the lateral ventricles (v). Anti-RC2 and anti-laminin immunolabeling of E14.5 coronal sections demonstrating radial glia cell morphology in cortices of a control animal (C, D) and a nestin-cre mutant (G, H) is shown. Attachment of the radial glia to the pial surface in the nestin-cre mutant does not appear altered (H, arrow), and glial processes display normal organization. Scalebars: E′, 20 μm; F-H, 50 μm.
Radial glia are precursor cells that give rise to both neurons and glial cell types (Doetsch, 2003) and have been shown to express αv integrins (Hirsch et al., 1994). To examine whether radial glia were also disorganized in the nestin-cre mutant, we stained E14.5 coronal forebrain sections for RC2, an early radial glia marker (Misson et al., 1988). E14.5 corresponds to the earliest time that hemorrhage was observed in the nestin-cre mutants. Control animals showed the expected radial pattern of glia in the ganglionic eminence of the neuroepithelium (Fig. 5B). However, in the itgβ8 nestin-cre mutants, radial glia were very disorganized in the ganglionic eminence (Fig. 5F). Because all nestin-cre mutants had visible hemorrhage in the ganglionic eminence at this time, we looked in areas of the developing cortex where hemorrhage had not yet occurred. Radial glial processes appeared normal and pial attachment did not appear altered in the nestin-cre mutants (Fig. 5G,H). These data suggest that radial glial disorganization closely coincides with, but may be a secondary consequence of, hemorrhage of the vasculature within the CNS of these mutants.
Adult itgβ8 nestin-cre mutants lack hemorrhages
Because itgβ8-null animals die before or shortly after birth, we used our conditional itgβ8-floxed allele to look at phenotypes in adult mutants. Adult itgβ8 nestin-cre mutants were examined 6-10 weeks after birth by Nissl stain (Fig. 6). Similar to control animals, this mutant showed no sign of cortical or thalamic hemorrhage compared with controls (n = 6) (Fig. 6D,E). Additionally, no cortical lamination defect was observed (Fig. 6F), which was not surprising, because radial glial attachment to the pial surface did not appear altered in the mutants during development. This suggests that the cerebrovascular defects observed in these animals at P0 were transient and were later repaired. The repair of the vascular defects observed at P0, however, was unexpected, because no itgβ8-null animal survived more than a few days postnatally.
Figure 6.
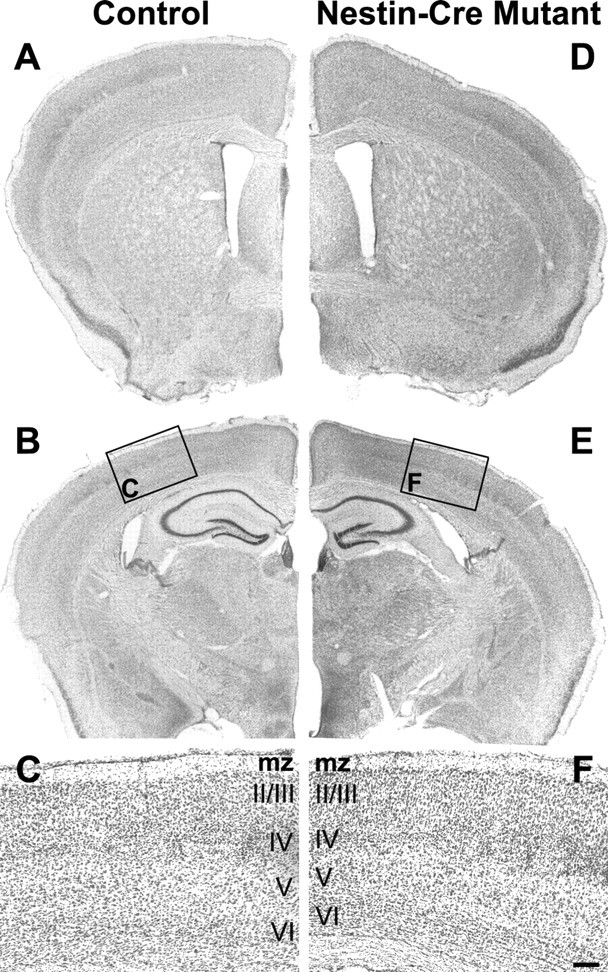
Adult itgβ8 nestin-cre mutants show no sign of brain hemorrhage. A, Six-week-old control. D, Six-week-old nestin-cre-targeted mutant coronal sections at the level of the lateral ventricles in the forebrain stained with Nissl. B, Control. E, Nestin-cre mutant coronal sections at the level of the hippocampus stained with Nissl. Note the lack of hemorrhage or other obvious defect in the cortex or thalamus. C, F, High magnification of cortical sections boxed in B and E. All cortical layers are present and in the correct order in the mutant. mz, Marginal zone. Scale bar: F, 100 μm.
