

Abstract
Introduction
Prostaglandins are important inflammatory mediators; PGE2 is the predominant prostaglandin in colorectal neoplasia and affects colorectal carcinogenesis. Prostaglandins are metabolites of omega-6 and omega-3 polyunsaturated fatty acids; their biosynthesis is the primary target of nonsteroidal anti-inflammatory drugs (NSAIDs), which reduce colorectal neoplasia risk.
Methods
We investigated candidate and tagSNPs in PGE2 synthase (PGES), PGE2 receptors (EP2 and EP4), and prostaglandin dehydrogenase (PGDH) in a case-control study of adenomas (n=483) vs. polyp-free controls (n=582) and examined interactions with NSAID use or fish intake, a source of omega-3 fatty acids.
Results
A 30% adenoma risk reduction was observed for EP2 4950G>A (intron 1; ORGA/AA vs. GG: 0.71; 95% CI: 0.52-0.99). For the candidate polymorphism EP4 Val294Ile, increasing fish intake was associated with increased adenoma risk among those with variant genotypes, but not among those with the Val/Val genotype, (p-interaction=0.02). An interaction with fish intake was also observed for PGES -664A>T (5’UTR; p-interaction=0.01). Decreased risk with increasing fish intake was only seen among those with the AT or TT genotypes (OR>2 t/wk vs. <1 t/wk: 0.56; 95% CI: 0.28-1.13). We also detected interactions between NSAIDs and EP2 9814C>A (intron 1) and PGDH 343C>A (intron 1). However, none of the observed associations was statistically significant after adjustment for multiple testing. We investigated potential gene-gene interactions using the Chatterjee 1df Tukey test and logic regression; neither method detected significant interactions.
Conclusions
These data provide little support for associations between adenoma risk and genetic variability related to PGE2, yet suggest gene-environment interactions with anti-inflammatory exposures.
Keywords: prostaglandins, colorectal cancer, colorectal polyps, NSAIDs, aspirin, fish, fat intake
Introduction
Aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) are effective chemopreventive agents for colorectal adenomas and cancer (1-4). The main targets of NSAIDs are the COX-1 and COX-2 proteins, which catalyze the first step in the prostaglandin synthesis pathway. This pathway metabolizes omega-6 (n-6) and n-3 poly-unsaturated fatty acids into prostaglandins, many of which are potent inflammatory mediators. Prostaglandin E2 (PGE2), one of the main downstream products of COX metabolism, is upregulated in colon cancer(5) and is associated with increased cell proliferation, survival, and motility (6-8). Apc Min mice treated with PGE2 have increased intestinal tumor number and size, compared to untreated mice (9). Conversely, PGE2 receptor knockout mice show reduced intestinal tumorigenesis (10-12).
PGE2 is produced by prostaglandin E2 synthase, of which there are three forms (13). One of these, microsomal prostaglandin E2 synthase-1 (PGES), is induced by pro-inflammatory stimuli and is preferentially coupled to COX-2 over COX-1 in several tissues (14). PGES is overexpressed in colorectal adenomas and cancer (13, 15), and transfection of PGES in concert with COX-2 resulted in faster growth, increased cell aggregation, and abnormal cell morphology (16), indicating that induction of PGES may be driving prostaglandin-related inflammatory processes.
PGE2 signals through four G-protein-coupled cell-surface receptors, referred to as EP1-4. The four receptor types have different downstream mechanisms of signal transduction, resulting in a wide variety of functions, although EP2 and EP4 have similar modes of action (17). Of the four EP receptors, EP2 and EP4 have been the most consistently linked to colorectal carcinogenesis (10, 12, 17, 18). APCΔ716/EP2-receptor-null mice had fewer intestinal polyps, reduced tumor growth, and reduced angiogenesis (10, 11, 19), indicating that PGE2 signaling through this receptor is an important contributor to colorectal neoplasia. Higher EP2 expression was associated with poorer colorectal cancer survival in a study of 99 Swedish cancer patients (18).
In mouse models, knocking out EP4, either through genetic deletion or through treatment with an EP4-receptor antagonist reduced the formation of aberrant crypt foci and decreased polyp formation (12). In an in vitro study, treatment of cells with an EP4 agonist countered the chemopreventive effects of NSAID treatment (20), indicating that PGE2 signaling through the EP4 receptor also plays an important role in the development of colorectal neoplasia. However, in mouse models of colitis, a condition predisposing to colorectal cancer, treatment with EP4 agonists ameliorates symptoms (21, 22), indicating that the full extent of the relationship between EP4 and colon carcinogenesis is not yet well understood.
Prostaglandins can be inactivated by 15-hydroxyprostaglandin dehydrogenase (PGDH) (23). APC Min -/- mice exhibit lower PGDH expression than APC Min +/- mice, and Pgdh knockout mice have greatly increased tumor development (24, 25). Human colon cancer cell lines exhibit low levels of PGDH expression compared to normal colon mucosa (24, 26), and expression of PGDH mRNA in human colon cancer tissues is greatly reduced compared to normal colon tissue (24-26). PGDH expression is also reduced in inflammatory bowel disease (27), a condition associated with markedly increased colorectal cancer risk.
Use of NSAIDs substantially reduces PGE2 levels (28-30), induces PGDH expression (24, 31, 32), and may decrease expression of EP2 (33), indicating that reduction of PGE2 production or signaling may be important mechanisms by which NSAIDs prevent colorectal neoplasia. Dietary factors also can influence prostaglandin levels. n-3 PUFAs, such as eicosapentaenoic acid, compete with arachidonic acid for metabolism by the prostaglandin pathway, resulting in production of 3-series prostaglandins that have reduced inflammatory potential (34-36). Thus, higher intakes of n-3 fatty acids, which are abundant in fatty fish, may also result in reduced risk of colorectal adenoma or cancer.
Given the known relevance of this pathway to colorectal carcinogenesis, we evaluated genetic variability in key proteins related to PGE2 synthesis, inactivation, and signaling in relation to colorectal polyp risk. We have previously shown that genetic variability in the prostaglandin synthesis pathway may alter the chemopreventive associations between adenoma risk and NSAID use or fish intake (a proxy for n-3 polyunsaturated fatty acid intake) (37-40). We and others have screened the coding regions of PGES, EP2, and EP4 for polymorphisms (41, 42). We investigated potential associations between tagSNPs in PGES, EP2, EP4, and PGDH, as well as two rare candidate non-synonymous SNPs (nsSNPs) in EP4, and risk of colorectal adenoma. We further explored potential interactions between these polymorphisms and use of aspirin or other NSAIDs and fish intake.
