

Summary
Notch signaling is essential for embryonic vascular development in mammals and other vertebrates. Here we show that mouse embryos with conditional activation of the Notch1 gene in endothelial cells (Notch1 gain of function embryos) exhibit defects in vascular remodeling, increased diameter of the dorsal aortae, and form arteriovenous malformations. Conversely, embryos with either constitutive or endothelial cell-specific Notch1 gene deletion also have vascular defects, but exhibit decreased diameter of the dorsal aortae and form arteriovenous malformations distinctly different from the Notch1 gain of function mutants. Surprisingly, embryos homozygous for mutations of the ephrinB/EphB pathway genes Efnb2 and Ephb4 exhibit vascular defects and arteriovenous malformations that phenocopy the Notch1 gain of function mutants. These results suggest that formation of arteriovenous malformations in Notch1 gain of function mutants and ephrinB/EphB pathway loss of function mutant embryos occurs by different mechanisms.
Keywords: angiogenesis, arteriovenous malformation, EphrinB2, EphB4, Notch signaling pathway, vascular morphogenesis
The Notch signaling pathway is an evolutionarily-conserved intercellular signaling mechanism, and mutations in Notch pathway components disrupt embryonic development in diverse multicellular organisms (Bray, 2006; Ehebauer et al., 2006). In mammals, four genes encode Notch family receptors (Notch1-Notch4), while five genes encode Notch ligands (Jag1, Jag2, Dll1, Dll3, Dll4). The signal induced by ligand binding is transmitted intracellularly by a process involving proteolytic cleavage of the receptor, followed by nuclear translocation of the intracellular domain of the Notch family protein (Notch-IC). Once in the nucleus, Notch-IC forms a complex with the RBPJ protein, a sequence-specific DNA binding protein that is the primary transcriptional mediator of Notch signaling. The Notch-IC/RBPJ complex then activates transcription of downstream target genes.
The Notch pathway is one of several conserved signaling pathways whose function is critically important for embryonic vascular development in mammals and other vertebrates (Gridley, 2007; Hofmann and Iruela-Arispe, 2007; Phng and Gerhardt, 2009; Roca and Adams, 2007). For example, mouse embryos with endothelial cell-specific deletion of the Rbpj gene (Krebs et al., 2004) or with loss of function mutations of the Dll4 gene (Duarte et al., 2004; Gale et al., 2004; Krebs et al., 2004) exhibit vascular remodeling defects that are accompanied by the presence of arteriovenous malformations, the aberrant fusion of arteries and veins without an intervening capillary bed.
The ephrinB/EphB receptor pathway is also essential for vascular development. While loss of function mutants of the ephrinB2 (Efnb2) (Wang et al., 1998) and EphB4 (Ephb4) (Gerety et al., 1999) genes exhibit embryonic vascular remodeling defects very similar to those exhibited by Notch pathway mutants, it had not been determined whether these ephrin pathway mutants form arteriovenous malformations. Similarly, it had been shown that ectopic activation of the Notch4 gene (Notch4GOF, for Notch4 gain of function) in endothelial cells and their progenitors leads to embryonic vascular defects (Uyttendaele et al., 2001). However, it had not been established whether ectopic activation of other Notch pathway receptors such as Notch1 could lead to similar vascular remodeling defects. Here we report that ectopic Notch1 activation in endothelial cells (Notch1GOF) leads to embryonic vascular remodeling defects and arteriovenous malformations that are distinct from those exhibited by Notch pathway loss of function mutants. We also show that Efnb2 and Ephb4 loss of function mutants form arteriovenous malformations, and that these arteriovenous malformations phenocopy the arteriovenous malformations present in Notch1GOF embryos.
Expression of the intracellular domain of the NOTCH4 protein in endothelial cell precursors under the control of the Vegfr2 promoter has previously been shown to cause lethal embryonic vascular remodeling defects (Uyttendaele et al., 2001). To determine whether endothelial cell-specific activation of the Notch1 gene would cause similar vascular defects, we crossed Tek-Cre (also known as Tie2-Cre) mice to Notch1-ICcond mice, a transgenic mouse line in which expression of the NOTCH1 intracellular domain (NOTCH1-IC) is activated by Cre recombinase. We compared the Tek-Cre; Notch1-ICcond (referred to hereafter as Notch1GOF, for Notch1 gain of function) embryos to mutant embryos with either constitutive (Notch1-/-) or endothelial cell-specific (Tek-Cre; Notch1flox/-) loss of function mutations in the Notch1 gene.
