

Abstract
Alzheimer's disease (AD) is the most common form of senile dementia. Aggregation of the amyloid- β42 peptide (Aβ42) and tau proteins are pathological hallmarks in AD brains. Accumulating evidence suggests that Aβ42 plays a central role in the pathogenesis of AD, and tau acts downstream of Aβ42 as a modulator of the disease progression. Tau pathology is also observed in frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) and other related diseases, so called tauopathies. Although most cases are sporadic, genes associated with familial AD and FTDP-17 have been identified, which led to the development of transgenic animal models. Drosophila has been a powerful genetic model system used in many fields of biology, and recently emerges as a model for human neurodegenerative diseases. In this review, we will summarize key features of transgenic Drosophila models of AD and tauopathies and a number of insights into disease mechanisms as well as therapeutic implications gained from these models.
Keywords: Alzheimer's disease, Tauopathies, Amyloid-β42, Microtubule associated protein tau, Drosophila
Introduction
The fruit fly Drosophila has been widely used to investigate many aspects of biology. Analysis of the Drosophila genome has revealed that approximately 70% of human disease-related genes have homologs in Drosophila (Fortini et al. 2000; Reiter et al. 2001). This observation suggests that Drosophila may also be used to study the pathomechanisms of human diseases and to identify disease modifier genes.
Recently, Drosophila has emerged as a powerful genetic model to study human neurological and neurodegenerative disorders. Many late-onset neurodegenerative diseases including Alzheimer's disease (AD), tauopathies, Parkinson's disease, Huntington's disease and other polyglutamine diseases, amyotrophic lateral sclerosis (ALS), and prion disease are characterized by accumulation of misfolded proteins (Soto 2003). Molecular genetic studies revealed the genes associated with familial forms of such diseases, and a transgenic overexpression approach has been used to model those diseases caused by toxic gain-of-function mechanisms. Overexpression of some of those genes in Drosophila neurons has been shown to recapitulate key features of human neurodegenerative diseases (Crowther et al. 2005; Deleault et al. 2003; Feany and Bender 2000; Finelli et al. 2004; Greeve et al. 2004; Iijima et al. 2004; Jackson et al. 1998; Jackson et al. 2002; Raeber et al. 1995; Ratnaparkhi et al. 2008; Warrick et al. 1998; Watson et al. 2008; Wittmann et al. 2001). A number of excellent reviews regarding modeling of human neurodegenerative diseases in the fly have been published (Bilen and Bonini 2005; Fortini and Bonini 2000; Khurana 2008; Lu and Vogel 2009; Marsh and Thompson 2004; Sang and Jackson 2005; Shulman et al. 2003). In this review, we will summarize key features of transgenic Drosophila models of AD and tauopathies and findings in pathogenic mechanisms and therapeutic implications gained from these models.
Drosophila models of Alzheimer's disease
The role of the Aβ peptide in the pathogenesis of Alzheimer's disease
AD is a fatal disorder and, in its later stages, global cognitive functions are disrupted and associated motor disabilities lead patients to become bedridden (Cummings 2003). Short-term memory impairment is detected in the early stage of the disease along with other psychiatric problems such as sleep disorders and increased agitation, which distinguishes AD from neurodegenerative conditions such as Parkinson's disease, tauopathies, or Huntington's disease (Cummings 2003; Selkoe 2002).
At the level of cellular pathology, extensive neuron loss and two characteristic hallmarks, senile plaques (SPs) and neurofibrillary tangles (NFTs), are observed in the AD brain (Selkoe 2001). SPs are extracellularly deposited protein aggregates that are referred to as amyloid deposits. Biochemical studies have revealed that the major components of SPs are two peptides, the 40 or 42 amino acid amyloid-β 40 or 42 (Aβ40 or Aβ42) (Glenner and Wong 1984; Masters et al. 1985). Although a small number of SPs are detected in normal aged brains, this lesion is relatively specific to AD. In contrast, NFTs, which are intracellular protein inclusions composed of the hyper-phosphorylated microtubule-associated protein tau (Lee et al. 1991), are observed in many other neurological diseases.
The majority of AD cases are sporadic, with disease onset after 65 years of age. Less than 10% of all AD cases are inherited in an autosomal dominant manner (Bertram and Tanzi 2005). Aβ peptides are physiological metabolites of the amyloid β-precursor protein (APP) and result from sequential cleavage by the β-secretase and γ-secretase complexes, whose catalytic subunits are Presenilin 1 (PS1) and Presenilin 2 (PS2) (Gandy 2005). Molecular genetic studies of early-onset familial AD (EOFAD) patients have identified causative mutations in the APP, PS1, and PS2 genes, and these mutations promote Aβ42 production, aggregation, and stabilize the protein against clearance (Tanzi and Bertram 2005). Therefore, accumulation of Aβ42 in the brain is generally accepted as causing AD (Hardy and Selkoe 2002). However, the mechanism by which the Aβ42 peptide initiates the pathogenesis of AD remains elusive.
Drosophila models to study human amyloid-β42 toxicity
Drosophila has a clear homolog of APP, APP-like protein (dAPPL) (Fig. 1) (Luo et al. 1992). Flies deficient for dAPPL exhibit behavioral abnormalities (phototaxis deficiency), which can be rescued by a human APP transgene, indicating a functional conservation between dAPPL and human APP (Luo et al. 1992). dAPPL is involved in synaptic differentiation (Torroja et al. 1999b), synaptic development (Ashley et al. 2005), and neurite arborization (Leyssen et al. 2005). dAPPL overexpression causes axonal transport defects (Torroja et al. 1999a). Similarly, overexpression of human APP induces axonal transport defects and increases cell death in the larval fly brain (Gunawardena and Goldstein 2001).
Fig. 1.
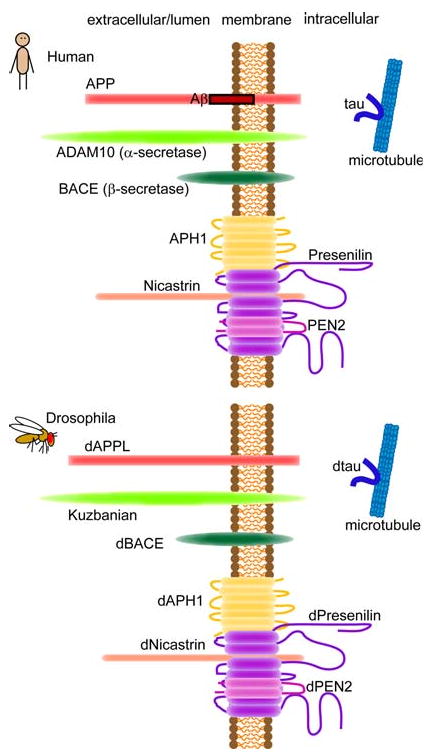
Schematic illustration of APP, α- and β-secretase, the components of γ-secretase complex and tau in human and Drosophila
dAPPL shares about 30% overall sequence identity with human APP, however, the region in dAPPL that corresponds to the Aβ peptide lacks significant homology with the human peptide (Luo et al. 1992).