Materials and Methods
Study Population
Participant recruitment has been previously described (4, 43, 44). Briefly, adenoma cases and colonoscopy-screened polyp-free controls were recruited through a large multiclinic gastroenterological practice in the Twin cities area of Minnesota from April 1991-April 1994. Eligibility criteria have been described elsewhere (43), participants recruited prior to colonoscopy and were aged 30-74 years, English-speaking residents of the Twin Cities metropolitan area with no known genetic syndrome associated with increased risk of colon neoplasia and no individual history of cancer (except non-melanoma skin cancer), prior colorectal polyps, or inflammatory bowel disease. The participation rate for all patients who underwent colonoscopy was 68%.
Information on use of aspirin or other NSAIDs, diet, physical activity, anthropometrics, demographics, and medical history was obtained via questionnaire. Participants reported the average weekly consumption of aspirin and other NSAIDs, and the duration of use. Study participants were provided with a list of 14 common aspirin brands and 24 common non-aspirin NSAIDs. Non-NSAIDs, that were to be excluded from responses (e.g., acetaminophen), were also listed for guidance.
Information about regular weekly fish intake over the year prior to polyp diagnosis or reference date was obtained using a food frequency questionnaire (FFQ). The FFQ was an adaptation of the Willett semiquantitative FFQ, which has been studied previously for validity and repeatability within the Nurses’ Health Study cohort (45), the Iowa Women's Health Study cohort (46), and the Health Professionals Follow-up Study cohort (47). Participants were asked four questions regarding their usual intakes of various types of fish, including canned tuna, fatty fish, and white fish. These responses were combined to form a total fish intake variable.
Candidate SNP selection
Polymorphisms that may affect protein levels or function are referred to as candidate polymorphisms. In our recent screening of PGES, EP2, and EP4 (41), only two non-synonymous polymorphisms in EP4, Thr176Ile (MAF 1%) and Val294Ile (MAF 3%), were found in Caucasians. Of these, Thr176Ile was predicted to affect protein function using the SIFT and PolyPhen algorithms (41). We genotyped both of these nsSNPs in the present study.
TagSNP selection
The coding regions, as well as 2KB 5’ and 3’ of PGES, EP2, EP4, and PGDH, were resequenced in 23 individuals of European descent and 24 individuals of African-American descent by the University of Washington-Fred Hutchinson Cancer Research Center Variation Discovery Resource (42). TagSNPs were selected from the European-descent population using the LD Select algorithm developed by Carlson and colleagues (48), with a cutoff minor allele frequency (MAF) of 4% (i.e., any variant that occurred twice among the European population) and an r2 value of 0.90. This resulted in the selection of 10 tagSNPs in PGES, 11 in EP2, 11 in EP4, and 32 in PGDH, estimated by the Genome Variation Server (49) to cover 85% of the common (≥4% MAF) variation in these loci (see Supplemental Table S1). A total of 51 SNPs successfully converted to the Illumina™ GoldenGate genotyping platform. Most of the polymorphisms that failed to convert (10/13) were “singleton” SNPs, not tagging for any other polymorphisms; the three failed SNPs that tagged “bins” of LD, as well as two candidate polymorphisms, were subsequently genotyped individually at FHCRC (see below).
Genotyping
The majority (51/56) of SNPs were genotyped using the Illumina™ GoldenGate bead-based genotyping technology at the Translational Genomics Institute (TGen, Phoenix, AZ). For polymorphisms that were not amenable to high-throughput methods, genotyping was conducted at FHCRC. These polymorphisms (EP4 Thr176Ile, EP4 Val294Ile, PGES -1254T>C, PGDH 12184A/-, and PGDH 19979A>T) were detected by allelic discrimination using the 5’ nuclease assay on a 7900HT sequence detection system (Applied Biosystems, Foster City, CA). The 5’ nuclease genotyping assays were validated by genotyping 92 individuals by both 5’ nuclease assay and RFLP. There were no discrepancies between the two assays. The 5 μl genotyping reactions contained Taqman universal master mix, primers, probes, and 2 ng of genomic DNA. Positive controls for all the genotypes as well as two negative controls were included on each plate.
Genotype Quality Control
Intraplate and interplate replicates at a rate of ~5% were included on all plates and in all batches. Blinded duplicates were also included on all plates as another quality control measure. Genotype data from 30 CEPH trios (Coriell Cell Repository, Camden, NJ) genotyped by the HapMap project were used to confirm reliability and reproducibility of the genotyping. Genotypes were excluded from analyses if SNPs had <85% call rate, <85% concordance with blinded or non-blinded duplicates, or Hardy-Weinberg Equilibrium p<0.0001 (see Supplemental Table S1). Eight tagSNPs were excluded due to quality control errors; leaving a total of 46 tagSNPs and two candidate SNPs in four genes included in the analyses presented here.
Statistical Analysis
Single SNP Analyses
Unconditional, logistic regression was used to estimate the odds ratio (OR) and corresponding 95% confidence interval (CI) for the associations between genotypes in PGES, EP2, EP4, and PGDH and risk of adenoma. Most SNPs were analyzed using indicator variables for the heterozygous and homozygous variant genotype groups (co-dominant or unrestricted model). If fewer than ten cases or controls had the homozygous variant genotype, we grouped the homozygous variant genotypes with the heterozygous genotypes for analysis (dominant model). In main-association models of genotypes, associations were adjusted for age and sex. Effect modification by NSAID use or fish intake was evaluated in two ways: 1) by the inclusion of multiplicative interaction terms in logistic regression models; and 2) by testing for a difference in trends within strata of genotype or NSAID/fish, coded as a continuous variable. Because use of NSAIDs or dietary intakes of fish may be associated with other known risk factors for colorectal neoplasia, we adjusted our NSAID- and fish-gene interaction analyses for age, sex, body mass index, dietary intakes of fiber, alcohol, and energy, postmenopausal hormone use, and smoking. Because of possible racial differences in genotype frequencies, our analyses were restricted to Caucasian individuals (97% of the total study population). To obtain tests for trend, the genotypes were treated as a continuous variable, with the wildtype genotypes coded as 0, the heterozygous as 1, and the homozygous variant as 2.
All statistical analyses were carried out using SAS v.9. As a secondary analysis, p-values were adjusted for multiple testing taking into account correlated tagSNPs using the p(ACT) method (50). We corrected for multiple testing on the gene level, i.e. all tests within a gene were corrected for the total number of SNPs within a gene.
Whole gene analyses
Haplotype analysis and principal components analysis (PCA) were used to evaluate the combined association of tagSNPs on a gene level (i.e. all SNPs in a gene were analyzed together). Haplotypes blocks were determined in Haploview (51) using the Gabriel method (52); the resulting blocks were individually analyzed in R v.2.8.0 using a modified version of the haplo.stats package (53), which allows adjustment for age and sex. All haplotypes with more than 5% frequency in the controls were included in the logistic regression models as a categorical variable; haplotypes with less than 5% frequency were grouped and included in the model as one variable. The most common haplotype among the controls was used as the referent group. Gene-level significance was determined using a score test of the haplotypes included in the model.
For PCA (54), we determined the number of principal components that explained at least 80% of the variance in adenoma risk and performed logistic regression using those major components, adjusted for age and sex. The gene-level significance was determined using a likelihood ratio test comparing a model that contained the principal components and one that did not.