Notch1GOF embryos exhibited phenotypes characteristic of defective vascular remodeling. Visualization of the vascular network by whole mount immunostaining with a monoclonal antibody to platelet endothelial cell adhesion molecule-1 (PECAM-1) demonstrated that, similarly to Notch1-/- (Figure 1b, f), Tek-Cre; Notch1flox/- (Figure 1c, g), and other Notch pathway loss of function mutant embryos (Krebs et al., 2004; Krebs et al., 2000), Notch1GOF embryos (Figure 1d, h) exhibited vascular defects in the embryo proper and failed to remodel the primary vascular plexus of the extraembryonic yolk sac to form the large and small vessels of the mature wild type yolk sac (Figure1 a, e). Histological analyses of the PECAM-1-stained embryos demonstrated that while the paired dorsal aortae were reduced in diameter or atretic in the two Notch1 loss of function mutant embryos (Figure 1j, k), the diameter of the dorsal aortae was increased in Notch1GOF embryos (Figure1l).
Figure 1. Vascular defects in Notch1-/-, Tek-Cre; Notch1flox/- and Notch1GOF mutant embryos.
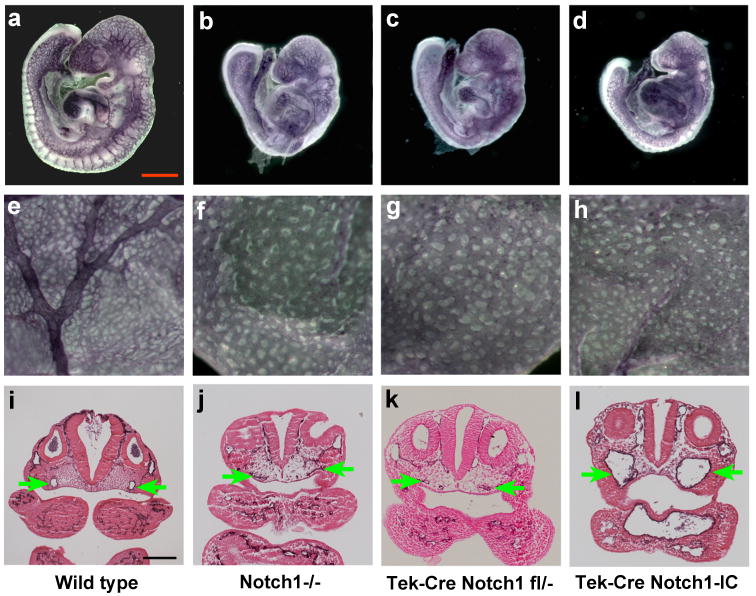
(a-d) PECAM-1 stained embryos. In the Notch pathway loss of function mutant embryos Notch1-/- (b), Tek-Cre; Notch1flox/- (c), and the Notch1GOF (Tek-Cre; Notch1-IC) embryo (d), the capillary network appears less intricate and more primitive than that of the wild type control (a). (e-h) PECAM-1 stained yolk sacs. Both the Notch pathway loss of function mutant embryos (f, g) and the Notch1GOF embryo (h) have failed to remodel the primary vascular plexus to form the large vitelline blood vessels observed in the wild type embryo (e). (i-l) Histological sections of PECAM-1-stained embryos at the level of the otic vesicle. In the wild type embryo (i), both dorsal aortae (green arrows) have open lumens and normal morphology. In the two Notch pathway loss of function mutant embryos Notch1-/- (j) and Tek-Cre; Notch1flox/- (k), the dorsal aortae are either reduced in diameter or atretic (i.e., contains no lumen). In contrast, in the Notch1GOF embryo (l) the lumens of the dorsal aortae are increased in diameter. All embryos shown are at E9.5.