Discussion
We generated a floxed allele of itgβ8 to examine the functions of integrins containing the β8 subunit in the CNS. Targeted deletion of itgβ8 from the embryonic neuroepithelium causes abnormal vascular development resulting in cerebral hemorrhage. Ablation of itgβ8 from embryonic endothelial cells or neurons does not result in abnormal development of the brain vasculature. Expression of integrins containing the β8 subunit appears to be required on non-neuronal cells within the neuroepithelium for proper blood vessel development in the CNS. Moreover, neuroepithelial-derived astroglia lacking itgβ8 are disorganized, particularly in relation to the vasculature within the forebrain. These findings complement our previous work on itgβ8-null mice and further define the cell types that must express β8 within the CNS to promote normal vascular development during embryogenesis. In addition, adult animals lacking itgβ8 in cells generated within the neuroepithelium did not have intracerebral hemorrhages, although hemorrhages occurred earlier during development. This result indicates that itgβ8 is not required postnatally for proper cerebral blood vessel function.
Comparison of itgβ8-null mutants to conditional itgβ8 nestin-cre mutants
All itgβ8-null animals that survive gestation die within hours after birth with severe cerebral hemorrhage throughout the forebrain. Although itgβ8 nestin-cre mutants also show widespread cortical hemorrhage the day of birth, the hemorrhage is not as severe as in itgβ8-null animals and they survive birth to become adults. This marked difference may be a result of several possibilities. First, the complete itgβ8-null animals may develop under abnormal environmental conditions, such as hypoxia-induced oxidative stress, because itgβ8 is required for proper vascular development of the placenta (Zhu et al., 2002). These environmental stress factor(s) may contribute indirectly by inducing cell death and brain tissue damage to increase the severity of hemorrhage in this mutant. Second, nestin-cre may have a slightly mosaic spatial expression pattern that prevents recombination of 100% of all neuroepithelial cells. The presence of a few cells that express β8 may reduce the severity of the hemorrhage observed at P0. Additionally, if itgβ8 mRNA is produced before the onset of cre expression (and thus itgβ8 deletion), enough β8 protein may be made to initiate aspects of proper vascular development. This latter possibility seems unlikely because the nescre8 mutant has a very similar phenotype to that of the neural-specific nestin-cre mutant used in the bulk of our analyses but expresses cre 2 d earlier (Petersen et al., 2002).
CNS function of integrin αvβ8
Null alleles and floxed alleles of itgβ8 and itgαv have been generated to elucidate the function of these integrin subunits in vivo. Null alleles of both itgαv and itgβ8 have two stages of lethality, with a majority of each mutant dying during embryogenesis and a minority surviving until birth with severe cerebral hemorrhage (Bader et al., 1998; McCarty et al., 2002; Zhu et al., 2002). This evidence strongly suggests that these two subunits function together as a heterodimer during CNS vascular development. Indeed, αv and β8 may function exclusively together during vascular development, because no vascular defects were observed in itgβ3/itgβ5 double knock-out mice (McCarty et al., 2002), itgβ6 knock-out mice (Huang et al., 1996), or nestin-cre-derived itgβ1-deficient mice (Graus-Porta et al., 2001).
