

Abstract
Intracellular accumulation of filamentous α-synuclein (α-Syn) aggregates to form Lewy bodies is a pathologic hallmark of Parkinson’s disease. To determine whether mitochondrial impairment plays a role in accumulation of α-Syn oligomer, we used 3D5 cell culture model of human neuronal type whereby conditional overexpression of wild-type α-Syn via the tetracycline off (TetOff) induction mechanism results in formation of inclusions that exhibit many characteristics of Lewy bodies. In the present study, we compromised mitochondrial function in 3D5 cells by using shRNA to knockdown peroxisome-proliferator activated receptor gamma coactivator-1α (PGC-1α), a key regulator of mitochondrial biogenesis and cellular energy metabolism and found that PGC-1α suppression at both protein and mRNA levels results in α-Syn accumulation (i.e. monomeric and oligomeric species in the TetOff-induced cells and monomeric only in the non-induced). These changes were accompanied with reduced mitochondrial potential as well as decreased levels of AKT, GSK3β (total and Ser9-phosphorylated) and p53 that are important for cell survival. The extent to which these proteins decreased following PGC-1α knockdown, in contrast to what was demonstrable with the viability assay, is greater in the induced than the non-induced. Together these findings indicate that such knockdown increases the propensity to accumulate α-Syn oligomers, but the accumulation appears to have very little toxic impact to the neuronal cells.
Parkinson’s disease (PD) is characterized by selective loss of dopaminergic neurons in the substantia nigra and accumulation of α-Syn aggregates. A plethora of cellular processes including oxidative stress and mitochondrial dysfunction are considered to play a role in PD pathogenesis [19, 23, 14]. Overexpression of mutant α-Syn was reported to result in synaptic degeneration and neuronal cell death [9]. Neurotoxins that target mitochondrial complex I, such as MPTP/MPP+ and rotenone have been used in animal and cell culture studies to generate experimental models of PD [25, 16, 3]. PD cybrid cell line model has been successfully used to explore the contribution of mitochondrial dysfunction and mtDNA gene mutations to PD pathogenesis [21]. However, a recent report suggests that the axonal transport of mitochondria was significantly reduced in the process of PD cybrids and in rotenone-treated SH-SY5Y cells [4].
Animal studies showed that knockout of PGC-1α gene in mice can cause neurodegeneration [15]. In addition, such PGC-1α knockout mice have increased sensitivity to oxidative stress mediators such as MPTP and kainic acid [20]. The PGC-1α gene encodes a transcription factor that coordinates mitochondria biogenesis and augments the expression of several genes implicated to protect against oxidative damage. It has been suggested that regulation of cellular oxidative capacity through enhancing mitochondrial biogenesis could be beneficial for neuronal recovery and survival in (human) neurodegenerative disorders [6, 11]. To date there is no information as to the consequence of suppressing mitochondrial biogenesis on α-Syn expression and aggregation. We addressed this issue by using lentivirus containing shRNAs-PGC-1α to transduce human neuronal 3D5 cells that overexpress wild-type human α-Syn via the tetracycline-off (TetOff) inducible mechanism [10]. Such α-Syn overproduction results in intraneuronal accumulation of α-Syn aggregates that form filamentous inclusions resembling Lewy bodies. In the present study, the 3D5 cell were maintained in DMEM supplemented with 10% fetal bovine serum, 400 μg/mL G418 (geneticin; EMD Biosciences), 1 μg/mL puromycin (Sigma) and 2 μg/mL tetracycline (Tet) and incubated at 37 °C with 5% CO2. They were exposed 24 hours after seeding to 10 μM retinoic acid (RA) in Neurobasal medium (Invitrogen) that is supplemented with B-27 supplement (Invitrogen), L-glutamine (2mM; Sigma), G418 (400 μg/mL), puromycin (1 μg/mL) and Tet (2 μg/mL). On the 10th days of RA treatment, subsets of cultures were incubated with the aforementioned medium without Tet (Fig 1A). Cultures maintained in Tet-containing medium are referred as non-induced, whereas those without Tet are induced. After 2 or 5 days, all the