In the non-amyloidogenic pathway, APP is cleaved within the Aβ domain by α-secretase precluding deposition of intact Aβ peptide. Kuzbanian (Kuz) is the Drosophila ortholog of α-secretase ADAM10 (Allinson et al. 2003; Rooke et al. 1996) and cleaves dAPPL (Fig. 1) (Carmine-Simmen et al. 2009). Very recently, Carmine-Simmen et al. identified Drosophila β-secretase-like enzyme (dBACE, Fig. 1), which has 25% identity to human BACE1 and 28% identity to human BACE2. dBACE does cleave human APP but not at β-site (Carmine-Simmen et al. 2009; Greeve et al. 2004). Interestingly, dBACE overexpression cleaves dAPPL and produces a fragment containing the region in dAPPL that corresponds to the Aβ peptide, which aggregate into intracellular fibrils, amyloid deposits, and cause age-dependent behavioral deficits and neurodegeneration (Carmine-Simmen et al. 2009). These results suggest that not only the biological functions but also amyloidogenic and non-amyloidogenic proteolytic processing may be conserved across species.
Transgenic flies to study human Aβ peptide-induced amyloid formation and neurodegeneration have been generated by several approaches using the GAL4/UAS system (Fig. 2) (Brand and Perrimon 1993). Drosophila has all the components of the γ-secretase complex (Fig. 1) (Periz and Fortini 2004; Takasugi et al. 2003), while Drosophila has very low β-secretase activity (Fossgreen et al. 1998; Yagi et al. 2000). To overproduce Aβ peptides from human APP in flies, Greeve et al. (2004) generated triple transgenic flies expressing human APP, human β-secretase, and Drosophila presenilin (dPsn) or dPsn with point mutations that correspond to the EOFAD mutants N141I, L235P, and E280A (Ye and Fortini 1999). These flies produced modest levels of Aβ peptides, including Aβ40 and Aβ42, and an additional Aβ peptide (δ-Aβ), as well as intracellular fragments of APP. The flies developed β-amyloid plaques and age-dependent neurodegeneration as well as semilethality. The neurodegeneration phenotype was enhanced in flies expressing dPsn carrying the EOFAD mutations, while the phenotype was suppressed by a genetic reduction in fly endogenous dPsn, suggesting that neurodegeneration was dependent on γ-secretase activity.
Fig. 2.
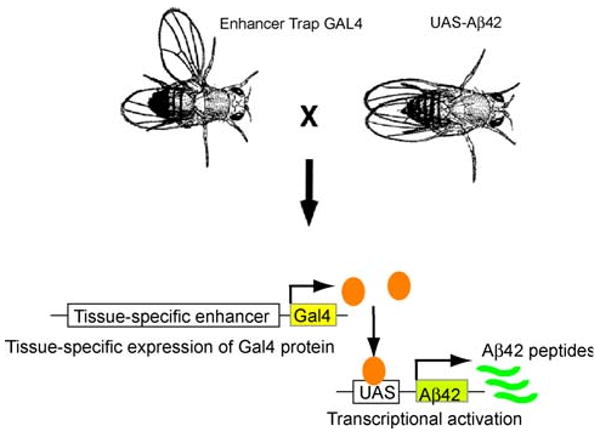
Spatial targeting of transgene expression in Drosophila. GAL4/UAS system. Driver lines expressing the transcriptional activator GAL4 in a tissue-specific fashion are crossed to UAS-lines with genomic inserts of a target gene fused to five GAL4-binding sites arrayed in tandem (5 × UAS) (shown here as UAS-Aβ42). Adapted from Development, (Brand and Perrimon 1993)
In contrast, we and others took a more direct approach to overproduce human Aβ42 peptides in fly neurons. To express human Aβ40 or Aβ42 in the secretory pathway, Aβ peptide sequences were directly fused to a signal peptide at the N-terminus. This artificial construct produced intact Aβ40 or Aβ42 peptides in the fly brain (Finelli et al. 2004; Iijima et al. 2004), which allowed us to evaluate the toxicity of Aβ40 and Aβ42 separately in the brain for the first time in an animal model. Both the Aβ40 and Aβ42 peptides accumulate during aging in the fly brain, but only Aβ42 formed amyloid deposits (Iijima et al. 2004). This result is consistent with previous observations of in vivo amyloid deposits in AD or Down syndrome patient brains, which have shown that Aβ42 first accumulates in amyloid plaques in the brain parenchyma (Iwatsubo et al. 1994).
Sensitive and quantitative assays using different cues, such as courtship and heat stress, as well as olfactory, taste, and visual cues, have been established to study memory in the fly (Heisenberg 2003; Quinn et al. 1974; Tully and Quinn 1985; Waddell and Quinn 2001). In the Pavlovian olfactory classical conditioning assay (Quinn et al. 1974; Tully and Quinn 1985), flies learn to avoid one of two odors because it is associated with an electric shock. During the training phase, flies are exposed to one odor together with an electric shock after which the second odor is delivered without electric shock. Temporally distinct memories such as learning, short-term memory, and middle-term memory can be tested at various time points after the training by allowing flies to choose between the two odors presented without electric shock.
Using this assay, we detected an age-dependent short-term memory defect in Aβ42 flies. High levels of Aβ40 expression also caused short-term memory defects, which suggest that an excess of Aβ40 was also toxic to synaptic plasticity. Sensory motor activity was not significantly affected in Aβ flies, suggesting that the observed defects were attributable to short-term memory defects.
At later stages, Aβ42, but not Aβ40, caused locomotor defects, premature death, and age-dependent neurodegeneration in the brain (Fig. 3). An electron microscopic analysis of Aβ42 fly brains revealed that most degenerating neurons showed necrotic cell death. Neither amyloid fibril, nor NFTs were detected in this model (Iijima et al. 2004). Finelli et al. (2004) showed that Aβ42 peptides expressed in fly eyes developed amyloid deposits and caused retinal degeneration. Using a similar approach, Crowther et al. (2005) expressed the wild-type and EOFAD-related Arctic mutant (E22G) Aβ42 peptides in Drosophila neural tissue, and showed that neuronal dysfunction and degeneration induced by the Arctic Aβ42 were more severe than that induced by wild-type Aβ42.
Fig. 3.
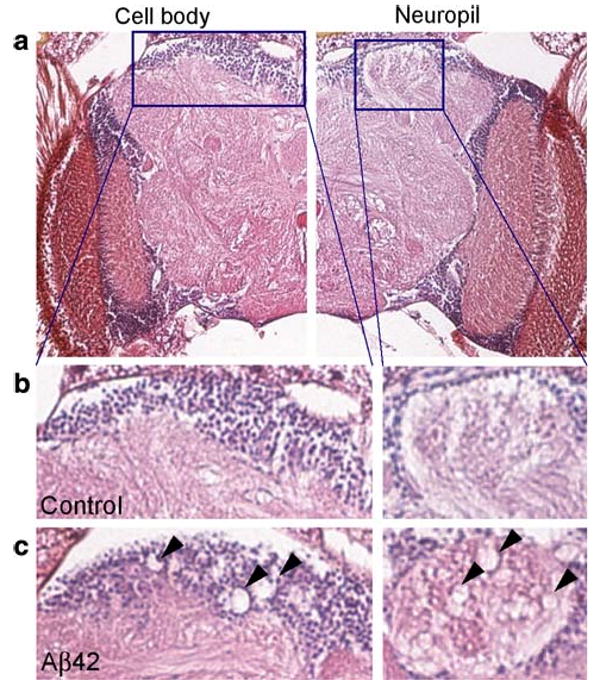
Cell body and neuropil degeneration in Aβ42 flies. a–c Neurodegeneration in Aβ42 flies at 25 days old. The cell body and neuropil region in the mushroom body are enlarged. Arrowheads in c indicate neurodegeneration. Adapted from PLoS One, (Iijima et al. 2008)
N-terminally truncated and, in particular, pyroglutamate (pE)-modified Aβ42 peptides have been suggested to be important in the initiation of pathological cascades because of their abundance, resistance to proteolysis, rapid aggregation, and neurotoxicity. The pE-modification of Aβ42 is catalyzed by glutaminyl cyclase. A recent report showed that transgenic Drosophila expressing pyroglutamate (pE)-modified Aβ42 (Aβ3(pE)-42), which were fed an inhibitor of glutaminyl cyclase, exhibited a reduced accumulation of Aβ3(pE)-42 (Schilling et al. 2008). In this model, expression constructs containing Aβ42N-terminally fused to the prepro-sequence of murine thyroliberin (TRH) were used, and liberation of Aβ was accomplished by prohormone convertase processing in the secretory pathway (Schilling et al. 2008). Neurotoxicity induced by Aβ3(pE)-42 in these flies has not been reported yet.