Both floxed alleles of itgβ8 and itgαv have been crossed to nestin-cre and tie2-cre (Figs. 1, 2) (McCarty et al., 2005; present study). Neither mutant obtained from the tie2-cre cross developed cerebral hemorrhage. Similarly, both mutants derived from the nestin-cre cross developed bilateral hemorrhage similar to, but less severe than, either null mutant. Our current study further defines non-neuronal cell types within the neuroepithelium responsible for proper capillary development within the CNS (Fig. 3). Moreover, our observation that astroglia in the itgβ8 nestin-cre mutant are disorganized (Fig. 5) implies that αvβ8 expressed on glial cell processes facilitates proper CNS blood vessel development. Cre expression driven by a glial lineage-specific promoter early during development would strengthen these findings; however, this may not be possible, because radial glia differentiate to form both glial cells and neurons (Doetsch, 2003). The itgαv-floxed mice were crossed to mice expressing cre driven by the human GFAP promoter (hGFAP) in an attempt to recombine primarily CNS glia originating from GFAP-positive radial glia. The resulting mutants, however, developed only a mild cerebral hemorrhage, most likely attributable to the late onset (∼E15) of cre expression (McCarty et al., 2005). Although GFAP-positive radial glia are known to give rise to astrocytes and other glial cell types, almost all cortical projection neurons, and some striatal neurons, also originate from this radial glial lineage (Doetsch, 2003). At least some of these neurons are also likely to have lost itgαv as a result of recombination mediated by hGFAP-cre; thus, they may contribute to any hemorrhage observed in this mutant. Our observation that itgβ8 nex-cre mutants do not develop cerebral hemorrhage (Fig. 3) indicates that cortical neurons do not contribute to the cerebral hemorrhage observed in itgβ8 nestin-cre mutants or itgβ8-null animals at P0. Together, this evidence suggests that deletion of αvβ8 on glial cells within the neuroepithelium is responsible for the hemorrhage observed in the itgβ8- and itgαv-null animals.
Because neither the itgαv- or itgβ8-null animals survive long after birth, use of the floxed itgβ8 allele has enabled investigation of the postnatal functions of β8. Surprisingly, we discovered that adult itgβ8 nestin-cre mutants do not have cerebral hemorrhage (Fig. 6). Although a majority of itgαv nestin-cre mutants also survive neonatal hemorrhage to become mature adults, a few do not fully repair the hemorrhage and die within a few weeks after birth (McCarty et al., 2005). The absence of premature death in itgβ8 nestin-cre mutants could reflect differences in the back-ground strains of these two mutants. Lastly, itgβ8 nestin-cre mutant animals begin to display abnormal gait in the hind limbs 8 weeks after birth (J. M. Proctor and L. F. Reichardt, unpublished observations). Although we are currently investigating this adult phenotype in more detail, it is worth noting that adult itgαv nestin-cre mutants also display abnormal gait with a similar time of onset (McCarty et al., 2005).