cultures were re-seeded and maintained for additional 4 days prior to lentivirus transduction using shRNAs-PGC-1α or non-target control (NT). Western blotting of cell lysates demonstrated a reduction of PGC-1α following PGC-1α knockdown (Figs 1B & C). The magnitude of such reduction is greater in the cultures with α-Syn-overexpression than those without (Figs 1D & E; 60% versus 40 % and 70% versus 55% after TetOff induction for 9 and 14, respectively, days), suggesting a difference in transduction efficiency following α-Syn-overexpression. We confirmed, at the mRNA level by quantitative real-time-PCR analysis, effective suppression of PGC-1α and demonstrated that the suppression has a greater impact on α-Syn-overexpressing cultures than their non-induced counterparts (Fig 1F). However, it remains to be determined how α-Syn-overexpression affects the efficiency of such viral transduction. It is plausible that overexpression of α-Syn may alter the susceptibility of the cells to viral infection because it is a lipid-binding protein after all. This scenario seems consistent with our finding that without such shRNA suppression, the level of PGC-1α in the α-Syn-overexpressing cultures is comparable to that detectable with their non-induced counterparts. It is also possible that this may result from destabilization of the cytoskeleton, since recent studies have demonstrated interactions among α-Syn, actin and tau [8, 18].
Fig. 1. Effect of PGC-1α knockdown on differentiated 3D5 cells following TetOff induction.

We undertook the culture paradigm illustrated in (A) to elicit α-Syn overexpression via TetOff induction for 9 and 14 days (9d & 14d) in 3D5 cells following their 10-day retinoic acid (RA) exposure (10 μM) beginning on the day after seeding. (B) Western blot analyses of the differentiated, non-induced (Non-Ind) or TetOff-induced (Ind) 3D5 cultures infected with PGC-1α shRNA-containing lentivirus (RNAi) or non-target control (NT), using GAPDH immunoreactivity to normalize sample loading, revealed that the RNAi strategy lowered PGC-1α level effectively. As depicted in (D) the PGC-1α knockdown of the 9-day induced cultures led to a significant reduction (p<0.0002, n=6) of PGC-1α when compared to their sibling cultures transduced similarly using non-targeted controls. The RNAi treatment of their non-induced counterparts also reduced PGC-1α significantly [p<0.005, n=5, error bar standard deviation (SD)], but the reduction is less than that observed in the induced. (C) Extension of α-Syn overexpression via TetOff induction to 14 days further augmented the susceptibility of differentiated 3D5 cells to PGC-1α suppression by the RNAi (p<0.0001, n=5), (E) as compared with their non-induced counterparts (p<0.001, n=5). Such increased sensitivity of the TetOff-induced cells to PGC-1α knockdown was verified by quantitative real-time PCR and subsequent normalization with GAPDH at the mRNA level (F). Knockdown of the PGC-1α gene using shRNA-containing lentivirus effectively reduced its mRNA level in the differentiated 3D5 cells after 9-day TetOff-induction (81% reduction as compared with the NT; p=0.02, n=3) but to a lesser extent in the non-induced (61% reduction; p=0.07. The data are represented as means ± SD of triplicate specimens that were derived from three experiments.
Western blotting also demonstrated that the cultures with PGC-1α knockdown accumulate more monomeric α-Syn than their NT controls, regardless of whether they were induced or not (Figs 2A & B). However, high-molecular-weight α-Syn species, referred to as oligomers, appeared only in the induced. Most importantly, the level of such species was higher following PGC-1α knockdown. The difference was greater in the cultures with 9-day induction than those derived from 14 days of induction (Figs 2C & D) but the cause for such discrepancy remains to be determined. Our quantitative real-time-PCR analysis showed that the level of α-Syn message is not affected by PGC-1α knockdown (Fig 2E), indicating that other mechanisms are involved in the accumulation of monomeric α-Syn. In this regards, it is worth noting that exposure to neurotoxins such as MPTP, rotenone and others have been shown to heighten the level of monomeric α-Syn in cultured cells [17, 12].
