

Abstract
The hormone corticotropin (ACTH) is employed as therapy for diverse neurological disorders, but the mechanisms for its efficacy remain unknown. ACTH promotes the release of adrenal steroids (glucocorticoids), and most ACTH effects on the central nervous system (CNS) have been attributed to activation of glucocorticoid receptors. However, in several human disorders, ACTH has therapeutic actions that differ qualitatively or quantitatively from those of steroids. This study tested the hypothesis that ACTH directly influences limbic neurons via the recently characterized melanocortin receptors and focused on the effects of ACTH on the expression of corticotropin-releasing hormone (CRH), a neuropeptide involved in neuroimmune functions and in certain developmental seizures. The results demonstrated that ACTH potently reduced CRH expression in amygdala neurons. This down-regulation was not abolished by experimental elimination of steroids or by blocking their receptors and was reproduced by a centrally administered ACTH fragment that does not promote steroid release. Importantly, selective blocking of melanocortin receptors prevented ACTH-induced down-regulation of CRH expression. Taken together, these data indicate that ACTH activates central melanocortin receptors to modulate CRH gene expression in amygdala, supporting the notion that direct, steroid-independent actions of ACTH may account for some of its established clinical effects on the CNS.
The hormone corticotropin (ACTH) has been used extensively to treat neurological disorders. In some auto-immune disorders (eg, opsoclonus-myoclonus) and in certain developmental seizure disorders (eg, infantile spasms), ACTH possesses increased efficacy or a different spectrum of effects compared to adrenal steroids.1–4 However, the mechanisms for these singular actions of ACTH on the central nervous system (CNS) have remained unresolved.1,5
Native ACTH is synthesized in the pituitary and functions to stimulate the adrenal cortex to release steroids into the blood stream as part of the neuroendocrine stress response. Neurons containing ACTH have been localized to the CNS, particularly the hypothalamus, and ACTH-immunoreactive cell bodies or fibers have also been described in amygdala, cerebral cortex, brainstem, and cerebellum.6,7 In contrast to pituitary ACTH, the functions of CNS ACTH have not been well-defined. Evidence from both human and animal studies has suggested that CNS ACTH may function as a neurotransmitter/neuromodulator.1,8 Indeed, central physiological roles for ACTH, including modulation of learning and memory processes and facilitation of arousal states, have been suggested, but the mechanisms for these actions of ACTH have remained unclear.1,8 Characterization of the melanocortin receptor family, consisting of several members possessing binding affinity for ACTH,9,10 has yielded insight regarding the likely sites and mechanisms of ACTH effects. However, the downstream effects of activation of these receptors and the specific molecular changes that can lead to the clinical effects of ACTH on CNS function have not been elucidated.
Pituitary ACTH synthesis and secretion are regulated by the peptide corticotropin-releasing hormone (CRH), and both hormones participate in the neuroendocrine response to stress. Furthermore, ACTH reduces CRH gene expression in hypothalamus as part of the neuroendocrine feedback loop of the stress response.11 ACTH and CRH are found in close proximity in the amygdala, and complex interactions between the ACTH- and CRH-expressing neuronal systems have been suggested:12–14 For example, ACTH-immunoreactive (ir) fibers have been demonstrated in the amygdaloid central nucleus, a major limbic locus of CRH-expressing neurons. Could regulation of CRH expression account for some of the actions of ACTH on CNS function?
Indeed, down-regulation of CRH by ACTH provides an attractive mechanism for some of the clinical effects of the latter. CRH has been implicated in neuroimmune functions,15–17 in learning and memory,18,19 and in developmental seizures.20,21 In the clinical context, CRH modulates immune function in a complex manner,15,22 and regulation of its actions may mediate some of the efficacy of ACTH in autoimmune disorders. Importantly, ACTH is much more potent than steroids in infantile spasms.3,4 Whereas the etiology of this disorder remains entirely unclear, evidence from human23,24 and animal20,21 data supports a potential role of stress-activated proconvulsant actions of CRH on limbic neurons. Thus, the all-or-none effects of ACTH on these seizures may involve reduction of CRH availability at limbic synapses.21,25
The goal of the current studies was to examine whether ACTH directly influenced CRH gene expression in the immature rat amygdala. Because steroids also regulate CRH expression in this region, this study focused on discerning steroid-independent effects, testing the hypothesis that ACTH suppresses CRH expression in amygdaloid central nucleus (ACe) directly, via activation of central melanocortin receptors.
Materials and Methods
Materials
ACTH (Acthargel; Rhone-Poulenc Rorer, Collegeville, PA) was administered into the peritoneal cavity (i.p.) at a high dose (80 I.U./kg), because of the limited blood–brain barrier penetration of the peptide.26,27 ACTH4–10, an analog that binds melanocortin receptors but does not induce steroid secretion from the adrenal, was a gift from Dr. J. Rivier. ACTH4–10 was dissolved in 0.9% saline and infused into the cerebral ventricles (i.c.v.) in a dose of 1 μg. RU 38486 (a gift from Roussel UCLAF, Romainville, France) was dissolved in 2% ethanol/98% polyethelene-glycol and injected i.p. (100 mg/kg).28 SHU9119 (courtesy of Dr. K. Yagaloff, Roche) was dissolved in 0.9% saline and infused i.c.v. (1 nmol).29
Experimental Design
Several experiments were carried out to test the hypothesis that ACTH alters CRH gene expression in ACe independently of the effects of steroids. These experiments were performed on developing rats (postnatal days 9–11) for several reasons: 1) ACTH is used frequently and in large doses in human infants during a stage of brain development generally equivalent to that of the immature rat in the second postnatal week.30 2) Previous work from our laboratory has characterized the levels and regulation of CRH mRNA expression in ACe during this age.21,31 Experimental groups were chosen to manipulate independently levels of ACTH and of circulating steroids (glucocorticoids). These groups and the respective hormonal profiles of each are schematized in the Table.