Comparison of Aβ42 flies to mouse models of Alzheimer's disease
There are many animal models of AD including nematodes, fly, mouse, rat, rabbit, canine, and non-human primates (Woodruff-Pak 2008). Interestingly, each model seems to recapitulate somewhat different aspects of AD. Most transgenic mice that overproduce Aβ successfully recapitulate several pathological lesions including extracellular amyloid deposits, behavioral deficits and memory defects, but they do not exhibit global neuronal loss (Gotz and Ittner 2008; McGowan et al. 2006). In contrast, progressive neurodegeneration was induced, and extensive intracellular accumulation of Aβ42 within neurons was observed in our transgenic Aβ42 flies (Iijima et al. 2008, 2004). Human AD brains show significant accumulation of intraneuronal Aβ42 (Gouras et al. 2000; Takahashi et al. 2002), and, a few mouse models of AD have shown intraneuronal accumulation of Aβ42 that was associated with memory defects (Oddo et al. 2003), significant neuron loss (Casas et al. 2004; Oakley et al. 2006), and axonopathy (Wirths et al. 2006). These results suggest that intraneuronal Aβ42 may contribute to neuronal dysfunction and degeneration in our fly model.
Another unique feature of our fly model is that the toxicity of Aβ40 and Aβ42 can be dissected in vivo. Since most of the mouse models of AD overexpress APP, which produces a series of Aβ species including Aβ40 and Aβ42 after proteolysis, it is difficult to study the toxicity of either species individually. In contrast, we found that while both Aβ40 and Aβ42 could cause memory defects, only Aβ42 caused neurodegeneration with amyloid deposits. Recently established transgenic mice separately expressing Aβ40 or Aβ42 have shown that extracellular amyloid deposit formation is induced by Aβ42, but not by Aβ40. However, no neuronal losses have been observed in Aβ42 mice (McGowan et al. 2005). The Aβ42 fly model thus provides a unique tool for studying the mechanism underlying intraneuronal Aβ42 accumulation and neurodegeneration.
Implications for anti-Aβ therapies from fly models
An increase in Aβ42 levels above a given threshold in the brain is generally regarded to be the primary event in AD pathogenesis, and approaches to develop disease-modifying therapies have focused on lowering Aβ42 levels. In contrast to EOFAD, in which Aβ42 production and/or aggregation is enhanced as a result of genetic factors, the mechanisms by which Aβ42 reaches pathological levels in the brains of late-onset AD (LOAD) patients are not well understood. The steady state level of Aβ42 reflects the balance between production and clearance of Aβ42, and an imbalance of these activities could be sufficient to raise Aβ42 levels. Thus, reducing Aβ42 levels can be achieved either by attenuating production, or by facilitating degradation and/or clearance of Aβ42 from the brain (Iwata et al. 2005; Tanzi et al. 2004).
Targeting β- and γ-secretases
In order to reduce the Aβ42 level in the brain, the prevention of amyloidogenic processing of APP by application of inhibitors of β-secretase (BACE) and γ-secretase is a viable option (Citron 2004). Both β- and γ-secretase inhibitor treatments ameliorate Aβ-induced toxicity, including semi-lethality and premature death, in triple transgenic flies expressing human APP, human β-secretase, and fly presenilin (dPsn) (Greeve et al. 2004). Recently, Rajendran et al. (2008) developed a β-secretase inhibitor peptide conjugated to a sterol moiety as a membrane anchor, which markedly increased the potency of the inhibitor. Feeding sterol-linked β-secretase inhibitor to the triple transgenic flies increases the survival rates, indicating a reduction in toxicity in vivo.
Drosophila has been used as a functional in vivo system to search for genetic and pharmacological modifiers of γ-secretase activity. Drosophila has all the components of the γ-secretase complex, which is responsible for intramembranous cleavage of several transmembrane proteins (Fig. 1), including APP, DCC, ErbB4, N-Cadherin, E-Cadherin, Notch, and Delta (Brunkan and Goate 2005; Periz and Fortini 2004; Takasugi et al. 2003). Psn is the enzymatic component of the γ-secretase complex, and EOFAD mutations in Psn affect the function of the γ-secretase complex in Drosophila (Ye and Fortini 1999). Ubiquilin 1 (UBQLN1) is a Psn interactor that promotes the accumulation of PS1 and regulates its endoproteolysis (Mah et al. 2000). The Drosophila ortholog of human UBQLN1, dUbqln, modifies the eye phenotype induced by overexpression of dPsn, indicating that the proteins have a functional interaction in vivo (Ganguly et al. 2008; Li et al. 2007). Using a transgenic reporter of γ-secretase-mediated APP processing (Guo et al. 2003), Gross et al. showed that dUbqln and the Drosophila homolog of X11 (Hase et al. 2002), an APP interacting protein that modulates Aβ production (Sano et al. 2006), affects APP processing (Gross et al. 2008).
In Drosophila, Notch signaling is required during development and functions in many cell fate specification events. The modifier screens of Psn-dependent Notch-related phenotypes identified many genes, and some of these modifiers genetically interact with APP (Mahoney et al. 2006; van de Hoef et al. 2009). Inhibitors of γ-secretase induce developmental defects in Drosophila that are remarkably similar to those caused by genetic reduction of Notch, indicating that the three-dimensional structure of the drug-binding site(s) in Drosophila γ-secretase is remarkably conserved vis-à-vis the same site(s) in the mammalian enzyme (Micchelli et al. 2003).
In summary, these reports suggest that fly models may be a sensitive and functional in vivo system for the validation of newly developed and the prescreening of drug candidates for β- and γ-secretases.
Enhancing Aβ-degrading enzymes
Several peptidases have been identified as candidate Aβ-degrading enzymes, including neprilysin (NEP) (Iwata et al. 2000), insulin-degrading enzyme (IDE) (Qiu et al. 1998), endothelin-converting enzyme 1 (ECE1) (Eckman et al. 2001), cathepsin D (Hamazaki 1996), and serine protease-α2-macroglobulin (Qiu et al. 1996). Interestingly, the expression of NEP is reduced in both LOAD patient brains and normally aging brains (Yasojima et al. 2001a; Yasojima et al. 2001b), suggesting that a reduction in Aβ-degrading activity may contribute to the onset and/or progression of LOAD.
Among the Aβ42 degrading enzymes, NEP has been identified as one of the major rate-limiting Aβ-degrading enzymes in the brain (Iwata et al. 2001, 2000). A deficiency in NEP accelerates formation of extracellular amyloid deposits (Farris et al. 2007), amyloid angiopathy (Farris et al. 2007), synaptic dysfunctions (Huang et al. 2006), and memory defects (Huang et al. 2006) caused by human Aβ in transgenic mice. Transgenic (Leissring et al. 2003; Poirier et al. 2006), viral (Iwata et al. 2004; Marr et al. 2003), or ex vivo (Hemming et al. 2007) delivery of NEP to brains of APP transgenic mice reduces extracellular amyloid deposits, synaptic dysfunction, and premature death. Thus, activation of NEP in the brain could be a potential disease modifying therapy for AD by reducing extracellular Aβ.
