

Abstract
There are a number of known genetic alterations found in men with nonobstructive azoospermia, or testicular failure, such as Y microdeletions and cytogenetic abnormalities. However, the etiology of nonobstructive azoospermia is unknown in the majority of men. The aim of this study was to investigate the possibility that unexplained cases of nonobstructive azoospermia are caused by nonsynonymous single-nucleotide polymorphisms (SNPs) in the coding regions of autosomal genes associated with sperm production and fertility. Using a candidate gene approach based on genetics of male infertility in mice, we resequenced nine autosomal genes from 78 infertile men displaying testicular failure using custom-made next-generation resequencing chips. Analysis of the data revealed several novel heterozygous nonsynonymous SNPs in four of nine sequenced genes in 14 of 78 infertile men. Eight SNPs in SBF1, three SNPs in LIMK2, two SNPs in LIPE, and one SNP in TBPL1 were identified. All of the novel mutations were in a heterozygous configuration, suggesting that they may be de novo mutations with dominant negative properties.
Keywords: genetics, male infertility, SBF1, SNP discovery, testis
A cluster of amino acid substitutions in SBF1 is associated at high frequency with testicular failure.
INTRODUCTION
The human population carries an enormous burden of male factor infertility. Approximately 1 man in 20 is infertile [1–3]. Of this group, up to 20% have azoospermia, while the rest have other sperm abnormalities resulting in subfertility. In general, men with azoospermia fall into two categories: two thirds are diagnosed with obstructive azoospermia and one third with nonobstructive azoospermia. Histopathological examination of testicular biopsies has revealed that spermatogenesis in obstructive azoospermic patients is normal but in nonobstructive patients can be divided into three general progressively abnormal categories: hypospermatogenic, maturation arrest, or Sertoli cell only.
The underlying causes of male factor infertility are poorly understood. Apart from environmental factors (toxin exposure, illness, etc.), male infertility has been linked to both congenital (e.g., Klinefelter syndrome and chromosome abnormalities, including translocations, deletions, and inversions) and genetic (e.g., CFTR mutations) defects [1–3]. Where an underlying cause can be positively correlated with the infertility, appropriate treatment, if available, or genetic counseling can be offered.
However, in a substantial proportion of cases (32% of infertile men, or 1.5% of the male population), the underlying causes are unknown (idiopathic infertility). Cytogenetic studies have been successful in revealing microdeletions of the azoospermia factor (AZF) region of the Y chromosome in approximately 10% of idiopathic azoospermic men as a strong correlate with male infertility, implicating the DAZ cluster of genes [1–3]. However, other cytogenetic abnormalities have correlated only with meiotic defects, probably mediated by chromosome pairing impairment [4, 5]. Most nonobstructive azoospermic men are believed to carry unknown autosomal mutations [6–9] based on studies of familial male infertility that often involves two or more siblings. Studies involving oligospermia have been somewhat easier to conduct because the sperm in the ejaculate often displays a characteristic phenotype that defines a clear trait that can be followed in families, usually in brothers [10–14]. In most cases (but not all), no other overt defects are apparent, suggesting that the pathologies leading to infertility are confined to the development and functioning of the testes. Those mutations that affect early events in gonad development and result in defects apparent in children have received more attention from the medical community, leading to identification of a number of mutations in the human population [15]. However, any mutations affecting testis function in adults remain obscure.
Studies of infertility and spermatogenesis in mice may provide the necessary molecular tools to broaden our understanding of infertility in humans, as more is known about male sterility in the mouse (Supplemental Table S1, all Supplemental Tables are available at www.biolreprod.org) [3]. A number of mutations affecting germ cell development have been identified. These tend to fall into four general categories: those affecting primordial germ cells and therefore causing sterility in both males and females, those affecting early stages of spermatogenesis (usually through a block in meiosis), those affecting spermiogenesis (i.e., the terminal stages of sperm differentiation), and those affecting sperm function. There is a strong similarity between the histopathology of these four kinds of mutations and that in nonobstructive azoospermic patients.
Genetic causes of male factor infertility can be broadly assumed to be a result of either de novo mutations inherited by the patient from one of his parents or from polymorphisms circulating in the population. In both cases, a search for potential causes must rely on statistical comparison of patient groups with a case-controlled fertile group.
We adopted a candidate gene approach to explore the potential of deleterious mutations in nine genes as potential causes of male factor infertility. Herein, we report the results of resequencing these genes in 78 infertile men presenting with nonobstructive testicular failure. Potentially deleterious nonsynonymous substitutions were found in four genes: SBF1, LIPE, LIMK2, and TBPL1. SBF1 appeared to be a mutation “hot spot,” with eight newly identified nonsynonymous single-nucleotide polymorphisms (nsSNPs). Two nonsynonymous SNPs, one in LIPE and the other in SBF1, were found in the fertile control group. All validated SNPs were present in heterozygous configuration, suggesting that de novo mutations inherited by infertile men from one parent may be a major cause of human male infertility.