Table.
Summary of Expected Plasma Hormone Levels
| Experimental Groups | ACTH | Corticosterone |
|---|---|---|
| Control | ↔ | ↔ |
| ACTH | ↑ | ↑ |
| Adrenalectomy (ADX) | ↑ | ↓ ↓ |
| ADX + ACTH | ↑ ↑ | ↓ ↓ |
| Sham ADX | ↔ | ↔ |
Experimental Groups
Controls. Several control groups were used so that, for each experiment, controls received vehicle or surgical procedure corresponding to that of the respective experimental groups. Controls were sacrificed concurrently with other groups at 2–4 PM to minimize potential diurnal variation in CRH mRNA levels, in endogenous plasma ACTH and steroids or in the sensitivity of CRH to regulation by ACTH or steroids. All animals were sacrificed 4 hours after ACTH administration, a time that has been found to be optimal, considering the half-life of CRH mRNA.28,31
ACTH i.p. recipients (n = 6) injected with ACTH and sacrificed 4 hours later. They were compared with i.p. vehicle controls.
ACTH i.c.v. (n = 7). ACTH4–10 was infused (1 μl) via stainless steel cannulae implanted into the lateral cerebral ventricle 24 hours prior to drug administration. This ACTH analog was chosen because it binds to melanocortin receptors but does not stimulate steroid secretion.
Adrenalectomy (n = 5). Animals underwent bilateral adrenalectomy 24 hours prior to experiments. They were also injected i.p. or infused i.c.v. with vehicle and sacrificed 4 hours later.
Adrenalectomy + ACTH i.p. (n = 7 ). Animals subjected to bilateral adrenalectomy were injected the following day with ACTH and sacrificed 4 hours later. (Controls were sham- adrenalectomized and administered vehicle i.p.)
RU 38486 (n = 5). Animals were injected i.p. with the glucocorticoid receptor antagonist RU 38486 and sacrificed 4 hours later.
RU 38486 + ACTH i.p. (n = 5). RU 38486 was injected to intact animals 35–40 minutes prior to administration of ACTH. Animals were sacrificed 4 hours after ACTH injection.
Adrenalectomy and SHU9119 (n = 5). Animals underwent bilateral adrenalectomy, resulting in elevated endogenous ACTH levels (because of elimination of endogenous steroid feedback). To block the actions of this endogenous ACTH, the melanocortin receptor-4 antagonist SHU9119 was infused i.c.v. on the following day, and animals were sacrificed 4 hours later.
For obvious technical reasons, experiments were performed in “batches.” However, each included both experimental animals and littermate controls, and tissue processing and data analysis were performed to include all experiments.
Animals and Tissue Preparation
Offspring of timed-pregnancy Sprague Dawley-derived rats (Zivic-Miller, Zelienople, PA) were used in this study. Dams were maintained in an NIH-approved facility on a 12 hour light/dark cycle (lights on at 7 AM). Cages were monitored every 12 hours for the presence of pups, and the date of birth was considered day 0; litters were culled within 24 hours of delivery to 12 pups. Rats were killed by decapitation, and brains were rapidly removed and frozen on dry ice for subsequent in situ hybridization. Trunk blood was collected at the time of sacrifice for measurement of plasma ACTH and corticosterone (the major glucocorticoid in rat) levels using commercial radioimmunoassay (RIA) kits (Inc-star, Stillwater, MN, and ICN, Irvine, CA, respectively). All experiments were carried out according to NIH guidelines for the care of experimental animals and were approved by the Institutional Animal Care Committee.
Adrenalectomy and Cannulae Implantation Surgery
Surgery was performed with the animals under halothane anesthesia 24 hours prior to experiments, to permit adequate recovery from the procedure. To eliminate endogenous steroids, bilateral adrenalectomy was performed via dorsal incisions. Sham adrenalectomy consisted of opening the peritoneal cavity without removal of the adrenals. Incisions were closed using acrylic glue (Krazy Glue; Elmer’s Products, Inc., Columbus, OH). The completeness of adrenalectomy was verified by visual inspection as well as by hormonal measurements: Animals with corticosterone levels >0.2 μg/dl were considered not fully adrenalectomized and were eliminated from the study. For i.c.v. infusions, stainless steel cannulae were implanted into the lateral cerebral ventricle using an infant-rat stereotaxic apparatus (Kopf, Tujunga, CA) and stereotaxic coordinates (P 0.7 mm, L 2 mm, and V 3.3 mm from bregma), as published elsewhere.32,33 When both adrenalectomy and cannula were required, surgical procedures were performed sequentially, during a single anesthesia session.
In Situ Hybridization Histochemistry for CRH mRNA
In situ hybridization (ISH) histochemistry was performed as described previously.31 Briefly, 20 μm coronal sections were collected on gelatin-coated slides and stored at −80°C. Sections were thawed, air dried, fixed in paraformaldehyde, dehydrated, and rehydrated through graded ethanols, then exposed to 0.25% acetic anhydride in 0.1 M triethanolamine (pH 8) and dehydrated. Prehybridization and hybridization steps were performed at 40°C in a humidified chamber. Following a 1 hour prehybridization, sections were hybridized overnight (20 hours) with a deoxyoligonucleotide probe complementary to the coding region of CRH mRNA and 3′-end-labeled with 35S-dATP. Sections were then washed and apposed to film (Hyperfilm β-Max; Amersham, Arlington Heights, IL) for 7–14 days. Selected sections were also dipped in emulsion (NTB-2; Eastman Kodak, Rochester, NY) and exposed for 3 or 4 weeks.