We have demonstrated previously that Notch pathway loss of function mutants, such as Dll4+/- embryos or embryos with endothelial cell-specific deletion of the Rbpj gene, form arteriovenous malformations (AVMs) (Krebs et al., 2004). We examined Notch1GOF embryos for the presence of AVMs by intracardiac ink injection at E9.5, and compared them to wild type embryos and embryos with loss of function Notch1 mutations. In wild type control embryos, ink injected into the proximal outflow tract of the heart entered the aortic sac and exited through the paired branchial arch arteries. The ink then entered the paired dorsal aortae and traversed the entire length of the embryo to exit through the umbilical artery (Figure 2a, Supporting Information Movie S1). All Notch1GOF mutant embryos exhibited the presence of AVMs. In Notch1GOF embryos (Figure 2d), injected ink exited the distal outflow tract via the aortic sac. Ink then entered an enlarged branchial arch artery, entered the descending dorsal aorta, and was shunted back into the heart via fusion of the dorsal aorta with the common cardinal vein (Figure 2d, Supporting Information Movie S2). By contrast, embryos with either constitutive (Notch1-/-) or conditional (Tek-Cre; Notch1flox/-) loss of function mutations in the Notch1 gene exhibit AVMs of a distinctly different type. In these embryos, ink injected into the proximal outflow tract of the heart exited via the aortic sac, then entered the venous circulation in the head of the embryo via small diameter anastamoses with the anterior cardinal vein (Figure 2b, c, Supporting Information Movie S3). The AVMs exhibited by Notch1-/- and Tek-Cre; Notch1flox/- embryos were identical to those we had observed previously in other Notch pathway loss of function mutants, such as Dll4+/- and Tek-Cre; Rbpjflox/- embryos (Krebs et al., 2004). Both the loss of function (Figure 2f, g) and gain of function (Figure 2h) Notch1 mutant embryos had an AVM caudal to the heart caused by fusion of the dorsal aorta with the common cardinal vein, shunting blood back into the heart through the sinus venosus. We have observed this type of AVM previously in another Notch pathway loss of function mutant, the Rbpj-/- embryo (Krebs et al., 2004). However, the distinct type of AVM in the anterior region of the embryo clearly distinguishes the Notch pathway gain of function mutants from the Notch loss of function mutants. While this work was in progress, two other groups also characterized vascular defects in different loss or gain of function Notch pathway mutants (Benedito et al., 2008; Kim et al., 2008; Trindade et al., 2008). Our results are concordant with the results of these other groups.
Figure 2. Arteriovenous malformations in Notch1GOF mutant embryos are distinct from those of Notch pathway loss of function mutant embryos.
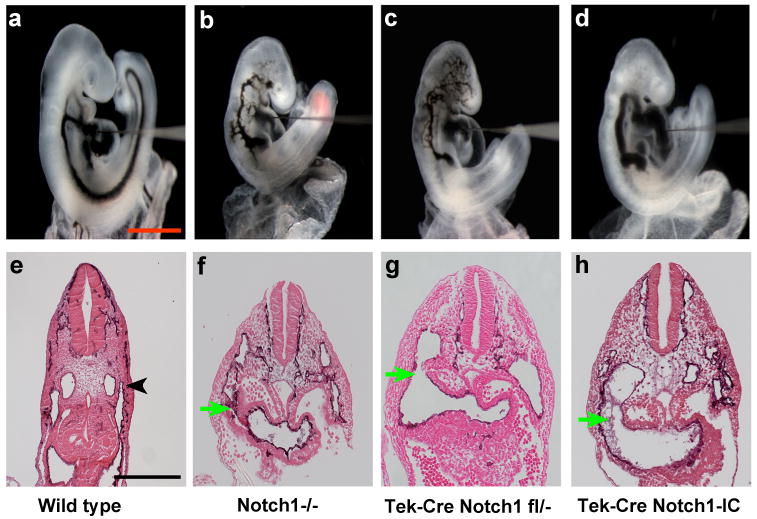
(a-d) India ink injected embryos. India ink was injected into the proximal outflow tract of the heart in order to visualize blood flow and arteriovenous malformations. (a) In the wild type embryo, ink injected into the heart exited through the branchial arch arteries, entered the paired dorsal aortae and traversed caudally the entire length of the embryo. (b, c) In the Notch pathway loss of function mutant embryos Notch1-/- (b) and Tek-Cre; Notch1flox/- (c), injected ink exited the distal outflow tract, then entered the venous circulation in the anterior of the embryo via small diameter anastamoses with the anterior cardinal vein. Caudally, injected ink was shunted back into the heart via fusion of the descending dorsal aorta with the common cardinal vein. (d) In the Notch1GOF (Tek-Cre; Notch1-IC) embryo, injected ink exited via an enlarged branchial arch artery, entered the descending dorsal aorta, and was shunted back into the heart via fusion of the dorsal aorta with the common cardinal vein. (e-f) Sections of PECAM-1 stained embryos just caudal to the heart. (e) In the wild type embryo, the dorsal aorta and common cardinal vein (black arrowhead) are distinct. (f-h) In both Notch loss of function and gain of function mutant embryos, the dorsal aorta has fused with the common cardinal vein (green arrow), shunting blood back into the heart through the sinus venosus. All embryos shown are at E9.5.