MATERIALS AND METHODS
Recruitment of Subjects
All infertile men were recruited from the Murray Koffler Urologic Wellness Centre, a part of Mount Sinai Hospital in downtown Toronto, ON. Infertility patients were first referred to the clinic by their general practitioner and were then further examined by the clinic urologists before they were asked to participate in this study. Semen parameters of patients recruited to the study are summarized in Figure 1; detailed clinical profiles are given in Supplemental Table S2. Patient age and racial/ethnic background were not significant factors for recruitment. All who agreed to participate signed an ethics agreement, which was approved by the University of Toronto and Mount Sinai Hospital ethics review boards. Participants had a 10-ml blood sample drawn in their arm, from which DNA was extracted by Biospecimen Repository, Pathology and Laboratory Medicine at Mount Sinai Hospital. All patient samples were tested for Y microdeletions with primer pairs specific for AZFa, b, and c; 41 samples were tested with two primer pairs for each AZF region (see Supplemental Table S4), while the remainder were tested with an expanded repertoire in a clinical multiplex assay. Eighty-six fertile controls, men whose biological children were not the result of assisted reproduction, were similarly recruited. Of these, 24 donated blood samples for DNA extraction, and the remainder provided a buccal swab (MasterAmp buccal swab kit). In the latter cases, DNA was extracted using the DNA extraction kit from Epicentre Biotechnologies. Preliminary verification of the PolyBayes (http://genome.wustl.edu/tools/software/polybayes.cgi) analysis was performed using the Polymorphism Discovery Resource collection 44 from Coriell. The sex and fertility status of the Coriell samples are unknown. The racial/ethnic profiles of the patient and control groups are given in Supplemental Table S3.
FIG. 1.
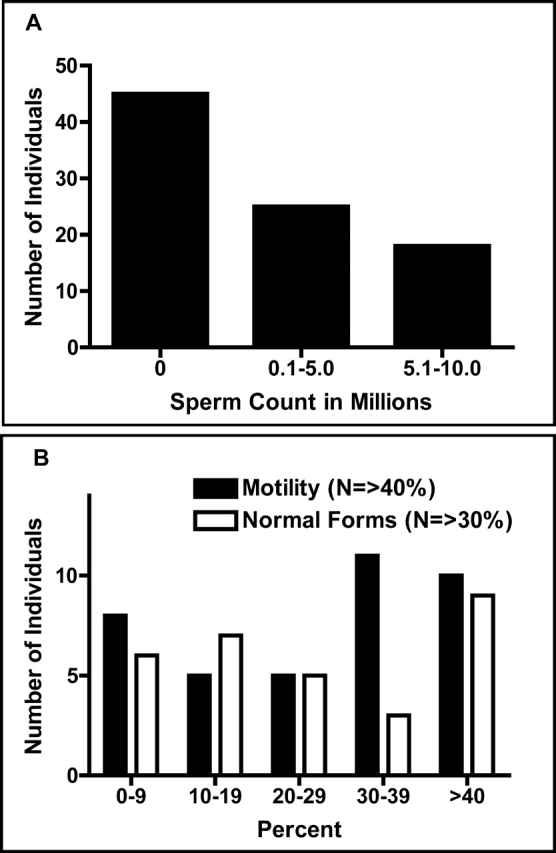
Semen parameters among infertile patients recruited to the study. Semen samples were collected by masturbation into a clean specimen container and analyzed using the World Health Organization protocols in less than 2 h for numbers of sperm (A) and motility/morphology (B).
Resequencing
Human DNA sequences of FUS, HMGB2, LIPE, SBF1, LIMK2, STYX, BCL2L2, PPP1CC, andTBPL1 were obtained from the Ensembl genome browser, release 34 (http://oct2006.archive.ensembl.org/index.html). The coding regions and the flanking intronic regions of these genes were used to custom design 88 Affymetrix GeneChip CustomSeq resequencing chips. Infertile and control sequences were amplified by PCR using Takara LA Taq polymerase. All primers used to amplify DNA for resequencing are listed in Supplemental Table S4. A separate section in the supplementary information lists primers used to amplify control DNA. Amplicon concentration from all PCR reactions was measured by spectrophotometer. Arrays were hybridized, washed, and stained according to the GeneChip CustomSeq resequencing array protocol. Washing and staining steps were performed by the GeneChip Fluidics Station. Stained chips were scanned using the Affymetrix GeneChip Scanner 3000. Sequences were analyzed using GeneChip Sequence Analysis Software, version 4.0 (http://www.affymetrix.com/support/technical/datasheets/gseq_datasheet.pdf).
Sequence Verification
All restriction enzymes used to digest relevant sequences were purchased from either Fermentas or New England Biolabs. All relevant infertile and control sequences were sequenced either by Macrogen, Seoul, South Korea, or The Centre for Applied Genomics, Toronto. In some cases, because of poor quality of cheek swab DNA, amplicons were subjected to 15 rounds of secondary amplification with nested primers (Supplemental Table S4).