Semiquantitative Analysis of CRH mRNA and Statistical Considerations
Semiquantitative analysis of CRH mRNA ISH signal was performed on digitized films using the ImageTool software program (UTHSC, San Antonio, TX) as described elsewhere.31,34 For balanced comparison among the different experimental groups, five anatomically matched sections per region per brain were analyzed. Unbiased methods for section sampling have been described in detail elsewhere,34 and all analyses were performed without knowledge of treatment group (blindly). 14C standards were used to ascertain that analyzed signals fell in the linear range, and values are expressed as relative optical density units (ROD). Differences among groups were determined using a one-way analysis of variance (ANOVA) or Student’s t test, as appropriate (Prism GraphPad, San Diego, CA), and significance levels were set at p < 0.05. Further analysis used Tukey’s multiple comparison posthoc tests to determine the source of the detected significance in the ANOVAs.
Results
Administration of Exogenous ACTH Down-Regulates CRH mRNA in ACe
CRH mRNA levels in ACe were significantly reduced by systemic administration of ACTH: 82 ± 4 and 53 ± 12 ROD in the control and ACTH groups, respectively (a 35% reduction; t = 2.67; df = 23; p < 0.02). Figure 1 shows CRH mRNA expression in representative photomicrographs of coronal brain sections derived from both groups; Figure 2A depicts semi-quantitative analyses of the intensity of the CRH mRNA signal.
Fig 1.

Differential regulation of CRH mRNA levels in ACe, hippocampus, and hypothalamic PVN by ACTH and adrenal steroids. Photomicrographs of coronal brain sections subjected to ISH for CRH mRNA. CRH mRNA expression in ACe (arrows, top row), PVN (arrows, second row), and hippocampus (third row) are shown. ACTH treatment decreased CRH mRNA signal over ACe. Adrenalectomy (ADX), by increasing endogenous ACTH levels, led to a similar reduction of CRH gene expression in ACe (top row). In contrast to the ACe, ADX resulted in up-regulation of CRH mRNA levels in PVN (middle row) and reduced CRH mRNA signal in hippocampus (third row) compared to control sections. The bottom row depicts representative darkfield photomicrographs of ACe (encircled region in left two panels) and PVN (right two panels). A marked reduction in the number of CRH-expressing cells is evident in ACTH-treated animals compared to controls. In PVN, increased CRH mRNA signal is visible in sections from an ADX animal. Scale bar = 1,500 μm in top two rows, 90 μm in third row, 200 μm in bottom row. Asterisks = basolateral nucleus; mpd, mpv = medial dorsal and medial ventral parvicellular cell groups of PVN, respectively.
Fig 2.

Effects of ACTH on CRH mRNA levels in ACe. (A) Semiquantitative analysis of CRH mRNA expression in ACe of immature rats subjected to selective hormone level manipulations. Signal was analyzed over ACe following ISH as discussed in Materials and Methods. (B) CRH mRNA levels in ACe correlated inversely with plasma ACTH. Thus, ACTH administration (ACTH i.p.) or augmentation of endogenous ACTH (ADX) or both (ACTH i.p. + ADX) significantly reduced CRH mRNA levels in ACe compared to both the i.p. vehicle-injected and the sham-adrenalectomy, vehicle-injected control groups. No correlation between plasma corticosterone levels and ACe-CRH mRNA was observed (C). *Significant difference from control and sham ADX; **significant difference from all other groups (p < 0.05). Values depict means ± SEM. ADX = adrenalectomy.
Glucocorticoid Receptor Activation Is Not Required for ACTH-Induced Down-Regulation of CRH mRNA in ACe
ACTH, given systemically, led to a significant reduction in CRH mRNA expression in ACe also in the absence of endogenous steroids (Fig 2A,C). Thus, in adrenalectomized rats—as in intact littermates—ACTH resulted in a significant (33%) suppression of CRH mRNA levels in ACe compared to vehicle-injected, sham-operated controls (56 ± 8 vs. 84 ± 4 ROD units, respectively; t = 3.1; df = 18; p < 0.01), indicating that endogenous steroids were not required for this effect. It should be noted that adrenalectomy—eliminating steroids and the consequent negative feedback on ACTH secretion—resulted in high levels of intrinsic, endogenous ACTH (Fig 2B). These ACTH levels, by themselves, were sufficient to down-regulate significantly CRH mRNA in ACe (34% reduction from sham-operated controls; see Fig 2A,B; t = 2.3; df = 18; p < 0.05). No further suppression of CRH expression resulted from adding exogenous ACTH to these animals, indicating that the high endogenous ACTH levels after adrenalectomy maximally activated melanocortin receptors, and further reduction of CRH expression was not possible via this mechanism. Additional reduction of CRH mRNA levels required an independent mechanism, i.e., blocking of the enhancing action of glucocorticoid receptor activation (see below).