Prior work had demonstrated that mouse embryos homozygous for null mutations of the ephrinB/EphB pathway genes Efnb2 or Ephb4 died by E9.5 from vascular defects (Gerety et al., 1999; Wang et al., 1998). We compared in detail the vascular defects exhibited by Efnb2-/- and Ephb4-/- embryos with the defects exhibited by Notch1GOF, Notch1-/-, and Tek-Cre; Notch1flox/- mutant embryos. Previous studies had demonstrated that the Efnb2 gene is a direct Notch target whose expression is activated by Notch signal reception (Grego-Bessa et al., 2007). We therefore expected that, if ephrinB2/EphB4-mediated signaling functions downstream of Notch signaling during embryonic vascular development in mice, the mutant phenotypes of the Efnb2-/- and Ephb4-/- embryos would resemble the vascular phenotypes exhibited by Notch pathway loss of function mutants. To our surprise, the phenotypes of the Efnb2-/- and Ephb4-/- mutants were virtually identical to the vascular phenotype exhibited by Notch1GOF embryos. Efnb2-/- and Ephb4-/- mutant embryos exhibited vascular remodeling defects in the embryo proper (Figure 3a, b), and did not remodel the yolk sac vascular plexus (Figure 3e, f). Efnb2-/- and Ephb4-/- embryos were examined for AVMs by intracardiac ink injection at E9.5, and all Efnb2-/- and Ephb4-/- mutant embryos exhibited AVMs of the same type exhibited by Notch1GOF embryos (Figure 3c, d, Supporting Information Movie S4). Histological analyses of PECAM-1-stained embryos revealed that, similarly to Notch1GOF embryos, Efnb2-/- and Ephb4-/- embryos exhibited dilated dorsal aortae (not shown) and the caudal AVM resulting from fusion of the dorsal aorta with the common cardinal vein (arrows in Figure 3g, h).
Figure 3. Vascular defects and arteriovenous malformations in Efnb2-/- and Ephb4-/- mutant embryos phenocopy those of Notch1GOF embryos.
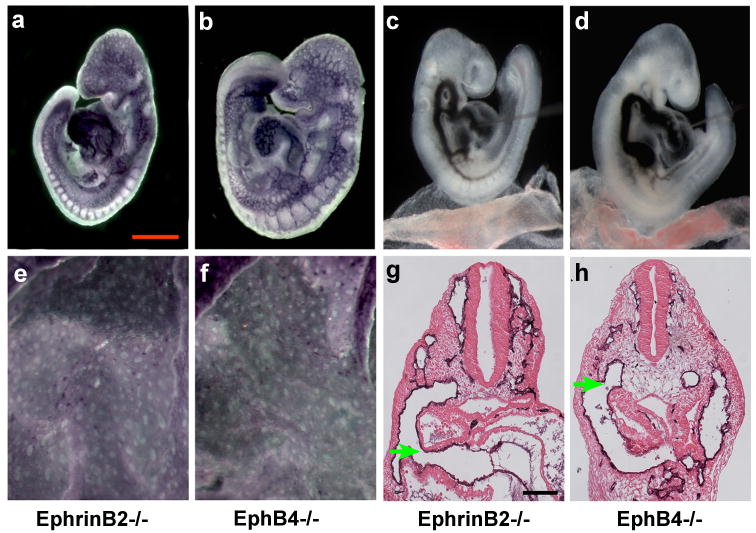
Efnb2-/- (a, c, e, g) and Ephb4-/- (b, d, f, h) mutant embryos at E9.5. (a, b) PECAM-1 stained embryos. (c, d) India ink injected embryos revealing arteriovenous malformations. (e, f) PECAM-1 stained yolk sacs exhibiting the absence of vascular remodeling. (g, h) Histological sections of PECAM-1-stained embryos. Vascular defects in both the Efnb2-/- and Ephb4-/- mutant embryos phenocopy those of Notch1GOF embryos.