Bioinformatics and Statistics
PolyBayes 3.0, freeware developed at Washington University [16], was used to analyze expressed sequence tags (ESTs) for 30 genes according to the instructions supplied with the software. Parameters for base quality and confidence limits were set arbitrarily at 13 and 0.01, respectively. The frequency of an SNP identified in this fashion was determined for the sequence depth at that particular location. For some very large genes or genes not present in large numbers in the EST database, the depth of the EST alignment was shallow. These analyses were ignored. The PolyBayes results for a cluster of SNPs in PPP1CC were verified by sequencing the Coriell collection using primers listed in Supplemental Table S4.
Protein sequences were obtained from the Ensembl genome browser. Information regarding domain structure and location within the proteins of interest was obtained through SMART [Simple Modular Architecture Research Tool] at EMBL (http://smart.embl-heidelberg.de/). All new and annotated nonsynonymous SNPS were analyzed by three bioinformatic programs: PolyPhen [17] (http://genetics.bwh.harvard.edu/pph/), BLOSUM62 (http://www.uky.edu/Classes/BIO/520/BIO520WWW/blosum62.htm), and SIFT [18] (http://blocks.fhcrc.org/sift/SIFT.html). In the latter case, alignments were performed with mammalian orthologues obtained from Ensembl, release 49, because for some genes no nonmammalian orthologues have been identified.
Statistical comparisons were performed between patient and control groups. Chi-square analysis was used.
Three-Dimensional Structure Prediction and Analysis
The three-dimensional structure of SBF1 was predicted using the Protein Homology/Analogy Recognition Engine (Phyre) (http://www.sbg.bio.ic.ac.uk/phyre/) [19, 20]. The human SBF1 protein contains 1893 residues, which exceeds the maximum number of residues (maximum, 1200) that can be modeled using the Phyre server. Therefore, the SBF1 amino acid sequence was submitted to Phyre in segments of approximately 1200 amino acid residues. To model the C-terminal Pleckstrin-type homology domain, only the 237 residues at the extreme C-terminus of SBF1 were submitted to Phyre. The resultant three-dimensional models were viewed and analyzed using PyMOL freeware (http://www.pymol.org/) [21].
RESULTS
Selection of Genes for Resequencing
At the time this study was undertaken, approximately 150 mouse genes had been identified through forward and reverse genetics as being important for spermatogenesis (Supplemental Table S1 gives an updated list). The method we planned to use, resequencing on custom-made Affymetrix resequencing chips, limited our query to 30 kilobase of DNA sequence. Therefore, we needed to reduce the number of genes that we could examine using the following three different criteria: 1) similarity of phenotype between mouse and human, 2) size of gene, and 3) potential for mutagenesis.
A careful examination of the phenotype revealed that null mutations in several of the candidate mouse genes also result in either embryonic lethality, additional female infertility, or some other pathology. These genes were eliminated from the list as being unsuitable candidates because the patients coming to the clinic were infertile but otherwise completely normal. Genes involved in female infertility were deemed poor candidates for population-based genetic causes of male infertility because of the reduced likelihood of transmission in the population. We focused our search on mutations causing defects in meiosis and postmeiotic aspects of spermiogenesis, given the symptomatic profile of many patients attending the infertility clinic (Fig. 1 and Supplemental Table S2). Finally, we performed an initial bioinformatic analysis of approximately 30 candidate genes using the PolyBayes SNP discovery program [16]. However, these data were based on analysis of ESTs in the database that were derived from a variety of sources, including tissue culture cells and tumors. The majority of SNPs identified with this approach were indels. We resequenced one particular cluster of SNPs found in PPP1CC, including an indel present in 14% of ESTs, in the Coriell Polymorphism Discovery Resource collection 44. None of the SNPs in this cluster were found in the Coriell DNAs. We concluded that the PolyBayes SNP discovery program was inappropriate for our needs, probably because of the error rate in the EST database. Therefore, we relied on phenotype and gene size as selection criteria. Table 1 lists the genes queried in this study.
TABLE 1.
Genes resequenced in infertile cohort.

SNP Discovery and Validation
Resequencing chips were designed and manufactured by Affymetrix based on the sequence data, deposited in Ensembl, release 34. Genomic DNA was extracted from blood samples donated by infertile patients recruited by the Murray Koffler Urologic Wellness Centre (Supplemental Table S2 summarizes the clinical evaluations). Amplicons for the exons represented on the chips were prepared and labeled according to the manufacturer's instructions. Each individual resequencing chip was hybridized with labeled amplicons for all nine genes from one patient. Primers used to generate the amplicons are listed in Supplemental Table S4.
Initial examination of the resequencing data revealed a very large number of novel SNPs, both synonymous and nonsynonymous. However, systematic validation using either restriction fragment length polymorphism (RFLP) or capillary sequencing indicated that the vast majority of SNPs on the resequencing chips were false positives. There is no way of knowing what the false-negative rate might be, although no additional SNPs were discovered through limited resequencing validation.