Adrenalectomy and ACTH Administration Determine Plasma ACTH and Steroid Levels
The expected changes in plasma stress hormones were observed after the experimental interventions reported here (Fig 2B,C). ACTH levels were significantly elevated in animals given exogenous ACTH (695 ± 50.7 pg/ml) and in adrenalectomized animals (296.3 ± 53.4 pg/ml) compared to controls (118.5 ± 16.37 pg/ml). In addition, additive effects of administration of exogenous ACTH and adrenalectomy were apparent, leading to plasma ACTH levels of 1475 ± 165.2 pg/ml, significantly higher than those of either treatment alone [F(24) = 12; p < 0.0001, one-way ANOVA]. Plasma corticosterone levels increased significantly after ACTH administration (6.8 ± 0.9 vs. 1.05 ± 0.19 μg/dl in controls), whereas corticosterone levels in adrenalectomized animals were negligible [F(28) = 42.1; p < 0.0001, one-way ANOVA].
ACTH Alters CRH mRNA Expression in ACe via a Central Effect
Similarly to systemically administered ACTH, the analog ACTH4–10 infused i.c.v. resulted in a significant down-regulation of CRH mRNA expression in ACe [45% reduction from i.c.v. vehicle-infused control levels (Fig 3); t = 4.04; p < 0.001]. The dose of ACTH4–10 used here was relatively small and was therefore unlikely to reach the adrenal when given i.c.v. In addition, this ACTH analog does not stimulate corticosterone secretion from the adrenal.1 Indeed, as shown in Figure 3, plasma corticosterone levels of ACTH4–10-infused rats were not higher than those of vehicle-treated controls, whereas systemic ACTH led to a marked increase in plasma corticosterone at the time of sacrifice (see Fig 2C). These data suggest that ACTH4–10 (and ACTH) reduced CRH expression in ACe via a central rather than a steroid-mediated peripheral mechanism.
Fig 3.
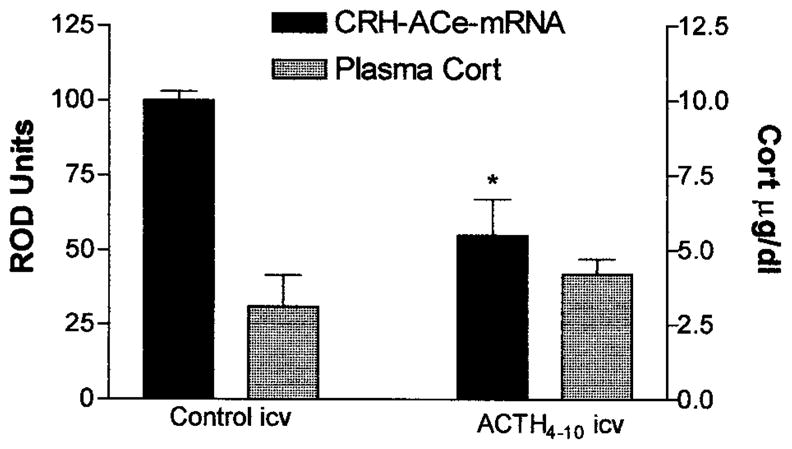
CRH mRNA expression in ACe is down-regulated by centrally administered ACTH analog that is devoid of glucocorticoid secretion. Semiquantitative analysis (see Materials and Methods) of CRH mRNA signal in sections of animals receiving an i.c.v. infusion of ACTH4–10, an analog binding melanocortin receptors but not stimulating corticosterone (Cort). Although infusion of ACTH4–10 did not induce Cort secretion (hatched bars, scale on right), this analog reduced ACe-CRH mRNA levels significantly (solid bars, left scale). Values are means ± SEM; *p < 0.05.
ACTH Influences CRH mRNA Levels in ACe by Activating Melanocortin Receptors, Independently of Glucocorticoid Receptor Occupancy
Further insight into the mechanism by which ACTH down-regulated CRH expression in ACe was gained by administering the melanocortin receptor-4 blocker SHU9119 and the glucocorticoid receptor antagonist RU 38486. As is evident from Figure 4A, in the setting of high endogenous plasma ACTH levels achieved by adrenalectomy, administration of SHU9119 blocked ACTH-induced reduction of CRH mRNA in ACe [90 ± 11 ROD in (SHU+ Adx) vs. 55 ± 12 ROD in adrenalectomy alone]. In contrast, when RU 38486 was given to intact animals, together with ACTH, it failed to block the effects of ACTH administration on CRH mRNA expression in ACe [30 ± 9 ROD in (ACTH + RU 38486), a 63% reduction from i.p.-injected controls; t = 5.3; df = 8; p < 0.001; Fig 4B]. These data indicate that activation of melanocortin receptors, but not of glucocorticoid receptors, is required for the actions of ACTH on CRH expression in ACe.
Fig 4.

Effects of ACTH on CRH mRNA expression in ACe require activation of MC4-Rs but not of glucocorticoid receptors. (A) High endogenous plasma ACTH levels were achieved by ADX, significantly reducing CRH mRNA levels (single asterisks). Administration of SHU9119, however, abolished ACTH-induced reduction of CRH mRNA. (B) In contrast, coadministered RU 38486 failed to block the effects of ACTH (i.p.) on CRH mRNA expression in ACe. In fact, CRH mRNA levels in ACTH+ RU 38486 treated rats were significantly lower from those given ACTH alone, as denoted by double asterisks. Values denote means ± SEM of values achieved using semiquantitative analysis (see Materials and Methods). i.c.v. Controls and i.p. controls received vehicle via the noted routes; ADX/icv were adrenalectomized and given i.c.v. vehicle. Diamonds over the i.p. control and ACTH groups indicate that they correspond to groups shown in Fig 2A.