In addition to extracellular Aβ, intraneuronal Aβ42 may contribute to AD pathogenesis. However, the protective effects of neuronal NEP expression on intraneuronal Aβ42 accumulation and neurodegeneration was not clear. To investigate the effects of NEP on intraneuronal Aβ42 accumulation and neuron loss induced by Aβ42 in vivo, we have recently established transgenic flies expressing human NEP and examined the effects of NEP on Aβ42-induced toxicity in the Aβ42 fly model (Iijima-Ando et al. 2008).
Expression of NEP significantly reduced the level of Aβ42 in both the detergent soluble and insoluble fractions of Aβ42 brains. Additionally, neuronal expression of NEP prevented formation of intraneuronal Thioflavin S-positive Aβ42 deposits in the cell body. Furthermore, NEP dramatically suppressed neuron loss induced by Aβ42. These protective effects were not observed in transgenic flies expressing an inactive mutant form of human NEP (NEPmut) with the amino acid substitution E585V in the zinc-binding motif of NEP (Hama et al. 2001; Shirotani et al. 2001), suggesting that the observed effects are attributable to the enzymatic activity of NEP (Iijima-Ando et al. 2008). These results are consistent with a recent study using APP transgenic mice (Spencer et al. 2008).
Interestingly, neuronal expression of NEP significantly shortened the lifespan of flies, in part through reduced CREB activity. NEP expression also caused axon degeneration in the aged fly brain. These results suggest that the transgenic fly model may also be used to detect potential side effects of a given manipulation (Iijima-Ando et al. 2008).
Targeting Aβ aggregation
Aβ is a hydrophobic and self-aggregation prone peptide (Lansbury and Lashuel 2006). In its native and soluble states, Aβ is an unfolded polypeptide consisting primarily of a random-coil structure (Kelly 2005; Rochet and Lansbury 2000), and its aggregation in vivo is affected by a combination of genetic (DeMattos et al. 2004), environmental (Cherny et al. 2001), and aging (Cohen et al. 2006a) factors. Aggregation of the Aβ42 peptide in the brain parenchyma is a hallmark of AD pathology, and the prevention of Aβ aggregation has been proposed as a therapeutic intervention in AD.
The azo-dye Congo Red binds to β-amyloid and inhibits fibrillization of Aβ42 in vitro. Feeding Congo Red to flies resulted in a significant increase in longevity, reduction in plaque formation, and delay in vacuolation in neurons, as well as preservation of the architecture of the brain and retinal tissues (Crowther et al. 2005). In a recent study, ligands were designed to specifically target aggregation of Aβ by binding and stabilizing the α-helical conformation of amino acids 13–26 in Aβ. These inhibitors reduced fibril formation of Aβ40 in vitro, and ameliorated Aβ42 toxicity to cells in culture and to hippocampal slice preparations. Additionally, feeding these inhibitors to flies expressing Aβ42 in the central nervous system prolonged lifespan, increased locomotor activity, and reduced neurodegeneration (Nerelius et al. 2009).
In addition to mature amyloid fibrils, Aβ can form a variety of misfolded structures in vitro, including multiple monomer conformers, different types of prefibrillar assemblies including small oligomers (Walsh et al. 2002), higher molecular weight complexes known as Aβ-derived diffusible ligands (ADDLs) (Lambert et al. 1998), oligomers composed of 15–20 monomers (Kayed et al. 2003), dodecameric oligomers (Aβ*56) (Lesne et al. 2006), and protofibrils (Harper et al. 1997). Some of these intermediate Aβ species can also be found in the cerebrospinal fluid and brains of AD patients (Georganopoulou et al. 2005; Kuo et al. 1996). Since aggregation of Aβ42 in the brain parenchyma is a hallmark of AD pathology (Thal et al. 2006), the neurotoxicity of Aβ42 was initially correlated with the tendency of Aβ42 to form insoluble mature amyloid fibrils (Murakami et al. 2003; Yankner et al. 1989). However, recent experimental data indicates that the soluble prefibrillar assemblies cause more severe synaptic dysfunctions, cognitive defects, and neurodegeneration than do the mature fibrils (Haass and Selkoe 2007).
A study using the Drosophila model in combination with computational predictions of Aβ42 aggregation has further demonstrated that there is a strong positive correlation between the magnitude of neurotoxicity, as manifested in locomotor deficits and reduced lifespan, and the propensity of Aβ42 to aggregate into protofibrils (Luheshi et al. 2007). These results indicate that soluble prefibrillar assemblies, but not mature amyloid fibrils, of Aβ42 are the primary neurotoxic species, which is consistent with the observation that the level of soluble Aβ is better correlated with the severity of cognitive impairment than the density of insoluble amyloid deposits (Lue et al. 1999; Naslund et al. 2000).
The complexity of Aβ42 aggregation and toxicity
In addition to the “quantitative” changes in toxicity, the structural diversity of Aβ42 species may also “qualitatively” influence the pathogenicity of Aβ42, as has been well established in the pathogenesis of prion disease mediated by various PrP species (Prusiner 2004). Deshpande et al. (2006) reported that both Aβ oligomers and ADDLs induced rapid and massive neuronal death, with ADDLs exhibiting their effects with a slightly slower time course. In contrast, Aβ fibrils induced progressive dystrophy and modest cell death. Recently, Chiang et al. reported that Aβ42 oligomers and larger aggregates have different effects on synaptic transmission and LTD at the neuromuscular junction of Drosophila larvae (Chiang et al. 2009).
We investigated the correlation between levels of aggregation of Aβ42 and memory defects and neurodegeneration through genetic manipulation of Aβ42 aggregation in the Drosophila model. We have established transgenic fly lines carrying human Aβ42 mutants with differing tendencies to aggregate (Iijima et al. 2008). Human Aβ42 with the Arctic mutation (E22G, Aβ42Arc), which causes EOFAD (Nilsberth et al. 2001), is more aggregation-prone than wild-type Aβ42 (Cheng et al. 2004; Johansson et al. 2006; Lord et al. 2006; Whalen et al. 2005). In contrast, an artificial mutation, L17P, Aβ42art, suppresses amyloid fibril formation (Fay et al. 1998; Morimoto et al. 2004; Murakami et al. 2003). Aβ42Arc accumulated in the insoluble fraction more readily than Aβ42. In contrast, Aβ42art accumulated primarily in the soluble fraction and was greatly reduced in the insoluble fraction.
Remarkably, Aβ42, Aβ42Arc, and Aβ42art each induced a distinct pathology. Aβ42Arc fly brains showed more extensive cell body loss than Aβ42 or Aβ42art brains. In contrast, the level of neuropil degeneration was greatest in Aβ42art flies. Thioflavin S staining, which labels aggregated Aβ42, revealed that the degenerated structures in Aβ42, Aβ42Arc, and Aβ42art flies were closely correlated with intraneuronal accumulation of each Aβ peptide (Fig. 4). Aβ42Arc accumulated primarily as deposits in the cell soma, while Aβ42art was distributed primarily in the neurites. Aβ42 was detected both in the cell body and in the neurites, but to a lesser extent than the mutants (Fig. 4). Thus, the differences in aggregation of Aβ42 correlated with qualitative shifts in pathology in the fly brain (Iijima et al. 2008).
Fig. 4.