Nonsynonymous SNPs discovered on the resequencing chips were validated by either RFLP or capillary sequencing of affected patient samples. Those SNPs that were found to be bona fide by one of these tests were then analyzed by RFLP or capillary sequencing in genomic DNA from racially/ethnically matched fertile volunteers. Of 87 nonsynonymous SNPs identified on the resequencing chips, 14 were validated in genomic DNA by RFLP or capillary sequencing. Moreover, many nonsynonymous SNPs were found multiple times (i.e., in several patient samples) on the chips but when validated were found to be present only in one individual. None of the homozygous SNPs could be validated. On the other hand, capillary sequencing did not reveal any SNPs not found on the chips, indicating that the false-positive rate was high, but the false-negative rate was not measurable. Despite the high false-positive rate of SNP detection on resequencing chips, we were able to identify several previously unknown nonsynonymous SNPs in genomic DNA from infertile men (Table 2). Sequence traces for the SNPs found in SBF1 are shown in Figure 2. Both forward and reverse sequence analyses gave identical results.
TABLE 2.
Nonsynonymous SNPs discovered in infertile cohort.

FIG. 2.

Sequence traces of 10 nonsynonymous SNPs found in SBF1. Genomic DNA from infertile patients with substitutions identified on the Affymetrix chips was subjected to capillary sequencing. Heterozygous readings are indicated with arrows. In one case (T1828A), a known synonymous substitution is also observed nearby (arrowhead).
SNP Frequency in Case-Matched Controls
Toronto is a highly multicultural city, with an immigrant population representing more than 180 different countries around the world. The patient sample reflects this racial/ethnic diversity. Therefore, we adopted a case-controlled approach to assess the SNPs in a fertile group. Only the 14 nonsynonymous SNPs that were validated in patient samples were assayed in the control group. One SNP (LIPE D519N) was found in control DNA. None of the other nsSNPs were found in the control DNA by either RFLP or capillary sequencing.
If the nsSNPs discovered in our study are de novo mutations, then it is possible that de novo SNPs may also be present in the fertile population and that by focusing on particular RFLPs we may have missed other substitutions in the fertile group. Therefore, we resequenced several exons of SBF1 in the fertile samples by capillary sequencing. SBF1 was chosen because of the large number of nsSNPs discovered in infertile samples in this gene, several of which were clustered (three in exon 14 and three in exons 27 and 28 [two of the latter in the same codon]; SBF1 is broken up into 40 exons), making resequencing of the entire gene in 86 individuals by capillary sequencing logistically challenging. Therefore, we focused on exons 14, 27, 28, 34, and 35, which accounted for seven validated nsSNPs in the infertile group. One nsSNP (R1264C) was discovered in exon 27 in one of 18 fertile control samples; all other control sequences were wild type in the coding sequence. Chi-square analysis indicates that the number of infertile men with nsSNPs in exons 14, 27, 28, and 34 is significantly different vs. case-matched controls (P = 0.02).
Two additional SBF1 SNPs (A300T and R1561G) were found in patients whose karyotypes were subsequently found to be abnormal (Supplemental Table S2, patients 45 and 55). These were excluded from statistical analysis. However, both SNPs were assessed to be damaging by two of three or three of three bioinformatic tests, respectively. Abnormal karyotypes have been associated with infertility [31]. Sequence traces for these two SNPs are shown in Figure 2, and modeling of one (R1561G) was included in the bioinformatic analysis (described herein).
A previously unidentified synonymous SNP was found in SBF1 (codon 1197, Y). This SNP (C/T) was present in both patient and control groups at similar frequencies (0.12 vs. 0.14, P = 0.33), indicating that the population structure of the patient and control groups is compatible. A second, less frequent (frequency, 0.03), synonymous SNP was also discovered in the control group but not the patient group at codon 1190 (L). Also present in both infertile and control groups was a previously identified polymorphism in intron 13 (rs2076714); statistical analysis of this SNP was hampered by the large number of ambiguous readings from the chips at this position, although the frequency of g/g homozygotes in both groups was indistinguishable by chi-square analysis (P = 0.23).
Chi-square analysis of the nsSNPs discovered in LIMK2 yielded nonsignificant results (P = 0.09), probably because the sample sizes of the infertile and control groups were too small to measure the significance of three SNPs. Capillary sequencing was performed on 82 controls for exons 7 and 9, where the three SNPs were discovered. No other SNPs were found in the controls.
Bioinformatic Predictions
All of the nonsynonymous SNPs discovered in our study were subjected to three bioinformatic analyses to assess the potential effect of the amino acid change. The first test, BLOSUM62, is a simple amino acid substitution matrix that assesses similarity based on chemical properties (acidity, polarity, etc.). Values of 0 or greater indicate some degree of compatibility, while negative values are deemed incompatible. The second test, PolyPhen, assesses the potential for damage based on known protein structure/function analyses and on conservation across a wide range of taxa. The third test, SIFT, also assesses the potential for damage based on phylogenetic comparisons. In our study, we relied on mammalian orthologues in the SIFT analysis, given the specialized nature of mammalian spermatogenesis. In addition, some genes (e.g., LIMK2) did not have any close orthologues outside of the mammalian phylogeny. Where two of three or three of three tests indicated that a particular nonsynonymous substitution was damaging, we assessed the SNP as deleterious. Figure 3 shows the location of several nonsynonymous SNPs in SBF1. Note the clustering of SNPs in two regions of the polypeptide: three SNPs in exon 14 and three in close proximity in exons 27 and 28, two of which alter the same codon. The predicted effect of the amino acid substitutions on the tertiary structure of the protein for several of the novel SNPs is shown in Figure 4. Note that the R1561G substitution (found in a patient with abnormal karyotype) is predicted to be damaging by the three bioinformatic programs applied to the structure and causes a shift in the orientation of the α-helical coils on the antiphosphatase domain. A similar shift in α-helical coil orientation is observed for the presumed benign nsSNP A1236T, while the presumed benign T1828A substitution has no apparent effect on protein structure. The nsSNP discovered in exon 27 of SBF1 in one fertile control DNA (R1264C) is predicted to be benign by two programs (PolyPhen and SIFT) and damaging by one (BLOSUM62).