Actions of ACTH on CRH Expression Are Region-Specific
The direct, steroid-independent down-regulation of CRH mRNA expression by ACTH was specific to ACe. Analysis of ACTH and steroid plasma levels and the corresponding CRH mRNA levels in hypothalamus and hippocampus (Fig 5) revealed distinct regional patterns: In hypothalamic paraventricular nucleus (PVN), CRH mRNA levels correlated inversely with plasma glucocorticoids, consistently with previous reports.35,36 ACTH administration, inducing high plasma corticosterone levels, reduced CRH mRNA expression 40% in PVN (290 ± 40 ROD in ACTH-treated vs. 480 ± 30 ROD in controls), whereas adrenalectomy, eliminating plasma glucocorticoids, enhanced PVN CRH mRNA levels 108% [830 ± 150 ROD in adrenalectomy vs. 400 ± 30 ROD in sham adrenalectomy controls; F(24) = 6.8; p < 0.01, one-way ANOVA]. No consistent correlation of PVN CRH mRNA expression with plasma ACTH levels was noted.
Fig 5.

Differential regulation of CRH mRNA expression in hippocampal regions of immature rats by glucocorticoids and ACTH. Semiquantitative analysis of signal over dentate gyrus (A), CA3 (B), and CA1 (C) was performed after ISH, as described in Materials and Methods. CRH mRNA levels in CA1 (the region rich in steroid receptors in the immature rat) were reduced by elimination of GCs (ADX), regardless of ACTH levels (C). CRH mRNA expression in other hippocampal regions was not appreciably altered by these hormonal manipulations.*Significant difference from i.p. vehicle-injected control and sham-ADX animals given i.p. vehicle (p < 0.05). Values are means ± SEM. ADX = adrenalectomy.
In hippocampus, exogenous ACTH did not alter CRH mRNA expression in any of the analyzed fields (CA1, CA3, and dentate gyrus; Fig 5). Steroid elimination via adrenalectomy significantly decreased CRH mRNA levels in CA1 neurons [F(25) = 10.44; p < 0.0001, one-way ANOVA], known to express a high density of glucocorticoid receptors, but not in the granule cell layer of the dentate gyrus.
Discussion
This study demonstrates direct actions of systemically or centrally administered ACTH on limbic neurons that are independent of adrenal steroids. ACTH-induced reduction of CRH expression in amygdala may provide a mechanism for the efficacy of ACTH in several human disorders. Importantly, these effects of ACTH involve activation of specific melanocortin receptors, which may provide new and important therapeutic targets.
ACTH has been used for treating a variety of neurological disorders. An established effect of this hormone includes activation of adrenal steroid secretion, and the latter has generally been considered as the major mechanism of action of ACTH. However, release of adrenal steroids leading to activation of steroid receptors cannot fully explain the spectrum and magnitude of the therapeutic effects of ACTH.
For example, both ACTH and steroids have been shown to be effective in treating certain childhood seizures,37,38 particularly infantile spasms.3,20,39 However, for this disorder, ACTH is a significantly more powerful agent than glucocorticoids.3,4 Indeed, both uncontrolled3 and prospective, blinded, and controlled studies4 have shown that ACTH in very large doses is significantly superior to large doses of steroids, controlling the seizures and normalizing the EEG in 86–88% of individuals compared to 35–40%.3,4,39 It should be noted that the levels of plasma steroids achieved after administration of the typical doses of prednisone should be sufficient to saturate neuronal glucocorticoid receptors.40 In addition, most studies demonstrate the superior efficacy of higher compared to relatively lower doses of ACTH3,4,41,42 (but see Hrachovy et al43), although these “lower” doses suffice to stimulate maximally secretion of steroids from the adrenal.
These clinical data raise several issues: First, if ACTH acts via steroids, then maximal doses of both agents should have similar effects. Second, once doses of ACTH that suffice to release endogenous steroids maximally are used, then higher doses should not have additional benefits. The enhanced potency of ACTH compared to steroids and the superiority of extremely high doses of ACTH compared to lower one are consistent with direct, steroid-independent actions of ACTH within the CNS. In addition, the poor penetration of systemically given ACTH through the blood–brain barrier26,27 may underlie the requirement for large systemic doses.
The studies described here support the notion of direct CNS actions of ACTH. Using doses analogous to those used clinically, we show that systemic ACTH influences gene expression in specific brain regions regardless of the presence or absence of adrenal steroids. We further show that these effects are generated by ACTH given either systemically or directly into the cerebral ventricles. These effects persist when glucocorticoid receptors are blocked, but not when a subtype of ACTH receptors (melanocortin receptors) is antagonized, showing that activation of these latter receptors is required and sufficient for these actions of ACTH. These findings are important for several reasons. First, they provide a mechanism for the direct actions of ACTH on neurons, in both therapeutic and physiological contexts. In addition, they demonstrate that ACTH may indeed be a native ligand for melanocortin receptor-4.