The work described here, as well as that of others (Benedito et al., 2008; Kim et al., 2008; Trindade et al., 2008), demonstrates that both loss and gain of function mutations of multiple Notch pathway components, as well as loss of function mutations in the ephrinB/EphB pathway genes Efnb2 or Ephb4, result in formation of AVMs in the mutant embryos. Formation of these large AVMs during embryonic development is likely due to an inability of the vascular beds to maintain distinct arterial and venous identities in these mutants. It was a surprising finding that Efnb2-/- and Ephb4-/- embryos exhibit vascular defects and AVMs similar to those exhibited by Notch pathway gain of function mutants, rather than Notch loss of function mutants. Similar findings were made by Kim and colleagues (Kim et al., 2008). Several studies have demonstrated upregulation of Efnb2 expression in a Notch signaling-dependent manner (Hainaud et al., 2006; Iso et al., 2006; Masumura et al., 2009; Yamanda et al., 2009). In the context of studying the role of Notch signaling during ventricular chamber development of the heart, Grego-Bessa and colleagues provided evidence that the Efnb2 gene is a direct Notch target (Grego-Bessa et al., 2007). Two evolutionarily conserved binding sites for the RBPJ protein, the transcriptional mediator of Notch signaling, are present in introns 1 and 2 of the Efnb2 gene, and these sites are functionally active in mouse embryos and in a human microvascular endothelial cell line (Grego-Bessa et al., 2007). However, the finding that Efnb2-/- and Ephb4-/- embryos exhibit vascular defects and AVMs similar to those exhibited by Notch pathway gain of function mutants, rather than Notch loss of function mutants, is contrary to the results expected if the ephrinB/EphB pathway simply acts in a linear pathway downstream of Notch signal reception. These data suggest independent mechanisms for formation of AVMs in Notch1GOF embryos and in Efnb2-/- and Ephb4-/- mutant embryos.
Methods
Mutant Mice
Previously described alleles used in these studies include targeted null mutations of the Notch1 (Swiatek et al., 1994), ephrinB2 (Efnb2) (Wang et al., 1998), and EphB4 (Ephb4) (Gerety et al., 1999) genes, a conditional null Notch1 allele (Yang et al., 2004), and the Tek-Cre transgenic line (Koni et al., 2001). Notch1-ICcond mice were supplied by author AVC. For construction of Notch1-ICcond mice, in which expression of the NOTCH1 intracellular domain (NOTCH1-IC) is activated by Cre recombinase, Notch1-IC sequences from plasmid p12 Notch1-IC were cloned behind the loxP-flanked βgeo/3X SV40pA cassette in the pCALL vector (Lobe et al., 1999). After Cre-mediated excision, Notch1-IC expression is driven by the chicken β-actin promoter with a cytomegalovirus enhancer (Niwa et al., 1991). The linearized construct was electroporated into embryonic stem cells, and βgeo-expressing clones were selected in G418. Germline transmission was obtained for two independent clones that behaved similarly. Animal maintenance and experimental procedures were performed in accordance with the NIH Guidelines for Animal Care and Use, and were approved by the Institutional Animal Care and Use Committee of the Jackson Laboratory.
Histology, Immunohistochemistry and Intracardiac Ink Injections
Histological analysis, immunohistochemistry for PECAM (CD31; BD Pharmingen) and intracardiac ink injections were performed as described previously (Krebs et al., 2004; Krebs et al., 2000).
Supplementary Material
Movie S1 Filling pattern of wild type embryo after India ink injection. Ink injected into the proximal outflow tract of the heart enters the aortic sac and exits through the branchial arch arteries. The ink then enters the dorsal aortae and traverses the entire length of the embryo to exit through the umbilical artery.
Movie S2 Filling pattern of Notch1GOF mutant embryo after India ink injection. Injected ink exits the distal outflow tract via the aortic sac. Ink then enters enlarged branchial arch arteries, enters the descending dorsal aortae, and is shunted back into the heart via fusion of the dorsal aorta with the common cardinal vein.
Movie S3 Filling pattern of Tek-Cre; Notch1flox/- mutant embryo after India ink injection. Injected ink exits via the aortic sac, then enters the venous circulation in the head of the embryo via small diameter anastamoses with the anterior cardinal vein. This pattern is representative of the filling pattern observed in all Notch pathway loss of function mutant embryos.
Movie S4 Filling pattern of Ephb4-/- mutant embryo after India ink injection. The filling pattern exhibited by both Ephb4-/- and Efnb2-/- (not shown) mutant embryos is identical to that exhibited by Notch1GOF mutant embryos.
Acknowledgments
This research was funded by grants from the NIH to TG and AVC.