Gene-gene interactions
We evaluated gene-gene interactions using two methods: 1) the Tukey 1df interaction test proposed by Chatterjee et al (55) and 2) logic regression (56). For the Chatterjee method, we performed the test of interaction in two ways: 1) by deriving scores for each parameter and then testing the maximum score against the normal distribution (i.e. asymptote-based testing); or 2) by permutation-testing. Logic regression is a model selection method that uses Boolean logic to find combinations of variables that predict the outcome variable (56). For the purposes of these comparisons, we ran models of various sizes to compare the fit and we also have performed Monte Carlo logic regression (57) to obtain a list of SNPs that show up frequently in the best fitting model, suggesting that these SNPs might be important predictors of disease risk. All gene-gene interaction analyses were conducted in R (version 2.8.0).
Results
Characteristics of the study population have been described previously (4, 43, 58) and are shown for the genotyped subset in Table 1 (n= 483 adenoma cases and 582 polyp-free controls. Briefly, the study population was mostly Caucasian (97%); adenoma cases tended to be older than controls and more likely to be male. Regular aspirin or other NSAID use was somewhat more common among controls than among adenoma cases (aspirin: OR 0.63, 95% CI 0.44-0.90; other NSAIDs: OR 0.50, 95% CI 0.31-0.82) (4). The quality control results for each SNP are shown in Supplemental Table S1.
Table 1.
Characteristics of the study populationa
| Adenomas N=483 Mean (SD) |
Controls N=582 Mean (SD) |
p-value |
|
|---|---|---|---|
| Age (years) | 58.0 (9.6) | 52.9 (11.0) | <0.0001 |
| Caloric Intake (kcal/day) | 2110.2 (766.9) | 2019.2 (708.9) | 0.05 |
| Alcohol Intake (gm/day) | 10.5 (17.0) | 6.6 (13.4) | <0.0001 |
| Dietary Fiber Intake (gm/day) | 21.9 (9.5) | 21.7 (9.6) | 0.79 |
| N (%) |
N (%) |
|
|
|---|---|---|---|
| Location of largest adenoma | |||
| Proximal | 102 (21.3) | NA | |
| Distal | 298 (62.2) | NA | |
| Rectal | 79 (16.5) | NA | NA |
| Sex | |||
| Male | 304 (62.9) | 229 (39.4) | |
| Female | 179 (37.1) | 353 (60.6) | <0.0001 |
| Regular Use of Aspirin or NSAIDs | |||
| Yes | 174 (36.0) | 258 (44.3) | |
| No | 309 (64.0) | 324 (55.7) | 0.006 |
| Smoking (pack-years) | |||
| 0 | 164 (34.7) | 278 (49.0) | |
| 1-25 | 148 (31.3) | 172 (30.3) | |
| >25 | 161 (34.0) | 117 (20.6) | <0.0001 |
| BMI | |||
| Normal/Underweight | 157 (33.2) | 227 (39.9) | |
| Overweight (25-29.9) | 202 (42.7) | 214 (37.6) | |
| Obese (30+) | 114 (24.1) | 128 (22.5) | 0.08 |
| Fish intake (servings/week) | |||
| <1 | 111 (23.2) | 123 (21.8) | |
| 1-2 | 237 (49.6) | 298 (52.7) | |
| >2 | 130 (27.2) | 144 (25.5) | 0.60 |
| Post-menopausal Hormone Use b | |||
| No | 103 (58.2) | 161 (45.6) | |
| Yes | 70 (39.6) | 181 (51.3) | |
| Not sure | 4 (2.3) | 11 (3.1) | 0.02 |
Numbers may not total to 1.00 due to rounding and missing values
Females only
Main SNP associations
Associations between risk of adenoma and tagSNPs in PGES, EP2, EP4, and PGDH are shown in Tables 2 and 3. We observed a marginally significant association with EP2 4950G>A (ORGA or AA vs. GG: 0.71; 95% CI: 0.52-0.99). There were no individuals with the variant genotype in the candidate SNP EP4 Thr176Ile in the study population, so data for that polymorphism are not shown. There was no association with the candidate SNP EP4 Val294Ile (Table 2). The heterozygous genotype for EP4 -1307G>A was associated with a 40% increase in adenoma risk compared to the most common genotype (ORGA vs. GG: 1.42; 95% CI: 1.06-1.91). An increase in risk was also observed for the homozygous variant genotype group; however, this was not statistically significant (Table 2).
Table 2.
Genetic variation in PGES, EP2, and EP4 and risk of colorectal adenomaa
| Gene | SNP | Genotypeb | Cases | Controls | OR | 95%CI | pc | p-trend |
|---|---|---|---|---|---|---|---|---|
| PGES | −1254T>C | TT | 435 | 526 | 1.00 | – | ||
| TC or CC | 40 | 40 | 1.28 | 0.79-2.07 | 0.32 | NA | ||
| −664A>T | AA | 397 | 468 | 1.00 | – | |||
| AT or TT | 86 | 114 | 0.94 | 0.67-1.30 | 0.69 | NA | ||
| 211A>G | AA | 313 | 381 | 1.00 | – | |||
| AG | 147 | 175 | 1.10 | 0.83-1.45 | ||||
| GG | 22 | 21 | 1.23 | 0.64-2.36 | 0.70 | 0.40 | ||
| 1152G>A | GG | 429 | 510 | 1.00 | – | |||
| GA or AA | 54 | 72 | 0.92 | 0.62-1.36 | 0.66 | NA | ||
| 3006G>A | GG | 459 | 546 | 1.00 | – | |||
| GA or AA | 24 | 36 | 0.86 | 0.49-1.51 | 0.60 | NA | ||
| 13425A>C | AA | 435 | 526 | 1.00 | – | |||
| |
|
AC or CC |
40 |