Fig. 2. PGC-1α knockdown enhanced α-Syn aggregation.

Following 9- (9d in A) or 14- (14d in B) day TetOff induction, transduction of RA-differentiated 3D5 cells using PGC-1α shRNA-containing lentivirus resulted in augmented assembly of α-Syn oligomers/aggregates. Quantitative analyses of these blots revealed that TetOff induction for 9 (C) and 14 (D) days caused a 2- (p=0.03; n=5) and 1.5- (p=0.03; n=5), respectively, fold increase in α-Syn aggregation. (E) RT-PCR analyses revealed that PGC-1α RNAi did not change the α-Syn message level which shows a 250–300 fold increase following TetOff induction. The mRNA quantification is normalized with GAPDH. The data are represented as means ± SD of triplicate specimens that were derived from three experiments.
To determine the consequence of PGC-1α knockdown on cell viability and mitochondrial potential we used a fluorescence-based LIVE/DEAD Viability/Cytotoxicity Kit (Invitrogen), MitoTracker Red CMXRos and fluorescence microscopy. The results of viability assay clearly showed reduced cell viability following PGC-1α knockdown (Figs 3A & B). In comparison to the NT control fewer cells remained attached to the coverslip after PGC-1α knockdown. Moreover, some of these cells displayed abnormal morphology (e.g. rounded cell bodies and retracted processes) indicating they are under stress (data not shown). The altered density and appearance were noted mainly after 3 days, but not less, of PGC-1α knockdown. MitoTracker staining revealed that PGC-1α knockdown results in weaker fluorescent signals (Figs 4A & B). Quantitative analysis of the cellular fluorescence from multiple fields showed that the α-Syn-overexpressing cells exhibit a significant 30% decrease (p=0.0003) in mitochondrial potential per cell following PGC-1α knockdown (n=165 cells) when compared to their NT controls (n=308 cells) (Fig 4C). The results are consistent with those reported by others indicating that impaired mitochondrial biogenesis is detrimental to the cell survival [22, 13].
Fig. 3. Calcein AM Cytotoxicity assessment.
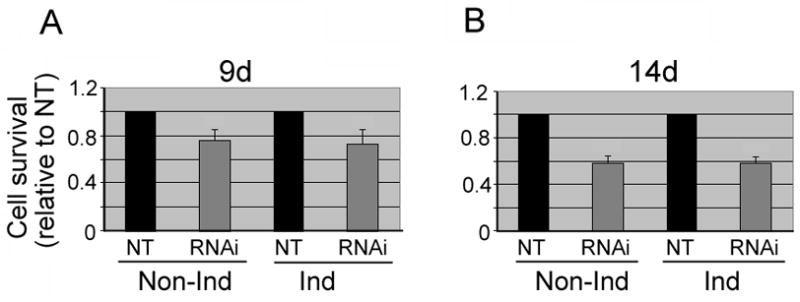
To assess the viability of the replica 3D5 cultures transduced with PGC-1α shRNA or its non-targeted controls, we used cell permeant calcein AM which is converted metabolically to fluorescent calcein thereby labeling live cells. (A) In comparison with their non-targeted controls (NT), the cells transduced after TetOff induction for 9 days showed reduced viability: 28% and 25% reduction in the induced and the non-induced, respectively, and (B) such reductions declined further (i.e. 41% and 42% for induced and non-induced, respectively) when α-Syn overproduction via TetOff induction was extended to 14 days. The data are represented as means ± SD of 84 fields selected for each group from four experiments. Comparisons were made between RNAi, and NT-controls.
Fig. 4. Reduced mitochondrial membrane potential from PGC-1α knockdown.