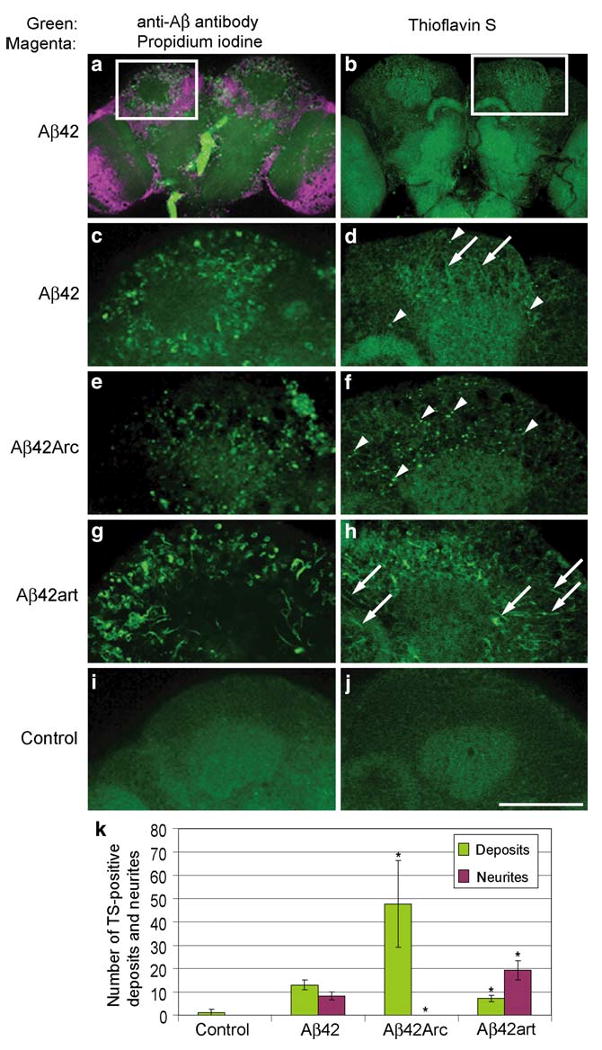
Distribution and aggregation of Aβ42, Aβ42Arc, and Aβ42art peptides in fly brains. a, c, e, g and i, Immunostaining of brains of 25 day-old flies with anti-Aβ antibody (green). In a, nuclei were stained with propidium iodide (magenta). b, d, f, h and j, Thioflavin S (TS) staining of brains of 25 day-old flies. Arrowheads and arrows indicate TS-positive deposits and neurites, respectively. No signal was detected in the control i, j. Scale bar in j: 50 μm. c and d are enlarged images of the boxed regions in a and b, respectively. k Numbers of TS-positive deposits and neurites were presented as averages ± SD (n = 6 hemispheres). Asterisks indicate significant differences from Aβ42 (P < 0.05, Student's t test). Adapted from PLoS One, (Iijima et al. 2008)
The increase in Aβ42 aggregation proneness associated with the pathogenic Arctic mutation (E22G, Aβ42Arc) correlated with more severe effects on memory, locomotor ability, and lifespan relative to the wild-type Aβ42. These data are consistent with the fact that Aβ42Arc causes EOFAD (Nilsberth et al. 2001) and indicates that aggregation proneness contributes to Aβ42 toxicity in vivo. In contrast, an artificial mutation (L17P) that decreased Aβ42 aggregation proneness suppressed the toxic effects on locomotor function and lifespan, but caused an earlier onset of memory defects, indicating that not all pathogenic effects of Aβ42 are directly correlated with aggregation proneness.
In summary, these results demonstrate that manipulation of the aggregation propensity of Aβ42 modified the pathogenicity of Aβ42 in vivo, and suggest that the partial prevention of Aβ42 amyloidgenesis by aggregation inhibitors may result in qualitative shifts in the pathogenic effects of Aβ42 (Iijima and Iijima-Ando 2008). Furthermore, the tendency of Aβ42, which is normally an unfolded polypeptide consisting primarily of a random-coil structure in the native, soluble state (Kelly 2005; Rochet and Lansbury 2000), to aggregate may be affected by a combination of genetic (DeMattos et al. 2004), environmental (Cherny et al. 2001), and aging factors (Cohen et al. 2006b), and the resultant Aβ42 conformers or species may contribute to the heterogeneous pathogenesis of AD (Cummings 2000).
Insights into pathomechanisms and genetic modifiers of Aβ42-induced toxicity from fly models
The power of genetics in Drosophila provides in vivo experimental platforms to examine whether gain or loss of function of a given gene can modify a disease phenotype in a time efficient and cost effective manner (Shulman et al. 2003). Since Drosophila generally does not possess as many redundant gene families as mammals, studying the consequence of a single gene disruption is easier in the fly. This enables us to systematically test hypotheses derived from biochemical or histopathological analysis of human disease tissues, genetic association studies performed on particular disease phenotypes, or findings in in vitro model systems. In addition to hypothesis testing, one of the most powerful advantages of using a Drosophila model is the ability to conduct large scale, genome-wide, forward genetic screens to identify novel genes and pathways that modify a given disease phenotype. The findings from this unbiased approach have great potential to open novel research areas.
Retinal toxicity has been widely used in genetic screens in Drosophila models to identify modifiers of neurode-generative disease. Expression of Aβ42 in the fly eye results in a visible reduction in eye size and a roughened eye surface due to the death of intrinsic photoreceptors and supporting cells (Cao et al. 2008). Using eye degeneration as the read-out phenotype, Cao et al. (2008) have tested the effects of 1,963 EP transposon insertions in the fly genome. The EP transposon has a GAL4 activated promoter and, depending upon the site and orientation of the insertion, the transposon will upregulate or downregulate gene activity (Rorth et al. 1998). Using this approach, 23 modifiers were identified, including genes involved in the secretory pathway (e.g., the human orthologs of carboxypeptidase D and the AP3 subunit δ-1), cholesterol homeostasis (AMP kinase γ-subunit), copper transport (ATP7), and ubiquitin/proteolysis (ubiquitin-conjugating enzyme E2Q and NEP 2), which have been implicated in AD pathogenesis. This screen also identified genes related to the regulation of chromatin structure as a potential pathway mediating Aβ42 toxicity. Loss-of-function mutations of several components of the Sin3A complex, which regulates transcription and chromatin remodeling through interactions with other transcription factors, including SAP130, and the histone deacetylases HDAC1 and 4, all enhanced the rough eye phenotype in Aβ42 flies (Cao et al. 2008).
Another screen for loss-of-function mutations that enhance or suppress the eye degeneration phenotype induced by Aβ42 identified that the Toll/NFκB signaling pathway as mediating human Aβ42 toxicity, suggesting the involvement of innate immunity/inflammatory pathway components in Aβ42 toxicity (Tan et al. 2008). In addition, recent study showed that Aβ42-induced toxicity was enhanced by the activation of autophagy and partially rescued by inhibition of autophagy, suggesting that autophagic-lysosomal injury is involved in Aβ42 toxicity (Ling et al. 2009).
Rival et al. conducted a genome wide gene-expression analysis and complementary genetic screen. Microarray analysis identified changes in the expression of oxidative stress-related genes induced by Aβ42 expression, and a subsequent genetic screen confirmed the importance of oxidative stress. The iron-binding protein ferritin and the H2O2 scavenger catalase are the most potent suppressors of wild-type and Arctic (E22G) Aβ42 toxicity, and oxidative stress mediated by the Arctic Aβ42 was reversed by ferritin despite an increase in the levels of Aβ42. Likewise, treatment with the iron-binding compound clioquinol increased the lifespan of flies expressing Arctic Aβ42 (Rival et al. 2009).