Brain vascular development and integrin αvβ8
The cerebral hemorrhage observed in the conditional itgβ8 nestin-cre mutants suggests a severe defect in vascular development and function. Analysis of mutant brains revealed aberrant capillary vessel morphology and abnormal clustering of endothelial cells (Fig. 4). However, vessels maintained an intact basement membrane and normal endothelial cell-associated pericytes (Fig. 4). The defect in glial alignment and possible lack of association with endothelial cells observed in the itgβ8 nestin-cre mutants (Fig. 5) suggest that αvβ8 regulates vascular morphogenesis through its expression on neuroepithelial cells, particularly glia. These data support a model in which αvβ8 adheres to ligand(s) within the extracellular matrix or ligand(s) expressed directly in brain capillary vessels to mediate glial cell-endothelial cell contact. This contact could provide instructive cues for proper vascular morphogenesis during early development and physical support during later aspects of CNS development. None of the known ligands for αvβ8, however, are required for vascular development of the CNS. For example, one-half of TGF-β1-deficient mice have a defective yolk sac vasculature, but the remaining one-half survive birth with no sign of cerebral hemorrhage and die because of a multifocal inflammatory disorder (Shull et al., 1992; Dickson et al., 1995). Knock-out mice of ECM ligands such as vitronectin develop normally and are fertile (Zheng et al., 1995). None of the laminin isoform knock-out mice develop cerebral hemorrhage (Li et al., 2003), although laminin-α5-deficient mice have a defective placental vasculature (Miner et al., 1998). Lastly, collagen IV-deficient mice die at midgestation, but the capillary networks of the embryo, yolk sac, and placenta appear normal (Poschl et al., 2004). Together, these data suggest that the αvβ8 ligand responsible for its regulatory function in CNS vasculature development has not been identified.
Strikingly similar to itgβ8-null mice, total deletion of neuropilin-1 (npn-1) results in defective yolk sac and cerebral vessel development and early embryonic death (Kawasaki et al., 1999). Neuropilin-1 is a known receptor for the VEGF165 isoform of VEGF-A (Breier et al., 1992; Soker et al., 1998). One putative mechanism by which αvβ8 expressed on glial cells could regulate vascular development is by indirectly regulating release of secreted factors, such as VEGF, through cross talk with endothelial cell receptors such as neuropilin-1. The VEGF165 binding site on neuropilin-1 has been shown to be necessary for proper capillary development in the embryonic mouse brain (Gu et al., 2003). Additionally, npn-1 mutants display aberrant endothelial cell clusters (Gerhardt et al., 2004) similar to those observed in the itgβ8-null and nestin-cre mutants. Increased capillary permeability and endothelial cell hyperproliferation are known effects of augmented VEGF expression (Cheng et al., 1997; Sundberg et al., 2001; Gora-Kupilas and Josko, 2005). Bromodeoxyuridine labeling of endothelial cells in the itgβ8-null animals demonstrates that these cells are in fact hyperproliferative in this mutant (Zhu et al., 2002). Through possible communication with neuropilin-1, αvβ8 may regulate expression or secretion of specific isoforms of VEGF, such as VEGF165, and thereby regulate vascular morphogenesis. Other molecules such as Eph receptors and semaphorins have been shown to interact with integrins and their signaling pathways (Zou et al., 1999; Pasterkamp et al., 2003; Serini et al., 2003) and thus may also play a role in regulating vascular development of the brain. We are currently pursuing the mechanism by which αvβ8 functions in the developing mouse brain.
Our current study defines neuroepithelial cells and glia as the primary cell types in which itgβ8 expression is essential for proper development of capillary growth in the embryonic brain. It is essential now to identify the ligand(s) for αvβ8 in the CNS and to develop reagents that will facilitate our understanding of the mechanism by which αvβ8 promotes proper vascular development. Use of the itgβ8-floxed allele in elucidating the signaling pathways through which integrin αvβ8 functions should help define the regulatory mechanisms necessary to establish an organized capillary network in the developing brain.
Footnotes
This work was supported by National Institutes of Health Grant R01-NS19090 (L.F.R). J.M.P. is the recipient of a predoctoral fellowship from the National Science Foundation. L.F.R. is an investigator of the Howard Hughes Medical Institute. We thank Klaus-Armin Nave for the nex-cre mice, Zhen Huang, James Linton, and Ben Cheyette for critical reading of this manuscript, and members of the Reichardt Laboratory for helpful discussions. The RC2 antibody, developed by Miyuki Yamamoto, was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biological Sciences, The University of Iowa (Iowa City, IA).