40 |
1.28
|
0.79-2.07 |
0.40 |
NA |
| EP2 | −1722A>G | AA | 171 | 222 | 1.00 | – | ||
| AG | 234 | 269 | 1.22 | 0.92-1.62 | ||||
| GG | 78 | 89 | 1.20 | 0.81-1.77 | 0.35 | 0.23 | ||
| −967G>A | GG | 436 | 531 | 1.00 | – | |||
| GA or AA | 47 | 51 | 1.14 | 0.74-1.78 | 0.55 | NA | ||
| −616G>C | GG | 367 | 444 | 1.00 | – | |||
| GC or CC | 116 | 137 | 1.01 | 0.75-1.36 | 0.96 | NA | ||
| 1690G>A | GG | 448 | 539 | 1.00 | – | |||
| GA or AA | 34 | 43 | 1.05 | 0.64-1.73 | 0.83 | NA | ||
| 4950G>A | GG | 400 | 448 | 1.00 | – | |||
| GA or AA | 83 | 134 | 0.71 | 0.52-0.99 | 0.04 | NA | ||
| 9814C>A | CC | 288 | 349 | 1.00 | – | |||
| CA | 159 | 197 | 1.00 | 0.76-1.32 | ||||
| AA | 36 | 35 | 1.10 | 0.66-1.85 | 0.93 | 0.80 | ||
| 12010G>A | GG | 342 | 406 | 1.00 | – | |||
| AG | 122 | 157 | 0.96 | 0.72-1.29 | ||||
| AA | 19 | 19 | 1.03 | 0.52-2.05 | 0.96 | 0.89 | ||
| 15674G>A | GG | 311 | 384 | 1.00 | – | |||
| AG | 145 | 177 | 1.01 | 0.76-1.33 | ||||
| |
|
AA |
27 |
21 |
1.41
|
0.76-2.62 |
0.55 |
0.48 |
| EP4 | Val294Ile | Val/Val | 476 | 565 | 1.00 | – | ||
| Val/Ile | 20 | 25 | 0.93 | 0.49-1.75 | 0.82 | NA | ||
| −1529G>A | GG | 186 | 238 | 1.00 | – | |||
| GA | 218 | 263 | 0.91 | 0.69-1.20 | ||||
| AA | 79 | 79 | 0.81 | 0.55-1.19 | 0.54 | 0.27 | ||
| −1408G>A | GG | 251 | 326 | 1.00 | – | |||
| GA | 199 | 223 | 0.87 | 0.66-1.13 | ||||
| AA | 32 | 31 | 0.84 | 0.49-1.45 | 0.53 | 0.28 | ||
| −1307G>A | GG | 169 | 166 | 1.00 | – | |||
| GA | 211 | 293 | 1.42 | 1.06-1.91 | ||||
| AA | 103 | 123 | 1.19 | 0.83-1.70 | 0.06 | 0.23 | ||
| −132C>G | CC | 401 | 475 | 1.00 | – | |||
| CG or GG | 82 | 106 | 0.89 | 0.64-1.24 | 0.49 | NA | ||
| 1455A>G | AA | 175 | 225 | 1.00 | – | |||
| AG | 203 | 254 | 0.90 | 0.68-1.20 | ||||
| GG | 86 | 88 | 0.81 | 0.56-1.19 | 0.54 | 0.27 | ||
| 2472A>G | AA | 137 | 182 | 1.00 | – | |||
| AG | 222 | 280 | 0.92 | 0.68-1.25 | ||||
| GG | 122 | 120 | 0.74 | 0.52-1.06 | 0.23 | 0.11 | ||
| 6374G>A | GG | 278 | 351 | 1.00 | – | |||
| GA | 181 | 199 | 0.86 | 0.66-1.13 | ||||
| AA | 24 | 32 | 1.13 | 0.63-2.03 | 0.46 | 0.63 | ||
| 8907G>A | GG | 219 | 278 | 1.00 | – | |||
| GA | 206 | 240 | 0.90 | 0.69-1.18 | ||||
| AA | 58 | 64 | 0.89 | 0.58-1.36 | 0.71 | 0.45 | ||
| 11851G>A | GG | 379 | 454 | 1.00 | – | |||
| GA or AA | 103 | 128 | 1.06 | 0.78-1.45 | 0.71 | NA | ||
| 13877A>C | AA | 257 | 315 | 1.00 | – | |||
| AC | 196 | 223 | 0.90 | 0.69-1.18 | ||||
| CC | 29 | 43 | 1.22 | 0.72-2.06 | 0.49 | 1.00 | ||
| 13981A>G | AA | 178 | 195 | 1.00 | – | |||
| AG | 220 | 289 | 1.27 | 0.95-1.68 | ||||
| GG | 85 | 98 | 1.05 | 0.72-1.53 | 0.23 | 0.50 |
Adjusted for age and sex.
The homozygous variant and heterozygous genotypes are grouped together if <10 cases or <10 controls were of the homozygous variant genotype.
Likelihood ratio test of a model containing dummy variables for each SNP + age and sex vs. a model containing only age and sex.
Table 3.
Genetic variation in PGDH and risk of colorectal adenomaa
| SNP | Genotypeb | Cases | Controls | OR | 95%CI | pc | p-trend |
|---|---|---|---|---|---|---|---|
| −1868G>A | GG | 359 | 437 | 1.00 | – | ||
| GA or AA | 124 | 145 | 1.10 | 0.82-1.48 | 0.51 | NA | |
| −1635G>A | GG | 121 | 164 | 1.00 | – | ||
| GA | 238 | 288 | 1.22 | 0.89-1.66 | |||
| AA | 122 | 128 | 1.31 | 0.91-1.88 | 0.30 | 0.14 | |
| 343C>A | CC | 412 | 511 | 1.00 | – | ||
| CA or AA | 70 | 71 | 1.31 | 0.90-1.91 | 0.16 | NA | |
| Gln52Gln | GG | 171 | 192 | 1.00 | – | ||
| GA | 225 | 283 | 0.97 | 0.73-1.29 | |||
| AA | 84 | 104 | 1.00 | 0.68-1.45 | 0.98 | 0.94 | |
| 782A>G | AA | 338 | 384 | 1.00 | – | ||
| AG | 129 | 187 | 0.76 | 0.57-1.01 | |||
| GG | 15 | 10 | 1.51 | 0.64-3.56 | 0.09 | 0.29 | |
| 1113G>A | GG | 374 | 464 | 1.00 | – | ||
| GA or AA | 109 | 118 | 1.14 | 0.84-1.56 | 0.41 | NA | |
| 1543G>A | GG | 137 | 155 | 1.00 | – | ||
| GA | 214 | 287 | 0.78 | 0.58-1.07 | |||
| AA | 131 | 139 | 0.95 | 0.67-1.35 | 0.24 | 0.73 | |
| 3580G>A | GG | 297 | 337 | 1.00 | – | ||
| GA | 161 | 220 | 0.84 | 0.64-1.10 | |||
| AA | 25 | 25 | 1.15 | 0.62-.13 | 0.37 | 0.53 | |
| 9442A>G | AA | 223 | 298 | 1.00 | – | ||
| AG | 210 | 217 | 1.21 | 0.92-1.58 | |||
| GG | 50 | 66 | 0.95 | 0.62-1.46 | 0.32 | 0.63 | |
| 9691C>A | CC | 247 | 286 | 1.00 | – | ||
| CA | 187 | 249 | 0.89 | 0.68-1.17 | |||
| AA | 48 | 47 | 1.29 | 0.81-2.05 | 0.28 | 0.77 | |
| 12184A/- | AA | 375 | 427 | 1.00 | – | ||
| A/- or -/- | 110 | 136 | 0.94 | 0.69-1.28 | 0.70 | NA | |
| 13316A>C | AA | 221 | 255 | 1.00 | – | ||
| AC | 208 | 259 | 0.95 | 0.72-1.24 | |||
| CC | 53 | 68 | 0.94 | 0.61-1.44 | 0.91 | 0.69 | |