FIG. 3.
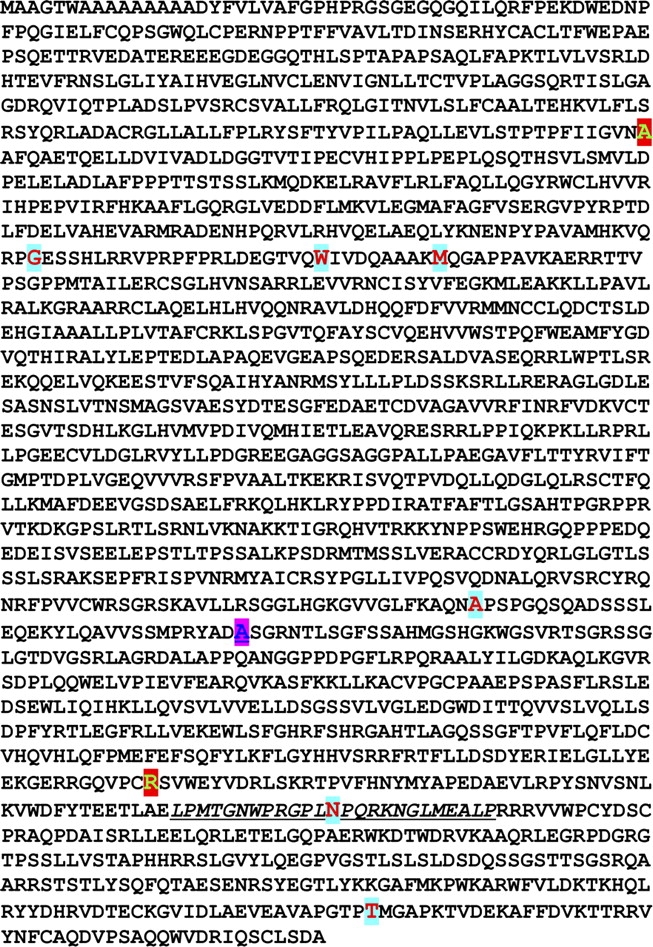
Amino acid sequence of SBF1. The sequence of SBF1 is shown, with amino acids found to be mutated in infertile men highlighted in large red font. Two different substitutions for A1268 (large blue font) were found. The differentially spliced exon 35 is underlined.
FIG. 4.

Predicted structural changes generated by nonsynonymous substitutions in SBF1. Predicted locations of mutated SBF1 residues present in infertile males. The three-dimensional structure of SBF1 was predicted using the Phyre server. A and B) A segment of the SBF1 molecules representing residues 1138–1636 (E value = 9.248842e−37). This segment contains a myotubularin-related domain (yellow), an antiphosphatase domain (blue), a SET-binding domain (green), and residues A1268 and R1587 (red). C) A segment representing residues 1792–1902 (E value = 4.841437e−17). This segment represents the Pleckstrin-type homology domain (cyan) at the extreme C-terminus of SBF1 and contains residue T1857 (red).
Using these criteria, the infertile group (excluding patients 45 and 55) contained two novel deleterious substitutions in SBF1 (one in LIPE and three in LIMK2). None of the deleterious substitutions were found in the control group. In addition, we assessed annotated nsSNPs in the same fashion and found several that fulfilled our “damaging” criteria (Table 3). While four previously annotated nsSNPs were found on the chips, only three were validated (LIPE Y100H, P156S, and R611C) and found to be present at expected frequencies for the general population. The latter two were predicted to be damaging substitutions.
TABLE 3.
Known nonsynonymous SNPs in genes queried in infertile cohort.

Prediction of the three-dimensional structure of human SBF1 using the Phyre server produced significant structures of two separate segments of the SBF1 molecule. The first segment comprises residues 1138–1636 and had an E value of 9.248842e−37 (Fig. 4, A and B). This segment contains a myotubularin-related domain (IPR010569), an antiphosphatase domain, and a suvar 3–9, enhancer-of-zeste, trithorax (SET)-binding domain. Residue A1268 is contained in this segment. Residue A1236 was not modeled, as it resided in a gap in the multiple sequence alignment on which the structure was based. The second segment, which models residues 1792–1902 represents nearly the entirety of the Pleckstrin-type homology domain (IPR011993) of SBF1 and had an E value of 4.841437e−17 (Fig. 4C). This segment contains T1828, which was also found to be mutated in one infertile patient. Residue R1561, mutated in patient 55, maps to the same segment as A1268 and is shown in Figure 4C.