Correspondence should be addressed to Louis F. Reichardt, Howard Hughes Medical Institute, University of California, San Francisco, Department of Physiology, 1550 4th Street, Room 284A, San Francisco, CA 94143-2611. E-mail: lfr@cgl.ucsf.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259940-09$15.00/0
References
- Abbott NJ, Romero IA (1996) Transporting therapeutics across the blood-brain barrier. Mol Med Today 2: 106-113. [DOI] [PubMed] [Google Scholar]
- Bader BL, Rayburn H, Crowley D, Hynes RO (1998) Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 95: 507-519. [DOI] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF (2003) FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 40: 501-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier G, Albrecht U, Sterrer S, Risau W (1992) Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development 114: 521-532. [DOI] [PubMed] [Google Scholar]
- Brockschnieder D, Lappe-Siefke C, Goebbels S, Boesl MR, Nave KA, Riethmacher D (2004) Cell depletion due to diphtheria toxin fragment A after Cre-mediated recombination. Mol Cell Biol 24: 7636-7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood DA (2004) Integrin activation. J Cell Sci 117: 657-666. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Nagane M, Huang HS, Cavenee WK (1997) Intracerebral tumor-associated hemorrhage caused by overexpression of the vascular endothelial growth factor isoforms VEGF121 and VEGF165 but not VEGF189. Proc Natl Acad Sci USA 94: 12081-12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ (1995) Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 121: 1845-1854. [DOI] [PubMed] [Google Scholar]
- Doetsch F (2003) The glial identity of neural stem cells. Nat Neurosci 6: 1127-1134. [DOI] [PubMed] [Google Scholar]
- Engelhardt B (2003) Development of the blood-brain barrier. Cell Tissue Res 314: 119-129. [DOI] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161: 1163-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Ruhrberg C, Abramsson A, Fujisawa H, Shima D, Betsholtz C (2004) Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev Dyn 231: 503-509. [DOI] [PubMed] [Google Scholar]
- Gora-Kupilas K, Josko J (2005) The neuroprotective function of vascular endothelial growth factor (VEGF). Folia Neuropathol 43: 31-39. [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U (2001) Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367-379. [DOI] [PubMed] [Google Scholar]
- Green LJ, Mould AP, Humphries MJ (1998) The integrin beta subunit. Int J Biochem Cell Biol 30: 179-184. [DOI] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD (2003) Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell 5: 45-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch E, Gullberg D, Balzac F, Altruda F, Silengo L, Tarone G (1994) Alpha v integrin subunit is predominantly located in nervous tissue and skeletal muscle during mouse development. Dev Dyn 201: 108-120. [DOI] [PubMed] [Google Scholar]
- Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese Jr RV, Sheppard D (1996) Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol 133: 921-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacem K, Lacombe P, Seylaz J, Bonvento G (1998) Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia 23: 1-10. [PubMed] [Google Scholar]
- Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, Fujisawa H (1999) A requirement for neuropilin-1 in embryonic vessel formation. Development 126: 4895-4902. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Meyers EN, Martin GR (1997) Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol 62: 159-168. [PubMed] [Google Scholar]
- Li S, Edgar D, Fassler R, Wadsworth W, Yurchenco PD (2003) The role of laminin in embryonic cell polarization and tissue organization. Dev Cell 4: 613-624. [DOI] [PubMed] [Google Scholar]
- McCarty JH, Monahan-Earley RA, Brown LF, Keller M, Gerhardt H, Rubin K, Shani M, Dvorak HF, Wolburg H, Bader BL, Dvorak AM, Hynes RO (2002) Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol 22: 7667-7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, Hynes RO (2005) Selective ablation of αv integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132: 165-176. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC (2004) Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36: 1073-1078. [DOI] [PubMed] [Google Scholar]