Melanocortin receptors, a family of transmembrane G-protein-coupled receptors, have been demonstrated in regions subserving ACTH-mediated functions (eg, substantia nigra for grooming and amygdala for learning and memory). In addition, activation of central melanocortin receptors plays a role in appetitive and grooming behavior.10,44 However, several endogenous members of the proopiomelanocortin family bind to melanocortin receptors, and whether ACTH is an important endogenous ligand of these receptors has not been determined. The current study demonstrates categorically that ACTH may activate melanocortin receptor-4. Indeed, in adrenalectomized rats, the effect of increased endogenous ACTH on CRH expression was abolished by administration of the melanocortin receptor-4 antagonist SHU9119 directly into the CNS. These data strongly support the notion that endogenous ACTH activates central melanocortin receptor-4 to influence CRH synthesis. Furthermore, the receptor implicated here in the effects of ACTH on CRH gene expression, melanocortin receptor-4, has been demonstrated in a number of brain regions, including hippocampus and all three major divisions of the central amygdaloid nucleus.45 It is therefore interesting to note that, unlike the hippocampus (expressing melanocortin receptors but devoid of endogenous ACTH) and the hypothalamus (containing ACTH but lacking melanocortin receptor-4), ACe is a site where both ACTH and melanocortin receptors coexist. Therefore, the current study is consistent with a scenario in which ACTH, released from fiber terminals in ACe, acts locally on melanocortin receptors located on CRH-expressing neurons. ACTH, given in clinical settings should have similar effects.
Interestingly, the 4–10 fragment of ACTH that does not release adrenal steroids also led to the depression of CRH gene expression. Earlier clinical studies have shown that shorter fragments, such as ACTH4–9, that failed to release adrenal steroids were not efficacious in treating infantile spasms.46,47 Currently available information explains the apparent discrepancy: New information about the binding of ACTH fragments to melanocortin receptors shows that, although the 4–10 fragment used here activates these receptors, the 4–9 fragment does not.10,48 Thus, the current data are consistent with activation of melanocortin receptor as the mechanism for the observed effects of ACTH on amygdala neurons.
Modulation of CRH synthesis may prove to be one of the mechanisms by which ACTH exerts its profound and diverse effects on CNS function. For example, CRH has been shown to play an important modulatory role in affective disorders49 and neuroimmune function.15,17,22 Cytokines including interleukins 1 and 2 up-regulate CRH expression in specific brain regions,16,50 and these cytokines, in turn, are induced by stress, by mechanisms that may involve CRH release.17 CRH released from central neurons interacts with immune responses both directly and by activation of steroid release.17,22 In addition, a role for CRH as an excitatory neuromodulator that can provoke severe seizures early in life has been emerging.20,21,51 These seizures originate in the amygdala and involve other limbic regions.51 A hypothesis for the involvement of CRH in infantile spasms has been proposed20,21 and is supported by abnormalities of the CRH-ACTH-steroid axis found in infants with this disorder.23,24 It has been proposed that the myriad etiologies of infantile spasms share the characteristic of activating the responses to stress, thus leading to enhanced CRH expression in the amygdala.20,31 This leads to excessive neuronal excitability25,33 and seizures.21,51 By repressing CRH expression, ACTH may decrease this hyperexcitability, ameliorating infantile spasms.20 Indeed, the time course and all-or-none effects of ACTH are consistent with such an effect.20,39,43,52
In summary, the current study provides a potential mechanism for the efficacy of ACTH for certain neuroimmune and developmental seizure disorders and highlights the ACTH-CRH system as a potential target for development of new therapeutic agents.
Acknowledgments
Supported by NIH grant RO1 NS28912 and R41 HD34975 (T.Z.B.), a system-wide University of California biotechnology-oriented predoctoral award (K.L.B.) and UCI undergraduate research (UROP) award (N.K.).
We thank Drs Bruce McEwen and Roger Cone for helpful critiques and discussions.
References
- 1.Pranzatelli MR. On the molecular mechanism of adrenocorticotrophic hormone in the CNS: neurotransmitters and receptors. Exp Neurol. 1994;125:142–161. doi: 10.1006/exnr.1994.1018. [DOI] [PubMed] [Google Scholar]
- 2.Pranzatelli MR, Huang YY, Tate E, et al. Monoaminergic effects of high-dose corticotropin in corticotropin-responsive pediatric opsoclonus-myoclonus. Mov Disord. 1998;13:522–528. doi: 10.1002/mds.870130323. [DOI] [PubMed] [Google Scholar]
- 3.Snead OC, III, Benton JW, Hosey LC, et al. Treatment of infantile spasms with high-dose ACTH: efficacy and plasma levels of ACTH and prednisone. Neurology. 1989;39:1027–1031. doi: 10.1212/wnl.39.8.1027. [DOI] [PubMed] [Google Scholar]