DISCUSSION
We resequenced nine genes in the DNA from 78 infertile men and found 14 novel nonsynonymous SNPs in four genes. The majority of mutations were found in one gene, SBF1. Only one of our newly identified polymorphisms (LIPE D519N) was found in DNA from control samples. In addition, a novel nsSNP was found in one fertile control DNA in SBF1 (R1264C). All of the mutations were in heterozygous configuration; if they are responsible for male factor infertility, it must be either as dominant negative alterations or a reflection of monoallelic expression. Recent evidence indicates that as many as 7% of human genes may be randomly silenced on one allele resulting in monoallelic expression [32].
Eight of the new nonsynonymous SNPs were discovered in one gene, SBF1. Despite its size (5682-base pair [bp] coding sequence), there are equal numbers of annotated SNPs in the coding sequence of this gene (22 annotated SNPs), including nonsynonymous, synonymous, and nonsense/frameshift mutations, relative to those found in the smaller LIPE gene (3232-bp coding sequence [22 annotated SNPs]). Thus, while this gene may be more highly mutable, it is not excessively so. The large number of nonsynonymous SNPs in the patient cohort suggests that this gene may have an important role in male infertility. The 22 annotated SNPs in SBF1 lack population data, while those in LIPE are present in the population at measurable frequencies. In a similar vein, LIMK2 may also be a hot spot for mutations in the infertile population, although the statistical support for the LIMK2 SNPs is less compelling.
Bioinformatic analysis using three different tools revealed that six of the SNPs in three genes, LIMK2, LIPE, and SBF1, could be classified as damaging by at least two of three methods. Two additional potentially damaging nsSNPs in SBF1 were found in patients with abnormal karyotypes (Supplemental Table S2). The predicted three-dimensional structure of SBF1 allowed the mapping of the location of two amino acid residues that were found to be mutated in the infertile male population, A1268 and T1828; both residues (as well as A1236, which was not modeled) are predicted to lie in close proximity to functional domains in the SBF1 protein, which could explain a loss in male fertility if the mutations were deleterious. It is unknown whether testis-specific functions depend on the residues with predicted benign substitutions.
In choosing the genes for analysis, we relied on mouse genetics. All of the genes identified in the mouse as being important for spermatogenesis were either null or hypomorphic alleles. Given the method for making targeted mutations, which typically uses male embryonic stem cells, the study of dominant negative alleles affecting spermatogenesis is a major challenge. One mouse “mutation” resulting in male infertility is the overexpressing Spz1 transgene [33]. In this case, early germ cell hyperproliferation leads to subsequent apoptosis, impaired spermiogenesis, and progressive germ cell loss. These phenotypic features are common sequelae in many mouse mutations affecting male fertility (Supplemental Table S1) and suggest that a common response pathway for dealing with abnormal cellular behavior may operate in the testis.
This kind of tissue response to molecular damage is reminiscent of neurodegenerative diseases, where a wide range of mutations in a variety of genes leads to neuronal cell death. SBF1 belongs to the myotubularin family of proteins; one member of the myotubularin family, MTMR2, is involved in Charcot-Marie-Tooth type 4B1 disease, while another member, MTM1, is associated with X-linked myotubular myopathy [34] (http://www.molgen.ua.ac.be/CMTMutations/Home/IPN.cfm). Many nonsynonymous disease-causing mutations in both MTM1 and MTMR2 have been identified. Approximately 66% (32 of 48) correspond to residues conserved with SBF1, suggesting that these proteins are sensitive to substitutions in multiple domains.
The myotubularin family of proteins are dual-specificity phosphatases and are able to dephosphorylate not just pTyr and pSer but also (at least in the case of MTMR2) PI3 [34]. SBF1 lacks phosphatase activity because of inactivating amino acid substitutions in the catalytic domain. It has been characterized as an “antiphosphatase” and presumably works by sequestering targets away from active phosphatases, thus having an important role in cell homeostasis. Loss of homeostasis in the testis, with accompanying cell death, would lead to reduced fertility.
The mouse mutations on which this study was based were recessive loss-of-function alleles. However, the SNPs identified in the infertile cohort were all heterozygous nonsynonymous substitutions and, if causative of infertility, would be acting either as dominant negative mutations or in a semidominant fashion as a result of monoallelic silencing of the wild-type allele [32]. Testing the potentially damaging nature of these substitutions would require either x-ray crystallographic analysis of protein structure or targeted mutations in mice, although the latter would be challenging because of the necessity of using female embryonic stem cells, which tend to suffer X-chromosome nondisjunction. One should also bear in mind the difference in not just size but also life span between humans and mice. The cellular milieu is very similar, and the length of time required for development of a mature sperm is approximately the same. However, the human testis churns out sperm for roughly 60 yr, while mouse testes rarely last beyond 2 yr because of the limited life span of the animal. Thus, a genetic lesion that has little effect in the mouse may, over time, begin to affect spermatogenesis in the human, leading to loss of tissue integrity and reduced fertility. In a similar vein, there are some mutations in the human that are without phenotype in mice. The most dramatic example of this is the Duchenne muscular dystrophy gene. Loss of function mutations in this gene, which result in the debilitating Duchenne muscular dystrophy in human males starting in later childhood, have no effect in mice [35], probably because they do not live long enough.