- Milner R, Frost E, Nishimura S, Delcommenne M, Streuli C, Pytela R, Ffrench-Constant C (1997) Expression of alpha vbeta3 and alpha vbeta8 integrins during oligodendrocyte precursor differentiation in the presence and absence of axons. Glia 21: 350-360. [PubMed] [Google Scholar]
- Miner JH, Cunningham J, Sanes JR (1998) Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol 143: 1713-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misson JP, Edwards MA, Yamamoto M, Caviness Jr VS (1988) Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res Dev Brain Res 44: 95-108. [DOI] [PubMed] [Google Scholar]
- Moyle M, Napier MA, McLean JW (1991) Cloning and expression of a divergent integrin subunit beta 8. J Biol Chem 266: 19650-19658. [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL (2002) The integrin alpha(v) beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol 157: 493-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura SL, Sheppard D, Pytela R (1994) Integrin alpha v beta 8. Interaction with vitronectin and functional divergence of the beta 8 cytoplasmic domain. J Biol Chem 269: 28708-28715. [PubMed] [Google Scholar]
- Nishimura SL, Boylen KP, Einheber S, Milner TA, Ramos DM, Pytela R (1998) Synaptic and glial localization of the integrin alphavbeta8 in mouse and rat brain. Brain Res 791: 271-282. [DOI] [PubMed] [Google Scholar]
- Park JA, Choi KS, Kim SY, Kim KW (2003) Coordinated interaction of the vascular and nervous systems: from molecule- to cell-based approaches. Biochem Biophys Res Commun 311: 247-253. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL (2003) Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature 424: 398-405. [DOI] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Hwang JK, Jan YN, Zhong W (2002) Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature 419: 929-934. [DOI] [PubMed] [Google Scholar]
- Pinkstaff JK, Detterich J, Lynch G, Gall C (1999) Integrin subunit gene expression is regionally differentiated in adult brain. J Neurosci 19: 1541-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U (2004) Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 131: 1619-1628. [DOI] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM (2000) High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 25: 139-140. [DOI] [PubMed] [Google Scholar]
- Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, Betsholtz C, Shima DT (2002) Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev 16: 2684-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, Tessier-Lavigne M, Taniguchi M, Puschel AW, Bussolino F (2003) Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424: 391-397. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T (1992) Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359: 693-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003) Signaling at the gliovascular interface. J Neurosci 23: 9254-9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M (1998) Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92: 735-745. [DOI] [PubMed] [Google Scholar]
- Sundberg C, Nagy JA, Brown LF, Feng D, Eckelhoefer IA, Manseau EJ, Dvorak AM, Dvorak HF (2001) Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery. Am J Pathol 158: 1145-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99-103. [DOI] [PubMed] [Google Scholar]
- Tuckwell DS, Humphries MJ (1997) A structure prediction for the ligand-binding region of the integrin beta subunit: evidence for the presence of a von Willebrand factor A domain. FEBS Lett 400: 297-303. [DOI] [PubMed] [Google Scholar]
- Venstrom K, Reichardt L (1995) Beta 8 integrins mediate interactions of chick sensory neurons with laminin-1, collagen IV, and fibronectin. Mol Biol Cell 6: 419-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA (2001) Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science 294: 339-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J (2000) Vascular-specific growth factors and blood vessel formation. Nature 407: 242-248. [DOI] [PubMed] [Google Scholar]
- Zheng X, Saunders TL, Camper SA, Samuelson LC, Ginsburg D (1995) Vitronectin is not essential for normal mammalian development and fertility. Proc Natl Acad Sci USA 92: 12426-12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF (2002) beta8 integrins are required for vascular morphogenesis in mouse embryos. Development 129: 2891-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JX, Wang B, Kalo MS, Zisch AH, Pasquale EB, Ruoslahti E (1999) An Eph receptor regulates integrin activity through R-Ras. Proc Natl Acad Sci USA 96: 13813-13818. [DOI] [PMC free article] [PubMed] [Google Scholar]