- 4.Baram TZ, Mitchell WG, Tournay A, et al. High-dose corticotropin (ACTH) versus prednisone for infantile spasms: a prospective, randomized, blinded study. Pediatrics. 1996;97:375–379. [PMC free article] [PubMed] [Google Scholar]
- 5.Riikonen R. Infantile spasms: some new theoretical aspects. Epilepsia. 1983;24:159–168. doi: 10.1111/j.1528-1157.1983.tb04875.x. [DOI] [PubMed] [Google Scholar]
- 6.Watson SJ, Richard CW, III, Barchas JD. Adrenocorticotropin in rat brain: immunocytochemical localization in cells and axons. Science. 1978;200:1180–1182. doi: 10.1126/science.206967. [DOI] [PubMed] [Google Scholar]
- 7.Pilcher WH, Joseph SA. Co-localization of CRF-ir perikarya and ACTH-ir fibers in rat brain. Brain Res. 1984;299:91–102. doi: 10.1016/0006-8993(84)90791-1. [DOI] [PubMed] [Google Scholar]
- 8.de Wied D. Behavioral effects of neuropeptides related to ACTH, MSH, and βLPH. Ann N Y Acad Sci. 1977;297:263–275. doi: 10.1111/j.1749-6632.1977.tb41859.x. [DOI] [PubMed] [Google Scholar]
- 9.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:543–546. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 10.Adan RAH, Gispen WH. Brain melanocortin receptors: from cloning to function. Peptides. 1997;8:1279–1287. doi: 10.1016/s0196-9781(97)00078-8. [DOI] [PubMed] [Google Scholar]
- 11.Sawchenko PE, Arias C. Evidence for short-loop feedback effects of ACTH on CRF and vasopressin expression in parvocellular neurosecretory neurons. J Neuroendocrinol. 1995;7:721–731. doi: 10.1111/j.1365-2826.1995.tb00814.x. [DOI] [PubMed] [Google Scholar]
- 12.Wu HC, Chen KY, Lee WY, Lee EHY. Antisense oligonucleotides to corticotropin-releasing factor impair memory retention and increase exploration in rats. Neuroscience. 1997;78:147–153. doi: 10.1016/s0306-4522(96)00533-7. [DOI] [PubMed] [Google Scholar]
- 13.Hauger RL, Irwin MR, Lorang M, et al. High intracerebral levels of CRH result in CRH receptor downregulation in the amygdala and neuroimmune desensitization. Brain Res. 1993;616:283–292. doi: 10.1016/0006-8993(93)90219-d. [DOI] [PubMed] [Google Scholar]
- 14.Joseph SA, Pilcher WH, Knigge KM. Anatomy of the corticotropin-releasing factor and opiomelanocortin systems of the brain. Fed Proc. 1985;44:100–107. [PubMed] [Google Scholar]
- 15.Tsagarakis S, Grossman A. Corticotropin-releasing hormone: interactions with the immune system. Neuroimmunomodulation. 1994;1:329–334. doi: 10.1159/000097184. [DOI] [PubMed] [Google Scholar]
- 16.Sapolsky R, Rivier C, Yamamoto G, Plotsky P, Vale W. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science. 1987;238:522–524. doi: 10.1126/science.2821621. [DOI] [PubMed] [Google Scholar]
- 17.Rothwell NJ, Luheshi G. Pharmacology of interleukin-1 actions in the brain. Adv Pharmacol. 1994;25:1–18. doi: 10.1016/s1054-3589(08)60428-7. [DOI] [PubMed] [Google Scholar]
- 18.Liang KC, Lee EH. Intra-amygdala injections of corticotropin releasing factor facilitate inhibitory avoidance learning and reduce exploratory behavior in rats. Psychopharmacology. 1988;96:232–236. doi: 10.1007/BF00177566. [DOI] [PubMed] [Google Scholar]
- 19.Lee EH, Huang AM, Tsuei KS, Lee WY. Enhanced hippocampal corticotropin-releasing factor gene expression associated with memory consolidation and memory storage in rats. Chin J Physiol. 1996;39:197–203. [PubMed] [Google Scholar]
- 20.Baram TZ. Pathophysiology of massive infantile spasms: perspective on the putative role of the brain adrenal axis. Ann Neurol. 1993;33:231–236. doi: 10.1002/ana.410330302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crofford LJ, Sano H, Karalis K, Friedman TC, Epps HR, Remmers EF, Mathern P, Chrousos GP, Wilder RL. Corticotropin-releasing hormone in synovial fluids and tissues of patients with rheumatoid arthritis and osteoarthritis. J Immunol. 1993;151:1587–1596. [PubMed] [Google Scholar]
- 23.Nalin A, Facchinetti F, Galli V, et al. Reduced ACTH content in cerebrospinal fluid of children affected by cryptogenic infantile spasms with hypsarrhythmia. Epilepsia. 1985;26:446–449. doi: 10.1111/j.1528-1157.1985.tb05678.x. [DOI] [PubMed] [Google Scholar]
- 24.Baram TZ, Mitchell WG, Snead OC, III, Horton EJ, Saito M. Brain adrenal axis hormones are altered in the CSF of infants with massive infantile spasms. Neurology. 1992;42:1171–1175. doi: 10.1212/wnl.42.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollrigel GS, Chen K, Baram TZ, Soltesz I. The proconvulsant actions of corticotropin-releasing hormone in the hippocampus of infant rats. Neuroscience. 1998;84:78–84. doi: 10.1016/s0306-4522(97)00499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholson WE, Liddle RA, Puett D, Liddle GW. Adrenocorti-cotropic hormone biotransformation, clearance, and catabolism. Endocrinology. 1978;103:1344–351. doi: 10.1210/endo-103-4-1344. [DOI] [PubMed] [Google Scholar]
- 27.Mezey E, Palkovitz M, de Kloet ER, et al. Evidence for pituitary-brain transport of a behaviorally potent ACTH analog. Life Sci. 1978;22:831–838. doi: 10.1016/0024-3205(78)90606-9. [DOI] [PubMed] [Google Scholar]
- 28.Yi SJ, Masters JN, Baram TZ. The effect of a specific glucocorticoid receptor antagonist on CRH gene expression in the neonatal rat hypothalamus. Brain Res Dev Brain Res. 1993;73:253–259. doi: 10.1016/0165-3806(93)90145-z. [DOI] [PubMed] [Google Scholar]