Deficiencies in the retromer sorting pathway have been linked to late-onset Alzheimer's disease (Small and Gandy 2006). Using transgenic flies expressing human APP and BACE, the role of the retromer sorting pathway in Aβ-induced neurodegeneration was studied. When APP and BACE are overexpressed, flies deficient in retromer complex components developed neuronal loss and Aβ aggregates (Muhammad et al. 2008). These results suggest that retromer deficiency observed in late-onset AD may contribute to the disease pathogenesis.
Recent reports suggest that, in EOAD caused by Psn mutations, more global perturbations in pathways involving other γ-secretase substrates should be considered in addition to Aβ42 toxicity (Baki et al. 2004; Marambaud and Robakis 2005; Marambaud et al. 2003; Saura et al. 2004). Recently, choosing from over 130 EOFAD mutations in Presenilin-1, Seidner et al. (2006) introduced 14 corresponding mutations at conserved residues in Drosophila Psn and assessed the Notch-cleavage activity of the mutants in transgenic flies. They found that the activity levels of the Psn mutants were tightly linked to their age-of-onset values, providing evidence that disease severity in humans primarily reflects differences in Psn lesions rather than contributions from unlinked genetic or environmental modifiers.
Drosophila models of tauopathies
Tau abnormalities are associated with AD and other neurodegenerative diseases
Neurofibrillary tangles are intracellular protein inclusions composed of abnormally hyperphosphorylated tau protein (Grundke-Iqbal et al. 1986a, b; Kosik et al. 1986; Wood et al. 1986). NFTs are associated with AD and a range of neurodegenerative diseases called tauopathies. These include corticobasal degeneration (CBD), Pick's disease (PiD), progressive supranuclear palsy (PSP), sporadic frontotemporal dementia (FTD), inherited frontotemporal dementia, and Parkinsonism linked to chromosome 17 (FTDP-17) (Lee et al. 2001).
Multiple tau gene mutations are identified in FTDP-17 patients, and tau polymorphisms appear to be genetic risk factors for other tauopathies (Lee et al. 2001). To date, tau mutations are not associated with any known form of familial AD. However, tau haplotypes driving slightly higher tau expression increase the risk of AD (Myers et al., 2005). More recent genetic association studies showed that tau variants associated with higher tau protein levels affect age onset of AD in the presence of Aβ pathology (Kauwe et al. 2008). These reports suggest that tau may play a role in the pathogenesis of AD as a modulator of disease progression.
Tau is a microtubule associated protein that is expressed in neurons and predominantly localizes in axons. Tau is also expressed in astrocytes and oligodendrocytes at lower levels, and, in some disease conditions, tau forms aggregates in these glial cells (Tashiro et al. 1997). In the adult human brain, alternative mRNA splicing produces six tau isoforms, which contain three repeats (3R tau) or four repeats (4R tau) of the microtubule-binding domain at the C-terminus. The major function of tau is to regulate the assembly and stability of microtubules, and 4R tau is approximately 40-fold more efficient at binding microtubules than 3R tau. In addition, 4R tau is more prone to form filaments than 3R tau (Lee et al. 2001).
Tau mutations that have been identified in FTDP-17 are missense, deletion, or silent mutations. These mutations change the relative tau isoform ratio, reduce the level of tau-microtubule binding, and/or increase tau aggregation. The autosomal dominant inheritance pattern of familial FTDP-17 and the prominent tau pathology in the brains of sporadic tauopathy patients suggest a gain-of-function mechanism in tau toxicity (Lee et al. 2001). However, the mechanisms by which tau toxicity mediates neuronal dysfunction and degeneration in the AD and other tauopathies are unknown.
The identification of pathogenic mutations in human Tau in familial FTDP-17 cases has greatly advanced the development of transgenic animal models of tauopathies. A variety of transgenic animal models including C. elegans, Drosophila, and mouse expressing human wild-type or FTDP-17-linked tau mutants have been generated (Gotz and Ittner 2008; Lee et al. 2005). These animal models recapitulate many important aspects of human diseases, and have been used to explore pathogenic mechanisms as well as to test and develop novel therapeutic strategies. We will briefly summarize Drosophila tauopathy models and a number of insights obtained from these models.
Modeling tauopathies in Drosophila
Human tau-induced neuronal dysfunction and degeneration have been successfully modeled in Drosophila. Tau-induced neurotoxicity in flies was first suggested by an anatomical study showing that transgenic expression of a fusion protein of bovine tau-green fluorescent protein in sensory neurons caused axonal degeneration (Williams et al. 2000). Subsequently, fly models of human tauopathy were created by expressing human wild-type or FTDP-17-linked tau mutants, R406W and V337M (Jackson et al. 2002; Wittmann et al. 2001). Expression of human wild-type or mutant tau in neurons caused an age-dependent progressive neurodegeneration characterized by nuclear fragmentation and vacuole formation in neurons of the cortex and neuropil (Nishimura et al. 2004; Wittmann et al. 2001). In addition, tau expression in fly eyes induced retinal toxicity in adult flies, which appears as a “rough” eye phenotype, characterized by a reduced external eye size and retina thickness, loss of the regular ommatidial organization and neuropil degeneration in the medulla (Jackson et al. 2002; Nishimura et al. 2004). Accumulation of disease-associated phospho- and conformational tau epitopes is observed in brains and eyes. Tau transgenic flies also have a reduced lifespan (Wittmann et al. 2001). Most pathological phenotypes induced by mutant tau are more severe than those induced by wild-type tau (Wittmann et al. 2001), suggesting that this fly model system recapitulates an important pathobiological aspect of tau.
In Drosophila genome, there is a single endogenous tau gene. The fly tau protein accumulates in axonal processes, as is the case with mammalian tau (Heidary and Fortini 2001). Interestingly, overexpression of the fly tau in neurons or eye induced apoptotic neuronal cell death (Chen et al. 2007).
Human tau- or fly tau-induced synaptic dysfunctions have also been studied in Drosophila larval motor neurons and neuromuscular junctions. Expression of tau within motor neurons causes morphological abnormalities in the presynaptic terminals and defects in synaptic transmission and microtubule-based axonal transport (Chee et al. 2005; Mudher et al. 2004). In addition, expression of human or fly tau in the mushroom body neurons, a center for olfactory learning and memory in flies, impairs associative olfactory learning and memory prior to the onset of neurodegeneration (Mershin et al. 2004).
Neurofibrillary tangle formation, a hallmark of AD pathology and tauopathies, is not observed in the fly neurons (Wittmann et al., 2001), indicating that tau toxicity is not conferred by large insoluble aggregates of tau in the Drosophila models. Studies in mouse models of tauopathy have shown that tau-induced memory defects and neuro-degeneration can be dissociated from NFT formation (Le Corre et al. 2006; Santacruz et al. 2005). These results suggest that some soluble, non-tangled form of phosphorylated tau is intrinsically toxic and that Drosophila models of tauopathies may recapitulate early, pretangle events of tau-associated neurodegeneration (Lee et al. 2005).
Insights into pathomechanisms and genetic modifiers of tau-induced toxicity from fly models
The rough eye phenotype is especially useful because the exterior eye size can be easily scored. Moreover, since the eye is not vital for fly survival, the tau eye phenotype provides a highly sensitive and robust readout for assessing phenotypic effects of genetic manipulations (Fig. 5) (Fortini and Bonini 2000). As summarized in this section, many groups have demonstrated that this system can lead to powerful insights into the pathophysiological mechanisms underlying tau toxicity.
Fig. 5.