| 13796C>T | CC | 273 | 322 | 1.00 | – | ||
| CT | 177 | 217 | 0.99 | 0.76-1.30 | |||
| TT | 33 | 43 | 0.90 | 0.54-1.50 | 0.92 | 0.77 | |
| 16362G>A | GG | 282 | 363 | 1.00 | – | ||
| GA | 168 | 182 | 1.16 | 0.88-1.53 | |||
| AA | 33 | 36 | 1.06 | 0.63-1.78 | 0.57 | 0.43 | |
| 17450A>G | AA | 394 | 445 | 1.00 | – | ||
| AG or GG | 88 | 135 | 0.79 | 0.58-1.09 | 0.15 | NA | |
| 18349G>A | GG | 379 | 452 | 1.00 | – | ||
| GA or AA | 104 | 129 | 0.96 | 0.71-1.31 | 0.81 | NA | |
| 19433G>A | GG | 243 | 275 | 1.00 | – | ||
| GA | 201 | 259 | 0.91 | 0.70-1.19 | |||
| AA | 37 | 48 | 0.88 | 0.54-1.44 | 0.76 | 0.47 | |
| 19979A>T | AA | 338 | 378 | 1.00 | – | ||
| AT | 135 | 166 | 0.96 | 0.72-1.28 | |||
| TT | 14 | 19 | 0.86 | 0.40-1.84 | 0.90 | 0.67 | |
| 20518A>G | AA | 160 | 208 | 1.00 | – | ||
| AG | 228 | 267 | 1.08 | 0.81-1.44 | |||
| GG | 95 | 107 | 1.12 | 0.78-1.62 | 0.79 | 0.50 | |
| 23196G>A | AA | 262 | 304 | 1.00 | – | ||
| AG | 180 | 243 | 0.87 | 0.67-1.14 | |||
| GG | 41 | 33 | 1.39 | 0.83-2.34 | 0.19 | 0.82 | |
| 26646G>A | GG | 320 | 381 | 1.00 | – | ||
| GA | 141 | 175 | 0.98 | 0.74-1.30 | |||
| AA | 22 | 26 | 1.09 | 0.58-2.06 | 0.95 | 0.94 | |
| 31659A>G | AA | 356 | 420 | 1.00 | – | ||
| AG | 111 | 144 | 0.90 | 0.67-1.22 | |||
| GG | 14 | 18 | 0.92 | 0.43-1.97 | 0.79 | 0.52 |
Adjusted for age and sex.
The homozygous variant and heterozygous genotypes are grouped together if <10 cases or <10 controls were of the homozygous variant genotype.
Likelihood ratio test of a model containing dummy variables for each SNP + age and sex vs. a model containing only age and sex.
For PGDH, we observed two associations of marginal statistical significance (Table 3). The heterozygous genotype of 782A>G was associated with decreased adenoma risk compared to wildtype (ORAG vs. AA: 0.76; 95% CI: 0.57-1.01) although the homozygous variant genotype was not associated with polyp risk. Similarly, the heterozygous genotype of 1543G>A was associated with a decreased risk of marginal statistical significance, compared to wildtype (ORGA vs. GG: 0.78; 95% CI: 0.58-1.07), whereas the homozygous variant genotype was not associated with altered risk. No associations were observed for tagSNPs in PGES.
Fish interactions
We observed several interactions between fish intake and tagSNPs in PGES, EP4, and PGDH. In PGES, a statistically significant interaction with fish intake was observed for PGES -664A>T (Table 4 and Figure 1). Fish intake was not associated with adenoma risk among those with the wildtype genotype (OR1-2/wk v. <1/wk: 0.93; 95% CI 0.65-1.34: OR >2/wk v. <1/wk: 1.31; 95% CI: 0.83-2.04); however, among those with the variant genotypes, higher fish intake was associated with decreased adenoma risk (OR>2/wk v. <1/wk: 0.56; 95% CI: 0.28-1.13; p-trend interaction=0.01).
Table 4.
Genetic variation in PGES, EP4, and PGDH, weekly fish intake, and risk of adenomaa
| Fish intake |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <1 serving per week |
1-2 servings per week |
>2 servings per week |
|||||||||||
| Gene | SNP | Cases |
Controls |
OR |
95% CI |
Cases |
Controls |
OR |
95% CI |
Cases |
Controls |
OR |
95% CI |
| PGES | −664A>T | ||||||||||||
| AA | 90 | 106 | 1.00 | – | 190 | 239 | 1.06 | 0.72-1.55 | 113 | 108 | 1.31 | 0.83-2.04 | |
| AT or TT | 21 | 17 | 1.74 | 0.81-3.76 | 47 | 59 | 1.01 | 0.59-1.71 | 17 | 36 | 0.56 | 0.28-1.13 | |
|
global p-interaction=0.02
SNP trendb p-interaction=0.01 |
|||||||||||||
| EP4 | Val294Ile | ||||||||||||
| Val/Val | 110 | 116 | 1.00 | – | 235 | 288 | 0.90 | 0.63-1.27 | 126 | 45 | 0.87 | 0.58-1.31 | |
| Val/Ile or Ile/Ile | 2 | 8 | 0.27 | 0.05-1.41 | 10 | 15 | 0.80 | 0.32-2.00 | 8 | 1 | 7.75 | 0.81-78.83 | |
|
global p-interaction=0.02
SNP trendb p-interaction=0.01 |
|||||||||||||
| PGDH | 19979A>T | ||||||||||||
| AA | 78 | 83 | 1.00 | – | 156 | 196 | 0.95 | 0.63-1.45 | 99 | 90 | 1.26 | 0.78-2.05 | |
| AT or TT | 30 | 38 | 1.06 | 0.56-2.00 | 86 | 87 | 1.25 | 0.78-2.03 | 33 | 53 | 0.64 | 0.35-1.18 | |
|
global p-interaction=0.03
SNP trendb p-interaction=0.03 |
|||||||||||||
| 31659A>G | |||||||||||||
| AA | 76 | 99 | 1.00 | – | 171 | 209* | 1.15 | 0.77-1.71 | 105 | 99 | 1.35 | 0.85-2.15 | |
| AG or GG | 34 | 24 | 1.86 | 0.96-3.61 | 65 | 89 | 1.04 | 0.64-1.70 | 25 | 45 | 0.75 | 0.40-1.44 | |
|
global p-interaction=0.03
SNP trendb p-interaction=0.02 |
|||||||||||||
Adjusted for age, BMI, total intakes of energy, alcohol, and fiber, sex, post-menopausal hormone use (women only), and smoking (pack-years)
Test for differences in fish intake trend among strata of genotype
Figure 1.