- 29.Kask A, Mutulis F, Muceniece R, et al. Discovery of a novel superpotent and selective melanocortin-4 receptor antagonist (HSO24): evaluation in vitro and in vivo. Endocrinology. 1998;139:5006–5014. doi: 10.1210/endo.139.12.6352. [DOI] [PubMed] [Google Scholar]
- 30.Gottlieb A, Keydar Y, Epstein HT. Rodent brain growth stages: an analytical review. Biol Neonate. 1977;32:166–176. doi: 10.1159/000241012. [DOI] [PubMed] [Google Scholar]
- 31.Hatalski CG, Guirguis C, Baram TZ. Corticotropin releasing factor mRNA expression in the hypothalamic paraventricular nucleus and the central nucleus of the amygdala is modulated by repeated acute stress in the immature rat. J Neuroendocrinol. 1998;10:663–669. doi: 10.1046/j.1365-2826.1998.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baram TZ, Schultz L. ACTH does not control neonatal seizures induced by administration of exogenous corticotropin-releasing hormone. Epilepsia. 1995;36:174–178. doi: 10.1111/j.1528-1157.1995.tb00977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brunson KL, Schultz L, Baram TZ. The in vivo proconvulsant effects of corticotropin releasing hormone in the developing rat are independent of ionotropic glutamate receptor activation. Brain Res Dev Brain Res. 1998;111:119–128. doi: 10.1016/s0165-3806(98)00130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eghbal-Ahmadi M, Avishai-Eliner S, Hatalski CG, Baram TZ. Differential regulation of the expression of corticotropin-releasing factor receptor type 2 (CRF2) in hypothalamus and amygdala of the immature rat by sensory input and food intake. J Neurosci. 1999;19:3982–3991. doi: 10.1523/JNEUROSCI.19-10-03982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sawchenko PE. Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in parvocellular neurosecretory neurons. Brain Res. 1987;403:213–224. doi: 10.1016/0006-8993(87)90058-8. [DOI] [PubMed] [Google Scholar]
- 36.Lightman SL, Young WS. Influence of steroids on the hypothalamic corticotropin releasing factor and proenkephalin mRNA responses to stress. Proc Natl Acad Sci USA. 1989;86:4306–4310. doi: 10.1073/pnas.86.11.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snead OC, III, Benton JW, Myers GJ. ACTH and prednisone in childhood seizure disorders. Neurology. 1983;33:966–970. doi: 10.1212/wnl.33.8.966. [DOI] [PubMed] [Google Scholar]
- 38.Holmes GL. Diagnosis and management of seizures in children. Philadelphia: W.B. Saunders; 1987. [Google Scholar]
- 39.Hrachovy RA, Frost JD, Kellaway P, Zion TE. Double-blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr. 1983;103:641–645. doi: 10.1016/s0022-3476(83)80606-4. [DOI] [PubMed] [Google Scholar]
- 40.Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 41.Lombroso CT. A prospective study of infantile spasms: clinical and therapeutic correlations. Epilepsia. 1983;24:135–158. doi: 10.1111/j.1528-1157.1983.tb04874.x. [DOI] [PubMed] [Google Scholar]
- 42.Koo B, Kwang PA, Logan WJ. Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic groups. Neurology. 1992;43:2322–2327. doi: 10.1212/wnl.43.11.2322. [DOI] [PubMed] [Google Scholar]
- 43.Hrachovy RA, Frost JD, Glaze DG. High-dose long-duration versus low-dose short duration corticotropin therapy for infantile spasms. J Pediatr. 1994;124:803–8066. doi: 10.1016/s0022-3476(05)81379-4. [DOI] [PubMed] [Google Scholar]
- 44.Fisher SL, Yagaloff KA, Burn P. Melanocortin and leptin signaling systems: central regulation of catabolic energy balance. J Recept Signal Transduct Res. 1999;19:203–216. doi: 10.3109/10799899909036646. [DOI] [PubMed] [Google Scholar]
- 45.Mountjoy KG, Mortrud MT, Low MJ, et al. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 46.Willig RP, Lagenstein I. Use of ACTH fragments in children with infantile spasms. Neuropediatrics. 1982;13:55–58. doi: 10.1055/s-2008-1059597. [DOI] [PubMed] [Google Scholar]
- 47.Pentella K, Bachman DS, Sandman CA. Trial of an ACTH 4–9 analogue in children with intractable seizures. Neuropediatrics. 1982;13:59–62. doi: 10.1055/s-2008-1059598. [DOI] [PubMed] [Google Scholar]
- 48.Gantz I, Miwa H, Konda Y, et al. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 49.Schulkin J, Gold PW, McEwen BS. Induction of corticotropin-releasing hormone gene expressions by glucocorticoids: implication for understanding the states of fear and anxiety and allostatic load. Psychoneuroendocrinol. 1998;23:219–243. doi: 10.1016/s0306-4530(97)00099-1. [DOI] [PubMed] [Google Scholar]
- 50.Raber J, Koob GF, Bloom FE. Interleukin-2 (IL-2) Induces corticotropin-releasing factor (CRF) release from the amygdala and involves a nitric oxide-mediated signaling; comparison with the hypothalamic response. J Pharmacol Exp Ther. 1995;272:815–824. [PubMed] [Google Scholar]
- 51.Baram TZ, Hirsch E, Snead OC, III, Schultz L. Corticotropin-releasing hormone-induced seizures in infant rats originate in the amygdala. Ann Neurol. 1992;31:488–494. doi: 10.1002/ana.410310505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baram TZ, Mitchell WG, Brunson K, Haden E. Infantile spasms: hypothesis-driven therapy and pilot human infant experiments using corticotropin-releasing hormone receptor antagonists. Dev Neurosci. 1999;21:281–289. doi: 10.1159/000017407. [DOI] [PMC free article] [PubMed] [Google Scholar]