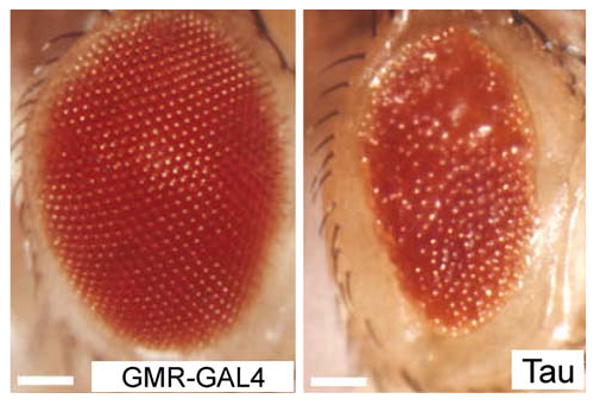
Neurotoxicity in the retina of tau transgenic flies. Compare normal eye structure (left) with transgenic wild-type tau-induced rough eye (right). The scale bars represent 10 μm. Adapted by permission from Macmillan Publishers Ltd: Nature Cell Biology, (Fulga et al. 2007), copyright (2007)
Roles of tau phosphorylation at disease-associated sites in tau toxicity
Tau protein is abnormally hyperphosphorylated and aggregated into NFT in the brains of AD and tauopathy patients. There are 79 putative Ser/Thr phosphorylation sites in the longest isoform of tau (441 amino acids), and at least 30 of those sites are phosphorylated in NFTs (Buee et al. 2000). In vitro studies have revealed that tau phosphorylation at some of these sites decreases microtubule binding or promotes tau aggregation (Gong et al. 2005). Since many of the mutations that cause FTDP-17 cluster around the microtubule binding domain and reduce microtubule binding or promote tau aggregation, an imbalance in phosphorylation and/or dephosphorylation of tau may initiate the abnormal metabolism and toxicity of tau in disease pathogenesis (Ballatore et al. 2007; Buee et al. 2000; Lee et al. 2001).
A number of studies have revealed that many kinases can phosphorylate tau in vitro (Gong et al. 2005). Among the 30 disease-associated phosphorylation sites, approximately half are targets for Serine/Proline (SP) or Threonine/Proline (TP) kinases (Gong et al. 2005). In vitro, these sites can be phosphorylated by glycogen synthase kinase-3β (GSK-3β), cyclin-dependent kinase-5, cyclin-dependent kinase-1 (Cdk1), cyclin-dependent kinase-2 (Cdk2), mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase (JNK). In contrast, other non-proline directed sites are phosphorylated by protein kinase A (PKA), protein kinase B (Akt), protein kinase C, calcium/calmodulin-dependent protein kinase II, or microtubule affinity-regulating kinase (MARK). These sites can be dephosphorylated by protein phosphatases 1, 2A, 2B, and 2C (Gong et al. 2005). In contrast, little is known about the role of kinases and phosphatases in the phosphorylation/dephosphorylation of specific tau sites, or the effects on aggregation and toxicity in vivo.
A forward genetic screen using Drosophila tauopathy models has identified kinases and phosphatases as modifiers of tau toxicity (Shulman and Feany 2003). In addition, candidate testing has revealed Cdk5 and GSK-3β as tau kinases (Jackson et al. 2002; Shulman and Feany 2003). Moreover, when fly GSK-3β is coexpressed with tau, large NFT-like aggregates form in photoreceptor neurons, and neurodegeneration is enhanced (Jackson et al. 2002), providing in vivo evidence that GSK-3β is involved in NFT formation.
To directly establish the role of tau phosphorylation at SP/TP sites in mediating toxicity, forms of tau with phosphorylation-incompetent or phosphorylation-mimicking Ser/Thr kinase sites were expressed (Nishimura et al. 2004; Steinhilb et al. 2007b). A tau construct in which all 14 SP/TP kinase target sites are mutated to alanine, thus rendering the protein phosphorylation incompetent, has greatly reduced neurotoxicity.(Steinhilb et al. 2007b). Conversely, a construct pseudo-phosphorylated at these 14 sites, by substitution of glutamate for the serines and threonines, substantially increases toxicity in both the fly eye and brain (Fulga et al. 2007; Steinhilb et al. 2007b). Moreover, single- or double-phosphorylation mutants, or a mutant with five altered residues (S202/205, T212, and T231/S235), did not exhibit substantially lower toxicity or accumulation of disease-associated conformational epitopes, compared to wild-type tau. These results suggest that the SP/TP sites work in concert to promote toxicity (Steinhilb et al. 2007a).
Among the non-proline directed phosphorylation sites, S262 and S356 have been shown to play critical roles in tau toxicity. Introducing alanine mutations at these sites dramatically reduced tau-induced neurodegeneration in fly eyes and brains (Nishimura et al. 2004). These sites are located in the microtubule binding domains and phosphorylation at these sites, especially S262, promotes dissociation of tau from microtubules in vitro (Biernat et al. 1993). These sites are phosphorylated by MARK kinases, which regulate microtubule dynamics, epithelial cell polarity, and neuronal differentiation under normal conditions and bind to NFT in AD brains (Lu 2009).
In the fly tauopathy model, overexpression of the Drosophila homolog of MARK (dMARK, also called partitioning defective-1, PAR-1) increases tau phosphorylation at S262 and S356 and enhances toxicity (Nishimura et al. 2004). Interestingly, dMARK was found to increase tau phosphorylation at multiple SP/TP sites, and mutating S262 and S356 to alanine (S2A tau) reduces phosphorylation at SP/TP sites. These results suggest that phosphorylation at S262 and S356 by dMARK enhances tau toxicity by promoting tau phosphorylation at the SP/TP sites by proline-directed kinases. However, a more recent study shows that S2A tau is resistant to GSK-3β induced phenotypic enhancement but not phosphorylation, suggesting that tau toxicity is not determined solely by phosphorylation (Chatterjee et al. 2009).
The upstream signaling mechanisms that regulate tau phosphorylation under normal and disease conditions are not fully understood. Recent studies have shown that the tumor suppressor protein LKB1 activates dMARK and promotes tau phosphorylation and toxicity in Drosophila. Because diverse stress stimuli, such as high osmolarity and human APP-induced neurotoxicity, can promote tau phosphorylation and toxicity through LKB1 and PAR-1 activation (Wang et al. 2007), the LKB1/PAR-1/tau phosphorylation cascade may be involved in AD pathogenesis.
Actin cytoskeleton
Intraneuronal actin-rich inclusions called Hirano bodies are found in many neurodegenerative diseases including AD and tauopathies (Hirano 1994), and also in the brain of mouse models of tauopathy (Fulga et al. 2007). Forward genetic screens with Drosophila tauopathy models have identified components of the actin cytoskeleton as modifiers of tau-induced retinal degeneration (Blard et al. 2007; Shulman and Feany 2003). Fulga et al. (2007) demonstrated that R406W tau-induced neurodegeneration is associated with accumulation of filamentous actin (F-actin)-containing rods in the Drosophila brain and retina. Actin directly interacts with tau in vivo, and the changes in actin structure occur downstream of tau phosphorylation. Furthermore, Aβ synergistically enhances the ability of wild-type tau to promote alterations in the actin cytoskeleton and neurodegeneration (Fig. 6).
Fig. 6.