Footnotes
Additional Supporting Information may be found in the online version of this article
Literature Cited
- Benedito R, Trindade A, Hirashima M, Henrique D, da Costa LL, Rossant J, Gill PS, Duarte A. Loss of Notch signalling induced by Dll4 causes arterial calibre reduction by increasing endothelial cell response to angiogenic stimuli. BMC Dev Biol. 2008;8:117. doi: 10.1186/1471-213X-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, Costa L, Henrique D, Rossant J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18:2474–2478. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehebauer M, Hayward P, Arias AM. Notch, a universal arbiter of cell fate decisions. Science. 2006;314:1414–1415. doi: 10.1126/science.1134042. [DOI] [PubMed] [Google Scholar]
- Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, Murphy AJ, Adams NC, Lin HC, Holash J, Thurston G, Yancopoulos GD. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerety SS, Wang HU, Chen ZF, Anderson DJ. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell. 1999;4:403–414. doi: 10.1016/s1097-2765(00)80342-1. [DOI] [PubMed] [Google Scholar]
- Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Perez-Pomares JM, de la Pompa JL. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- Hainaud P, Contreres JO, Villemain A, Liu LX, Plouet J, Tobelem G, Dupuy E. The role of the vascular endothelial growth factor-Delta-like 4 ligand/Notch4-ephrin B2 cascade in tumor vessel remodeling and endothelial cell functions. Cancer Res. 2006;66:8501–8510. doi: 10.1158/0008-5472.CAN-05-4226. [DOI] [PubMed] [Google Scholar]
- Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res. 2007;100:1556–1568. doi: 10.1161/01.RES.0000266408.42939.e4. [DOI] [PubMed] [Google Scholar]
- Iso T, Maeno T, Oike Y, Yamazaki M, Doi H, Arai M, Kurabayashi M. Dll4-selective Notch signaling induces ephrinB2 gene expression in endothelial cells. Biochem Biophys Res Commun. 2006;341:708–714. doi: 10.1016/j.bbrc.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Kim YH, Hu H, Guevara-Gallardo S, Lam MT, Fong SY, Wang RA. Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development. 2008;135:3755–3764. doi: 10.1242/dev.022475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–2473. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A. Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- Masumura T, Yamamoto K, Shimizu N, Obi S, Ando J. Shear Stress Increases Expression of the Arterial Endothelial Marker EphrinB2 in Murine ES Cells via the VEGF-Notch Signaling Pathways. Arterioscler Thromb Vasc Biol. 2009 doi: 10.1161/ATVBAHA.109.193185. In press. Published online Oct. 1, 2009. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21:2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- Trindade A, Kumar SR, Scehnet JS, Lopes-da-Costa L, Becker J, Jiang W, Liu R, Gill PS, Duarte A. Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood. 2008;112:1720–1729. doi: 10.1182/blood-2007-09-112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyttendaele H, Ho J, Rossant J, Kitajewski J. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci U S A. 2001;98:5643–5648. doi: 10.1073/pnas.091584598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- Yamanda S, Ebihara S, Asada M, Okazaki T, Niu K, Ebihara T, Koyanagi A, Yamaguchi N, Yagita H, Arai H. Role of ephrinB2 in nonproductive angiogenesis induced by Delta-like 4 blockade. Blood. 2009;113:3631–3639. doi: 10.1182/blood-2008-07-170381. [DOI] [PubMed] [Google Scholar]
- Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 2004;269:81–94. doi: 10.1016/j.ydbio.2004.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1 Filling pattern of wild type embryo after India ink injection. Ink injected into the proximal outflow tract of the heart enters the aortic sac and exits through the branchial arch arteries. The ink then enters the dorsal aortae and traverses the entire length of the embryo to exit through the umbilical artery.
Movie S2 Filling pattern of Notch1GOF mutant embryo after India ink injection. Injected ink exits the distal outflow tract via the aortic sac. Ink then enters enlarged branchial arch arteries, enters the descending dorsal aortae, and is shunted back into the heart via fusion of the dorsal aorta with the common cardinal vein.
Movie S3 Filling pattern of Tek-Cre; Notch1flox/- mutant embryo after India ink injection. Injected ink exits via the aortic sac, then enters the venous circulation in the head of the embryo via small diameter anastamoses with the anterior cardinal vein. This pattern is representative of the filling pattern observed in all Notch pathway loss of function mutant embryos.
Movie S4 Filling pattern of Ephb4-/- mutant embryo after India ink injection. The filling pattern exhibited by both Ephb4-/- and Efnb2-/- (not shown) mutant embryos is identical to that exhibited by Notch1GOF mutant embryos.