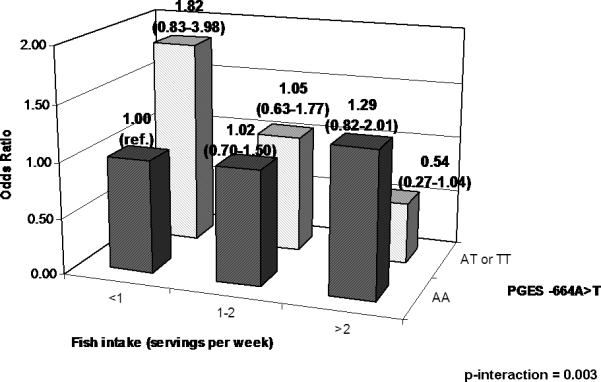
PGES -664T>A, weekly fish intake, and risk of adenoma.
For the candidate polymorphisms EP4 Val294Ile, among the Val/Val genotype group, increasing fish intake was associated with a possibly decreased risk of adenoma (OR>2/wk v. <1/wk: 0.87; 95% CI: 0.58-1.31), whereas among those with at least one Ile allele, higher fish intake was associated with substantially increased adenoma risk (p-trend interaction=0.01; Table 4). However, the EP4 Val294Ile is a rare polymorphism and the numbers of subjects with a variant allele were very small (n= 44), indicating that this interaction requires confirmation in a larger population.
For PGDH 31659T>C (3’ UTR), no association with fish intake was observed among those with the common genotype. Those with the heterozygous or homozygous variant genotypes had a reduced adenoma risk with increasing weekly fish intake (p-trend interaction=0.02; Table 4). No interactions with fish intake were observed for EP2. None of the observed interactions remained statistically significant when adjusted for multiple comparisons.
NSAID interactions
We also evaluated NSAID interactions, but we observed no strong interactions. For two SNPs (EP2 9814C>A [intron 1] and PGDH 343C>A [intron 1]), there were marginal or statistically significant interactions (see Supplemental Table S2). We analyzed NSAID interactions using the wildtype non-NSAID users as the referent group. For the EP2 SNP, the risk reduction associated with regular NSAID use was more pronounced among those with at least one of the more common alleles. However, for PGDH 343C>A among NSAID non-users, the variant genotypes were associated with increased risk (ORCA or AA vs. CC: 1.98; 95% CI: 1.18-3.33); among regular NSAID users, decreased adenoma risk was observed for all genotypes (p-interaction=0.03). No interactions with NSAID use were observed for SNPs in PGES or EP4. None of the interactions remained statistically significant when adjusted for multiple comparisons.
Gene-level analyses
Haplotype and PCA analysis results are shown in Supplemental Tables S3 and S4, respectively. There were no statistically significant associations observed for any of the four genes.
Gene-gene interaction analyses
The results of Chatterjee gene-gene interaction testing are shown in Supplemental Table S5. No statistically significant gene-gene interactions were detected. The results of logic regression modeling are shown in Supplemental Tables S6-S8. In Supplemental Table S6, the variables that were included in models of various sizes are shown. The most commonly occurring pairs of SNPs in 100,000 iterations of Monte Carlo Logic Regression are shown in Supplemental Table S7. None of the pairs of SNPs occur with ≥1% frequency in 100,000 iterations, indicating that important SNP-SNP interactions are unlikely in these genes. The most commonly occurring SNPs are shown in Supplemental Table S8. The most commonly occurring single SNPs in 100,000 iterations of Monte Carlo Logic Regression were EP4 -1307G>A, which occurred with a frequency of 13.3% and EP2 4950G>A, which occurred with a frequency of 7.3%; these were the only SNPs that occurred with a frequency greater than 5%. Our finding that no single SNP was selected for inclusion in more than 15% of fitted models indicates that none of the SNPs we genotyped in these four genes is strongly related to risk of adenoma. This reflects the main SNP association findings, in which we detected very few statistically significant main associations.
Discussion
We investigated genetic variability in a biochemical pathway that has been unequivocally linked to colorectal carcinogenesis and has not yet been previously studied in the context of colorectal adenoma. Our results indicate that genetic variability related to PGE2 signaling is generally not related to risk of colorectal adenoma. However, some data from this study suggest that genetic variability in PGES, EP2, EP4, or PGDH may alter the anti-inflammatory effects of specific fatty acids or NSAID use. We hypothesized that NSAID use or high fish intake would be associated with lower risk of colorectal neoplasia, predominantly among those with genetic variants that are likely to increase PGE2 production or signaling. We observed a statistically significant association for a tagSNP in EP2 and in EP4, but no other associations between tagSNPs in PGES or PGDH and risk of colorectal adenomas. Further, we observed several statistically significant or marginally significant interactions with NSAID use or fish intake, indicating that the consequences of genetic variability in prostaglandin synthesis may be further modified by underlying dietary or pharmacologic pro- or anti-inflammatory profiles. For the tagSNPs that were associated with adenoma risk or interacted with inflammatory exposures, we investigated their LD with any potentially functional SNPs (Supplemental Table S9) by evaluating SNP locations, regulatory regions, and conservation across species. However no SNPs with clear functional significance were identified.
To our knowledge, no previous studies have examined associations between genetic variants in PGES, EP2, EP4, and PGDH and risk of adenoma. However, although our findings require confirmation in additional studies, our results suggest similar patterns to previous reports on interactions between polymorphisms in COX-1, COX-2, and PGIS, in which the inverse associations with NSAID use or fish intake were limited to certain genotype groups (37-40). Two EP2 tagSNPs included in this study, -616G>C and -166G>A, were associated with aspirin-intolerant asthma in Korean individuals (59). Although that study was small and 77 polymorphisms were tested without correction for multiple testing, those results indicate that polymorphisms in EP2 may be a predictor of who is likely to benefit from (or even experience adverse reactions from) aspirin. Two EP2 SNPs included in the present study (-1722A>G and -616G>C) were not associated with hypertension in a study of 266 Japanese subjects (60) and also were not associated with adenoma risk in the present study. In a recent whole genome association study of Crohn's disease, an inflammatory bowel condition, SNPs upstream of EP4 linked to altered EP4 expression were associated with increased risk (61). However, these SNPs were not included in the present study because they are over 200KB upstream of EP4. In a study of lymphoma risk, three polymorphisms in PGES that were not included in this study (but one of which was tagged in our study by another SNP), were not associated with risk of lymphoma (62).
This study has several limitations. First, a large number of statistical tests were performed, increasing the likelihood of false positives. Although we corrected for multiple testing, we chose to report the uncorrected p-values. However, given both the importance of PGE2 for colorectal carcinogenesis and the fact that no one has yet reported on potential associations with tagSNPs in these genes for colorectal neoplasia risk, we decided to report all our findings for possible replication by other groups. Conversely, due to the relatively small number of study subjects, it is possible that true associations or gene-environment interactions were missed.