The need to identify causative factors in male infertility is highlighted by the widespread use of assisted reproductive technologies, in particular intracytoplasmic sperm injection, for its treatment. All of the health burden in assisted reproductive technology (ART) is borne by women and children, with not just increased risk of birth defects but also obstetrical complications such as hypertension and preeclampsia [36–39]. As ART becomes standard practice around the world, the probability that the next generation of infertile men is augmented by male offspring inheriting mutant alleles from their infertile fathers merely compounds the problems associated with this complicated technology. Until now, many mutation discovery projects such as the present work have been aimed at a small subset of genes. However, emerging technology such as Solexa resequencing (www.illumina.com) may make whole-genome SNP discovery a viable (i.e., affordable) possibility. Identification of the mutation hot spots that lead to male infertility will make the analysis of risk factors through in-depth, controlled, animal studies more feasible.
Supplementary Material
Acknowledgments
The authors wish to thank the anonymous participants who donated tissue samples for this study.
Footnotes
1Supported by a Strategic Initiative grant from the Canadian Institutes of Health Research to S.V.
REFERENCES
- Carrell DT, De Jonge C, Lamb DJ.The genetics of male infertility: a field of study whose time is now. Arch Androl 2006; 52: 269–274. [DOI] [PubMed] [Google Scholar]
- Krausz C, Giachini C.Genetic risk factors in male infertility. Arch Androl 2007; 53: 125–133. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Lamb DJ.Genetic dissection of mammalian fertility pathways. Nat Cell Biol 2003; 4(suppl):S41–S49. [DOI] [PubMed] [Google Scholar]
- Paoloni-Giacobino A, Kern I, Rumpler Y, Djlelati R, Morris M, Dahoun S.Familial t(6;21)(p21.1;p13) translocation associated with male only sterility. Clin Genet 2000; 58: 324–328. [DOI] [PubMed] [Google Scholar]
- Thielemans BF, Spiessens C, D'Hooghe T, Vanderschueren D, Legius E.Genetic abnormalities and male infertility: a comprehensive review. Eur J Obstet Gynecol Reprod Biol 1998; 81: 217–225. [DOI] [PubMed] [Google Scholar]
- Gianotten J, Westerveld GH, Lescho NJ, Tanck MWT, Lilford RJ, Lombardi MP, van der Veen F.Familial clustering of impaired spermatogenesis: no evidence for a common genetic inheritance pattern. Hum Reprod 2004; 19: 71–76. [DOI] [PubMed] [Google Scholar]
- van Golde RJT, Van der Avoort IAM, Tuerlings JH, Kiemeney LA, Meuleman EJ, Braat DDM, Kremer JAM.Phenotypic characteristics of male subfertility and its familial occurrence. J Androl 2004; 25: 819–823. [DOI] [PubMed] [Google Scholar]
- Lilford R, Jones AM, Bishop DT, Thornton J, Mueller R.Case-control study of whether subfertility in men is familial. BMJ 1994; 309: 570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meschede D, Lemcke B, Behre H, De Geyter C, Nieschlag E, Horst J.Clustering of male infertility in the families of couples treated with intracytoplasmic sperm injection. Hum Reprod 2000; 15: 1604–1608. [DOI] [PubMed] [Google Scholar]
- Baccetti B, Capitani S, Collodel G, DiCairano G, Gambera L, Moretti E, Piomboni P.Genetic sperm defects and consanguinity. Hum Reprod 2001; 16: 1365–1371. [DOI] [PubMed] [Google Scholar]
- Benzackin B, Gavelle F, Martin-Pont B, Dupuy O, Liève N, Hugues JN, Wolf JP.Familial sperm polyploidy induced by spermatogenesis failure. Hum Reprod 2001; 16: 2646–2651. [DOI] [PubMed] [Google Scholar]
- Carrell D, Wilcox A, Udoff L, Thorpe C, Campbell B.Chromosome 15 aneuploidy in the sperm and conceptus of a sibling with variable familial expression of round headed sperm syndrome. Fertil Steril 2001; 76: 1258–1260. [DOI] [PubMed] [Google Scholar]
- Chemes H, Puigdomenech E, Carizza C, Olmedo S, Zanchetti F, Hermes R.Acephalic spermatozoa and abnormal development of the head-neck attachment: a human syndrome of genetic origin. Hum Reprod 1999; 14: 1811–1818. [DOI] [PubMed] [Google Scholar]
- Tuerlings J, van Golde R, Oudakker A, Yntema H, Kremer J.Familial oligoasthenoteratozoospermia: evidence of autosomal dominant inheritance with sex-limited expression. Fertil Steril 2002; 77: 415–418. [DOI] [PubMed] [Google Scholar]
- Layman L.Human gene mutations causing infertility. J Med Genet 2002; 39: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth GT, Korf I, Yandell MD, Yeh RT, Gu Z, Zakeri H, Stitziel NO, Hillier L, Kwok PY, Gish WR.A general approach to single-nucleotide polymorphism discovery. Nat Genet 1999; 23: 452–456. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, III, Kondrashov A, Bork P.Prediction of deleterious human alleles. Hum Mol Genet 2001; 10: 591–597. [DOI] [PubMed] [Google Scholar]
- Ng P, Henikoff S.Predicting deleterious amino acid substitutions. Genome Res 2001; 11: 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett-Lovsey RM, Herbert AD, Sternberg MJE, Kelley LA.Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 2008; 70(3):611–625. [DOI] [PubMed] [Google Scholar]
- Kelley LA, MacCallum RM, Sternberg MJ.Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol 2000; 299: 523–544. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. 2002. World Wide Web (URL: http://www.pymol.org). (April 2008).
- Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, de Kretser D, Metcalf D, Kontgen F, Adams JM, Cory S.Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci U S A 1998; 95: 12424–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, Chung P, de Rooij DG, Akhmedov A, Ashley T, Ron D.Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J 2000; 19: 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronfani L, Ferraguti M, Croci L, Ovitt CE, Schöler HR, Consalez GG, Bianchi ME.Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development 2001; 128: 1265–1273. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Koshimizu U, Miyazaki J, Nakamura T.Impaired spermatogenic ability of testicular germ cells in mice deficient in the LIM-kinase 2 gene. Dev Biol 2002; 241: 259–272. [DOI] [PubMed] [Google Scholar]
- Chung S, Wang S, Pan L, Mitchell G, Trasler J, Hermo L.Infertility and testicular defects in hormone-sensitive lipase deficient mice. Endocrinology 2001; 142: 4272–4281. [DOI] [PubMed] [Google Scholar]
- Varmuza S, Jurisicova A, Okano K, Hudson J, Boekelheide K, Shipp EB.Spermiogenesis is impaired in mice bearing a targeted mutation in the protein phosphatase 1cγ gene. Dev Biol 1999; 205: 98–110. [DOI] [PubMed] [Google Scholar]
- Firestein R, Nagy PL, Daly M, Huie P, Conti M, Cleary ML.Male infertility, impaired spermatogenesis, and azoospermia in mice deficient for the pseudophosphatase Sbf1. J Clin Invest 2002; 109: 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart MJ, Dixon JE.The archetype STYX/dead-phosphatase complexes with a spermatid mRNA-binding protein and is essential for normal sperm production. Proc Natl Acad Sci U S A 2002; 99: 2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Penttila TL, Morris PL, Teichmann M, Roeder RG.Spermiogenesis deficiency in mice lacking the Trf2 gene. Science 2001; 292: 1153–1155. [DOI] [PubMed] [Google Scholar]
- Ferlin A, Arredi B, Carlo Foresta C.Genetic causes of male infertility. Reprod Toxicol 2006; 22: 133–141. [DOI] [PubMed] [Google Scholar]
- Gimelbrant A, Hutchinson JN, Thompson BR, Chess A.Widespread monoallelic expression on human autosomes. Science 2007; 318: 1136–1140. [DOI] [PubMed] [Google Scholar]
- Hsu SH, Hsieh-Li HM, Li H.Dysfunctional spermatogenesis in transgenic mice overexpressing bHLH-Zip transcription factor, Spz1. Exp Cell Res 2004; 294: 185–198. [DOI] [PubMed] [Google Scholar]
- Begley M, Dixon J.The structure and regulation of myotubularin phosphatases. Curr Opin Struct Biol 2005; 15: 614–620. [DOI] [PubMed] [Google Scholar]
- Hansen J, Floss T, Van Sloun P, Ernst-Martin Füchtbauer EM, Vauti F, Arnold HH, Schnütgen F, Wurst W, von Melchner H, Ruiz P.A large-scale, gene-driven mutagenesis approach for the functional analysis of the mouse genome. Proc Natl Acad Sci U S A 2003; 100: 9918–9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Kurinczuk J, Bower C, Webb S.The risk of major birth defects after intracytoplasmic sperm injection and in vitro fertilization. N Engl J Med 2002; 346: 725–730. [DOI] [PubMed] [Google Scholar]
- Allen V, Wilson R, Cheung A.Pregnancy outcomes after assisted reproductive technology. J Obset Gynaecol Can 2006; 28: 220–233. [DOI] [PubMed] [Google Scholar]
- Schieve L, Meikle S, Ferre C, Peterson H, Jeng G, Wilcox L.Low and very low birth weight in infants conceived with use of assisted reproductive technology. N Engl J Med 2002; 346: 731–737. [DOI] [PubMed] [Google Scholar]
- Wang J, Knottnerus AM, Schult G, Norman R, Chan A, Dekker G.Surgically obtained sperm, and risk of gestational hypertension and pre-eclampsia. Lancet 2002; 359: 673–674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.