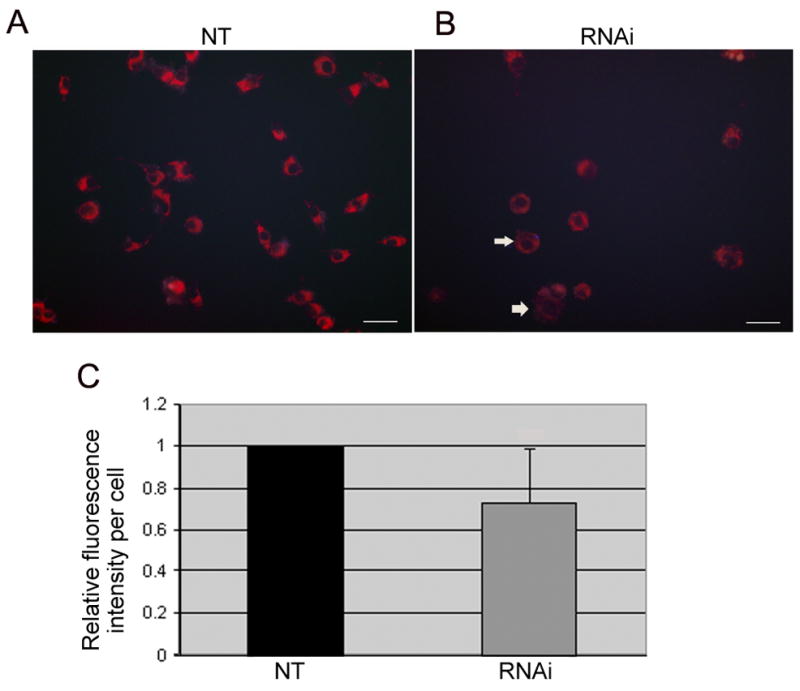
We used MitoTracker Red CMXRos to probe the 3D5 cultures for the change in mitochondrial membrane potential (MMP) subsequent to their RNAi treatments. (A) Bright red fluorescent signal indicative of roboust MMP was demonstrated in the non-target controls (NT). (B) In contrast, the fluorescent intensity appeared much lower in the RNAi-treated (arrows) suggesting compromised MMP. Quantitative analysis of the 3D5 cells using the MCID imaging software revealed that PGC-1α knockdown leads to a significant reduction in MitoTracker fluorescence (P=0.0003, n=165 cells) in contrast with that of the NT-control (n=308 cells). Values are presented as means ± SD.
We next investigated whether PGC-1α knockdown or α-Syn overexpression has any effects on GSK3β and AKT. It has been reported that GSK3β plays a role in regulating PGC-1α protein stability [2]. Activation of AKT leads to phosphorylation of Ser9 and stabilization of PGC-1α, whereas GSK3β inhibition facilitates PGC-1α degradation [5]. Results from our Western blot analysis demonstrated that the level of total AKT as well as that of total and Ser9 phosphorylated GSK3β are lower in samples with PGC-1α knockdown than NT controls (Figs 5A–5F). The changes, greater in the α-Syn-overexpressing cells than their non-induced counterparts, may reflect an attempt of these cells trying to stabilize their remaining PGC-1α proteins. With respect to the relationship between α-Syn and GSK3β expressions, a recent study showed that transgenic expression of α-Syn increases the level of GSK3β [24]. It was also demonstrated in several experimental models of neurotoxin exposure that α-Syn is essential for GSK3β activation and tau phosphorylation [7]. We have compared our NT controls with and without α-Syn overproduction and found that the induced tend to express more ATK and GSK3β (total & Ser9 phosphoryalted) although the difference is not statistically significant.
Fig. 5. Down-regulation of AKT, GSK-3β and P-GSK-3β-Ser9 from PGC-1α RNAi.