Intakes of specific types of fish were not measured in this study. Because fish vary in their content of n-3 polyunsaturated fatty acids, total fish intake is not a precise measure of n-3 polyunsaturated fatty acid intake. Further, we did not have a quantitative measure of dietary n-6 polyunsaturated fatty acids nor of fish oil or other fatty acid-containing supplements. This limited our power to detect true interactions between dietary fatty acids and genetic variability related to PGE2 signaling on risk of colorectal adenoma.
This study has several strengths. First, all controls who participated in this study underwent colonoscopy prior to enrollment. Because colorectal polyps are common in older adults, an unscreened population-based control group would include undiagnosed adenoma cases, resulting in disease misclassification and an attenuation of any true associations. In addition, by using tagSNPs as well as rare non-synonymous SNPs to investigate associations between these target genes and risk of colorectal adenoma, we achieved comprehensive coverage of these genes, increasing the likelihood of observing any existing true association. The prostaglandin synthesis pathway is an important one for colorectal carcinogenesis and our complete coverage of the genes and their 5’ and 3’ regions, as well as some lifestyle factors that are likely to influence prostaglandin synthesis, provides a more complete investigation of this pathway.
In summary, the results from the present study provide some limited evidence that genetic variability in genes related to PGE2 levels and signaling may be modifiers of the relationship between NSAID use or fish intake and risk of colorectal adenoma.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Roberd Bostick and Lisa Fosdick for their contributions to the initial establishment of this study.
Sources of financial support: Grants R01 CA114467. R01 CA112516, and R03 CA123577
References
- 1.Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. New Engl J Med. 2003;348:891–9. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 2.Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. New Engl J Med. 2003;348:883–90. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 3.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–13. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 4.Bigler J, Whitton J, Lampe JW, Fosdick L, Bostick RM, Potter JD. CYP2C9 and UGT1A6 genotypes modulate the protective effect of aspirin on colon adenoma risk. Cancer Res. 2001;61:3566–9. [PubMed] [Google Scholar]
- 5.Rigas B, Goldman IS, Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med. 1993;122:518–23. [PubMed] [Google Scholar]
- 6.Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998;58:362–6. [PubMed] [Google Scholar]
- 7.Qiao L, Kozoni V, Tsioulias GJ, et al. Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochim Biophys Acta. 1995;1258:215–23. doi: 10.1016/0005-2760(95)00100-q. [DOI] [PubMed] [Google Scholar]
- 8.Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276:18075–81. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Wang H, Shi Q, et al. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6:285–95. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Sonoshita M, Takaku K, Sasaki N, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–51. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 11.Yang L, Yamagata N, Yadav R, et al. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111:727–35. doi: 10.1172/JCI16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mutoh M, Watanabe K, Kitamura T, et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 2002;62:28–32. [PubMed] [Google Scholar]
- 13.Kamei D, Murakami M, Nakatani Y, Ishikawa Y, Ishii T, Kudo I. Potential role of microsomal prostaglandin E synthase-1 in tumorigenesis. J Biol Chem. 2003;278:19396–405. doi: 10.1074/jbc.M213290200. [DOI] [PubMed] [Google Scholar]
- 14.Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68-69:383–99. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimatsu K, Golijanin D, Paty PB, et al. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin Cancer Res. 2001;7:3971–6. [PubMed] [Google Scholar]
- 16.Murakami M, Naraba H, Tanioka T, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–92. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 17.Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–53. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 18.Gustafsson A, Hansson E, Kressner U, et al. EP1-4 subtype, COX and PPAR gamma receptor expression in colorectal cancer in prediction of disease-specific mortality. Int J Cancer. 2007;121:232–40. doi: 10.1002/ijc.22582. [DOI] [PubMed] [Google Scholar]
- 19.Seno H, Oshima M, Ishikawa TO, et al. Cyclooxygenase 2- and prostaglandin E(2) receptor EP(2)-dependent angiogenesis in Apc(Delta716) mouse intestinal polyps. Cancer Res. 2002;62:506–11. [PubMed] [Google Scholar]
- 20.Pozzi A, Yan X, Macias-Perez I, et al. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem. 2004;279:29797–804. doi: 10.1074/jbc.M313989200. [DOI] [PubMed] [Google Scholar]
- 21.Kabashima K, Saji T, Murata T, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–93. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang GL, Nieves A, Im WB, Old DW, Dinh DT, Wheeler L. The prevention of colitis by E Prostanoid receptor 4 agonist through enhancement of epithelium survival and regeneration. J Pharmacol Exp Ther. 2007;320:22–8. doi: 10.1124/jpet.106.111146. [DOI] [PubMed] [Google Scholar]
- 23.Ensor CM, Tai HH. 15-Hydroxyprostaglandin dehydrogenase. J Lipid Mediat Cell Signal. 1995;12:313–9. doi: 10.1016/0929-7855(95)00040-w. [DOI] [PubMed] [Google Scholar]
- 24.Backlund MG, Mann JR, Holla VR, et al. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005;280:3217–23. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myung SJ, Rerko RM, Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:12098–102. doi: 10.1073/pnas.0603235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan M, Rerko RM, Platzer P, et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci U S A. 2004;101:17468–73. doi: 10.1073/pnas.0406142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otani T, Yamaguchi K, Scherl E, et al. Levels of NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase are reduced in inflammatory bowel disease: evidence for involvement of TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2006;290:G361–8. doi: 10.1152/ajpgi.00348.2005. [DOI] [PubMed] [Google Scholar]
- 28.Anderson GD, Hauser SD, McGarity KL, Bremer ME, Isakson PC, Gregory SA. Selective inhibition of cyclooxygenase (COX)-2 reverses inflammation and expression of COX-2 and interleukin 6 in rat adjuvant arthritis. J Clin Invest. 1996;97:2672–9. doi: 10.1172/JCI118717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giagoudakis G, Markantonis SL. Relationships between the concentrations of prostaglandins and the nonsteroidal antiinflammatory drugs indomethacin, diclofenac, and ibuprofen. Pharmacotherapy. 2005;25:18–25. doi: 10.1592/phco.25.1.18.55618. [DOI] [PubMed] [Google Scholar]
- 30.Winde G, Schmid KW, Schlegel W, Fischer R, Osswald H, Bunte H. Complete reversion and prevention of rectal adenomas in colectomized patients with familial adenomatous polyposis by rectal low-dose sulindac maintenance treatment. Advantages of a low-dose nonsteroidal anti-inflammatory drug regimen in reversing adenomas exceeding 33 months. Dis Colon Rectum. 1995;38:813–30. doi: 10.1007/BF02049838. [DOI] [PubMed] [Google Scholar]
- 31.Frenkian M, Segond N, Pidoux E, Cohen R, Jullienne A. Indomethacin, a COX inhibitor, enhances 15-PGDH and decreases human tumoral C cells proliferation. Prostaglandins. 2001;65:11–20. doi: 10.1016/s0090-6980(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 32.Quidville V, Segond N, Pidoux E, Cohen R, Jullienne A, Lausson S. Tumor growth inhibition by indomethacin in a mouse model of human medullary thyroid cancer: implication of cyclooxygenases and 15-hydroxyprostaglandin dehydrogenase. Endocrinology. 2004;145:2561–71. doi: 10.1210/en.2003-0915. [DOI] [PubMed] [Google Scholar]
- 33.Fujino H, Chen XB, Regan JW, Murayama T. Indomethacin decreases EP2 prostanoid receptor expression in colon cancer cells. Biochem Biophys Res Commun. 2007;359:568–73. doi: 10.1016/j.bbrc.2007.05.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.James MJ, Gibson RA, Cleland LG. Dietary polyunsaturated fatty acids and inflammatory mediator production. Am J Clin Nutr. 2000;71:343S–8S. doi: 10.1093/ajcn/71.1.343s. [DOI] [PubMed] [Google Scholar]
- 35.Rose DP, Connolly JM. Omega-3 fatty acids as cancer chemopreventive agents. Pharmacol Ther. 1999;83:217–44. doi: 10.1016/s0163-7258(99)00026-1. [DOI] [PubMed] [Google Scholar]
- 36.Roynette CE, Calder PC, Dupertuis YM, Pichard C. n-3 polyunsaturated fatty acids and colon cancer prevention. Clin Nutr. 2004;23:139–51. doi: 10.1016/j.clnu.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Ulrich CM, Bigler J, Sparks R, et al. Polymorphisms in PTGS1 (=COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2004;13:889–893. [PubMed] [Google Scholar]
- 38.Ulrich CM, Whitton J, Yu JH, et al. PTGS2 (COX-2) -765G > C promoter variant reduces risk of colorectal adenoma among nonusers of nonsteroidal anti-inflammatory drugs. Cancer Epidemiol Biomarkers Prev. 2005;14:616–9. doi: 10.1158/1055-9965.EPI-04-0510. [DOI] [PubMed] [Google Scholar]
- 39.Poole EM, Bigler J, Whitton J, Sibert JG, Potter JD, Ulrich CM. Prostacyclin synthase and arachidonate 5-lipoxygenase polymorphisms and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2006;15:502–8. doi: 10.1158/1055-9965.EPI-05-0804. [DOI] [PubMed] [Google Scholar]
- 40.Poole EM, Bigler J, Whitton J, et al. Genetic variability in prostaglandin synthesis, fish intake, and risk of colorectal polyps. Carcinogenesis. 2007 doi: 10.1093/carcin/bgm026. [DOI] [PubMed] [Google Scholar]
- 41.Bigler J, Sibert JG, Poole EM, Carlson CS, Potter JD, Ulrich CM. Polymorphisms predicted to alter function in prostaglandin E2 synthase and prostaglandin E2 receptors. Pharmacogenet Genomics. 2007;17:221–7. doi: 10.1097/FPC.0b013e3280119d50. [DOI] [PubMed] [Google Scholar]
- 42. UW-FHCRC Variation Discovery Resource.
- 43.Potter JD, Bostick RM, Grandits GA, et al. Hormone replacement therapy is associated with lower risk of adenomatous polyps of the large bowel: the Minnesota Cancer Prevention Research Unit Case-Control Study. Cancer Epidemiol Biomarkers Prev. 1996;5:779–84. [PubMed] [Google Scholar]
- 44.Ulrich CM, Kampman E, Bigler J, et al. Colorectal adenomas and the C677T MTHFR polymorphism: evidence for gene-environment interaction? Cancer Epidemiol Biomarkers Prev. 1999;8:659–68. [PubMed] [Google Scholar]
- 45.Willett WC, Sampson L, Stampfer MJ, et al. Reproducibility and validity of a semiquantitative food frequency questionnaire. Am J Epidemiol. 1985;122:51–65. doi: 10.1093/oxfordjournals.aje.a114086. [DOI] [PubMed] [Google Scholar]
- 46.Munger RG, Folsom AR, Kushi LH, Kaye SA, Sellers TA. Dietary assessment of older Iowa women with a food frequency questionnaire: nutrient intake, reproducibility, and comparison with 24-hour dietary recall interviews. Am J Epidemiol. 1992;136:192–200. doi: 10.1093/oxfordjournals.aje.a116485. [DOI] [PubMed] [Google Scholar]
- 47.Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol. 1992;135:1114–26. doi: 10.1093/oxfordjournals.aje.a116211. discussion 1127-36. [DOI] [PubMed] [Google Scholar]
- 48.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–20. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.SeattleSNPs Program for Genomic Applications. Genome Variation Server. 2008 2008. [Google Scholar]
- 50.Conneely KN, Boehnke M. So Many Correlated Tests, So Little Time! Rapid Adjustment of P Values for Multiple Correlated Tests. Am J Hum Genet. 2007:81. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 52.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 53.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–34. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gauderman WJ, Murcray C, Gilliland F, Conti DV. Testing association between disease and multiple SNPs in a candidate gene. Genet Epidemiol. 2007;31:383–95. doi: 10.1002/gepi.20219. [DOI] [PubMed] [Google Scholar]
- 55.Chatterjee N, Kalaylioglu Z, Moslehi R, Peters U, Wacholder S. Powerful multilocus tests of genetic association in the presence of gene-gene and gene-environment interactions. Am J Hum Genet. 2006;79:1002–16. doi: 10.1086/509704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kooperberg C, Ruczinski I, LeBlanc ML, Hsu L. Sequence analysis using logic regression. Genet Epidemiol. 2001;21(Suppl 1):S626–31. doi: 10.1002/gepi.2001.21.s1.s626. [DOI] [PubMed] [Google Scholar]
- 57.Kooperberg C, Ruczinski I. Identifying interacting SNPs using Monte Carlo logic regression. Genet Epidemiol. 2005;28:157–70. doi: 10.1002/gepi.20042. [DOI] [PubMed] [Google Scholar]
- 58.Morimoto LM, Newcomb PA, Ulrich CM, Bostick RM, Lais CJ, Potter JD. Risk factors for hyperplastic and adenomatous polyps: evidence for malignant potential? Cancer Epidemiol Biomarkers Prev. 2002;11:1012–8. [PubMed] [Google Scholar]
- 59.Kim SH, Kim YK, Park HW, et al. Association between polymorphisms in prostanoid receptor genes and aspirin-intolerant asthma. Pharmacogenet Genomics. 2007;17:295–304. doi: 10.1097/01.fpc.0000239977.61841.fe. [DOI] [PubMed] [Google Scholar]
- 60.Sato M, Nakayama T, Soma M, et al. Association between prostaglandin E2 receptor gene and essential hypertension. Prostaglandins Leukot Essent Fatty Acids. 2007;77:15–20. doi: 10.1016/j.plefa.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 61.Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoeft B, Becker N, Deeg E, Beckmann L, Nieters A. Joint effect between regular use of non-steroidal anti-inflammatory drugs, variants in inflammatory genes and risk of lymphoma. Cancer Causes Control. 2008;19:163–73. doi: 10.1007/s10552-007-9082-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.