We probed lysate preparations of the differentiated 3D5 cultures, with or without TetOff induction, by immunoblotting analysis for (A) AKT and (B) GSK-3β as well as (C) its phosphorylated form (p-GSK-3β-Ser9). Immunoreactivities of AKT, GSK-3β and p- GSK-3β-Ser9appeared down-regulated following PGC-1α knockdown of non-induced cells and such reduction declined further after TetOff induction. This was verified quantitatively by densitometric analyses using GAPDH to normalize sample loading as illustrated for (D) AKT [20% reduction detectable with the non-induced (p=0.06) but 53% decrease after TetOff induction (p=0.02)], (E) GSK-3β [57% drop discernible with the non-induced (p<0.001) while 80% decline in the induced (p<0.0001)] and (F) p-GSK-3β-Ser9 [54% decrease detected in the non-induced but 74% reduction after TetOff induction (p<0.001)]. Values represent means ± SD derived from immunoblots of the PGC-1α RNAi-treated and their corresponding NT controls from 4 experiments.
In our previous study we have demonstrated that α-Syn-overexpression has no impact on the viability of the 3D5 cells and on the levels of total phosphorylated p53, a tumor suppressor gene that is important for regulating cellular senescence [10]. However, it was reported by others that wild-type α-Syn transfected neuroblastoma cells have reduced levels of p53 when compared with that of their mock-transfected counterparts, and such responses were abolished after exposing the neuronal cultures to 6-hydroxydopamine [1]. To determine if p53 were involved in cell death observed in our PGC-1α knockdown cultures we analyzed its expression at the protein level. Consistent with our previous studies, no significant difference in p53 level is evident between the non-induced and the induced of NT-transduced controls. Furthermore, we showed a decrease in the level of total p53 in the PGC-1α-suppressed cultures suggesting that p53 is a downstream target of PGC-1α (Figs 6A & B). Similar to that observed in our analysis of AKT, GSK3β and PGC-1α, the extent of p53 decrease caused by PGC-1α knockdown was greater in the α-Syn-overexpressing cells than the non-induced. This is in contrast to the results obtained from our viability assays in which PGC-1α knockdown led to a similar extent of decrease in the survival of both induced and non-induced cultures when compare to respective NT controls. Additional investigations are necessary to elucidate the molecular mechanism(s) contributing to the observed decrease of AKT, GSK3β and p53. The lack of coordination between cell survival and the level of α-Syn oligomer raises the possibility that increased accumulation of α-Syn oligomer per se, as observed in our induced plus knockdown, is not cytotoxic. However, the long-term impact of α-Syn oligomerization in neuronal cells remains unclear. In this regard, it is worth noting that Parkinson’s disease is associated with aging and its neurodegeneration proceeds over decades.
Fig. 6. Down-regulation of p53 in differentiated 3D5 cells upon PGC-1α RNAi treatment.
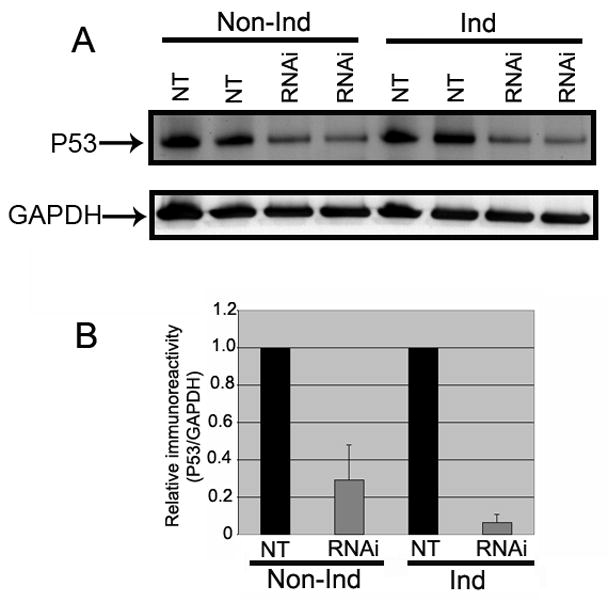
(A) Cell lysates were probed with p53 antibodies. The p53 protein expression was lowered in PGC-1α RNAi treated cells than Non-targeted controls (NT) in both induced and non-induced conditions. (B) Quantitative analysis of p53 Western blots demonstrated that the p53 level was significantly decreased in PGC-1α RNAi treated induced groups (n=4) than non-targeted controls (n=4). Induced groups: (90% reduction, p<0.0001); Non-induced groups: (71% reduction, p<0.001]. All data were normalized using GAPDH. Values are represented as means ± SD.
Acknowledgments
We thank Ms. Katrina Willams and Dr. Brian M. Necela at Mayo Clinic for their assistance in Quantitative Real time PCR analysis, Dr. Daniel P. Kelly at Washington University for providing PGC-1α antibody. This study was funded by Grant No. P50NS40256 from the National Institute of Health and by the Mayo Foundation (SH Yen).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alves Da Costa C, Paitel E, Vincent B, Checler F. Alpha-synuclein lowers p53-dependent apoptotic response of neuronal cells. Abolishment by 6-hydroxydopamine and implication for Parkinson’s disease. J Biol Chem. 2002;277:50980–50984. doi: 10.1074/jbc.M207825200. [DOI] [PubMed] [Google Scholar]
- 2.Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:7101–7111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinefelter G, Cookson MR, Greenamyre JT. Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis. 2006;22:404–420. doi: 10.1016/j.nbd.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Borland MK, Mohanakumar KP, Rubinstein JD, Keeney PM, Xie J, Capaldi R, Dunham LD, Trimmer PA, Bennett JP., Jr Relationships among molecular genetic and respiratory properties of Parkinson’s disease cybrid cells show similarities to Parkinson’s brain tissues. Biochim Biophys Acta. 2009;1792:68–74. doi: 10.1016/j.bbadis.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark J, Simon DK. Transcribe to Survive: transcriptional control of antioxidant defense programs for neuroprotection in Parkinson’s disease. Antioxid Redox Signal. 2009;11:509–528. doi: 10.1089/ars.2008.2241. [DOI] [PubMed] [Google Scholar]
- 6.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 7.Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23:2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esposito A, Dohm CP, Kermer P, Bahr M, Wouters FS. Alpha-synuclein and its disease-related mutants interact differentially with microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis. 2007;26:521–531. doi: 10.1016/j.nbd.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko LW, Ko HH, Lin WL, Kulathingal JG, Yen SH. Aggregates assembled from overexpression of wild-type alpha-synuclein are not toxic to human neuronal cells. J Neuropathol Exp Neurol. 2008;67:1084–1096. doi: 10.1097/NEN.0b013e31818c3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Lee DW, Rajagopalan S, Siddiq A, Gwiazda R, Yang L, Beal MF, Ratan RR, Andersen JK. Inhibition of prolyl hydroxylase protects against MPTP-induced neurotoxicity: Model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson’s disease. J Biol Chem. 2009;284:29065–29076. doi: 10.1074/jbc.M109.000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 15.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT. An in vitro model of Parkinson’s disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci. 2002;22:7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sousa VL, Bellani S, Giannandrea M, Yousuf M, Valtorta F, Meldolesi J, Chieregatti E. Alpha-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol Biol Cell. 2009;20:3725–3739. doi: 10.1091/mbc.E08-03-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 20.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 21.Trimmer PA, Bennett JP., Jr The cybrid model of sporadic Parkinson’s disease. Experimental neurol. 2009;218:320–325. doi: 10.1016/j.expneurol.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 23.Wooten GF. Neurochemistry and neuropharmacology of Parkinson’s disease. In: Watts RL, Koller WC, editors. Movement disorders: neurologic principles and practice. 1997. pp. 153–160. [Google Scholar]
- 24.Yuan Y, Jin J, Yang B, Zhang W, Hu J, Zhang Y, Chen NH. Overexpressed alpha-synuclein regulated the nuclear factor-kappaB signal pathway. Cell Mol Neurobiol. 2008;28:21–33. doi: 10.1007/s10571-007-9185-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG. Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson’s disease. J Pharmacol Exp Ther. 2007;322:913–922. doi: 10.1124/jpet.107.124669. [DOI] [PubMed] [Google Scholar]