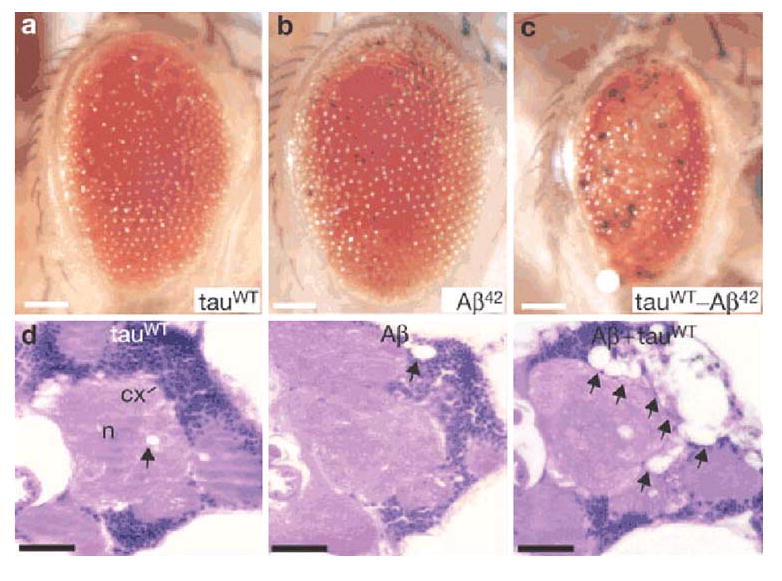
Aβ and tau interact in a synergistic manner to promote neuronal degeneration. a–c Retinal toxicity in flies expressing wild-type tau a, Aβ b and double-transgenic flies expressing wild-type tau and Aβ c. d Hematoxylin and eosin-stained frontal brain sections from flies expressing wild-type tau, Aβ or coexpressing wild-type tau and Aβ indicates that Aβ substantially enhances wild-type tau-induced neurodegeneration. Vacuolization of the neuropil (n) and cortex (cx) is indicated by arrows. Adapted by permission from Macmillan Publishers Ltd: Nature Cell Biology, (Fulga et al. 2007), copyright (2007)
Cell-cycle activation
Abnormal accumulations of cell-cycle proteins are observed in post-mitotic neurons in AD and tauopathies (Herrup and Yang 2007). Khurana et al. (2006) demonstrated that abnormal expression of cell-cycle markers is recapitulated in the brain of wild-type and R406W tau flies. An extensive genetic analysis showed that cell-cycle activators enhanced, and cell-cycle inhibitors suppressed, tau-induced neurodegeneration. Furthermore, this study showed that cell-cycle activation was downstream of tau phosphorylation and that activation of TOR (target of rapamycin kinase) by tau overexpression induced neurodegeneration in a cell-cycle-dependent manner (Khurana et al. 2006).
Oxidative stress
Oxidative stress occurs early in the progression of Alzheimer disease (Moreira et al. 2008). Genetic reduction in antioxidant defense enzymes enhanced, whereas activation of these enzymes or administration of the anti-oxidant α-tocopherol (vitamin E) suppressed, tau-induced neurotoxicity in the fly brain (Dias-Santagata et al. 2007). These manipulations did not affect tau phosphorylation but did alter tau-induced activation of the JNK pathway and cell-cycle activation. Since Aβ causes oxidative stress, these findings support the hypotheses that oxidative stress and activation of cell-cycle components may play a role in AD (Klein and Ackerman 2003).
Autophagy and lysosomal function
AD and tauopathies are associated with the accumulation of misfolded proteins, and the factors that regulate the clearance of these aggregation-prone proteins could be therapeutic targets. For example, TOR inhibition promotes autophagy and upregulation of TOR is observed in the AD brain (Williams et al. 2006). Berger et al. (2006) have shown that rapamycin promotes autophagy in a number of fly models of neurodegeneration, including a tauopathy model. Interestingly, the effect of rapamycin was more prominent to the toxicity induced by R406W tau than wild-type tau, suggesting that rapamycin treatment led to preferential degradation of non-microtubule-bound (and aggregation-prone) forms. In addition to autophagy, a study demonstrated that loss of Drosophila benchwarmer (bnch), a lysosomal sugar carrier, strongly enhances tau neurotoxicity, suggesting the importance of lysosomal activity in tau degradation (Dermaut et al. 2005).
Aminopeptidase
Each neurodegenerative disease selectively targets certain vulnerable neuronal populations, and the underlying mechanisms are largely unknown. In AD and tauopathies, the cerebellum is relatively resistant to neurodegeneration. A recent cross-species, functional genomic approach has identified a potential role for the puromycin-sensitive aminopeptidase (PSA) in selective neurodegeneration in tauopathy (Karsten et al. 2006). Several genes, including PSA, are prominently upregulated in the cerebellum, but not in other regions of the brain, in a transgenic tau P301L-expressing mouse. The upregulation of these genes in a relatively resistant brain region may indicate a protective role against tau-induced neurodegeneration (Karsten et al. 2006). Using the Drosophila tauopathy model, the effects of manipulation of these genes was tested. Overexpression of PSA suppressed, whereas PSA loss-of-function exacerbated, tau-induced neurodegeneration. Furthermore, human PSA was found to degrade tau. Finally, PSA was shown to be differentially expressed between human cerebellum and frontal cortex in both frontotemporal dementia cases and controls (Karsten et al. 2006). These data highlight the utility of using both mouse and fly models for genetic screening and functional assessment of modifiers of neurodegeneration.
Conclusions
Drosophila have proven to be excellent models for human neurodegenerative diseases as a powerful tool for both hypothesis-testing approaches and non-biased genome-wide screens (Bilen and Bonini 2005; Fortini and Bonini 2000; Khurana 2008; Lu and Vogel 2009; Marsh and Thompson 2004; Sang and Jackson 2005; Shulman et al. 2003). The fly models of neurodegenerative disease can be used to systematically evaluate the effects of known genetic and environmental risk factors, and candidates of susceptibility genes identified by genetic association studies with humans or that revealed by functional genomics with mouse models. Also, the genes discovered in the forward genetic approach in fly models may help reveal disease susceptibility genes in humans. Furthermore, because of the lack of a stringent blood–brain barrier in the fly, which allows compounds to easily gain access to the nervous system, fly models are also excellent in vivo model for the testing and screening of therapeutic compounds (Marsh and Thompson 2006). We anticipate that, in addition to genetic screens, drug screens using the fly models will play a significant role in the development of therapeutic avenues for AD and tauopathies in the future.
Acknowledgments
We apologize to those authors whose work we could not cite because of space constraints. We would like to dedicate this manuscript to the memory of our friend, Goemon Ando. This work was supported by start-up funds from the Farber Institute for Neurosciences, a pilot research grant from the Thomas Jefferson University, and grants from Gilbert Foundation/the American Federation for Aging Research, the Alzheimer's Association (NIRG-08-91985) and the National Institute of Health (R01AG032279).
Contributor Information
Kanae Iijima-Ando, Email: Kanae.Iijima-Ando@jefferson.edu, Laboratory of Neurogenetics and Pathobiology, Department of Biochemistry and Molecular Biology, Farber Institute for Neurosciences, Thomas Jefferson University, 900 Walnut Street, JHN410, Philadelphia, PA 19107, USA.
Koichi Iijima, Email: Koichi.Iijima@jefferson.edu, Laboratory of Neurodegenerative and Metabolic Diseases, Department of Biochemistry and Molecular Biology, Farber Institute for Neurosciences, Thomas Jefferson University, 900 Walnut Street, JHN410, Philadelphia, PA 19107, USA.
References
- Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res. 2003;74:342–352. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- Ashley J, Packard M, Ataman B, Budnik V. Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J Neurosci. 2005;25:5943–5955. doi: 10.1523/JNEUROSCI.1144-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 2004;23:2586–2596. doi: 10.1038/sj.emboj.7600251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neuro-degeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. The genetic epidemiology of neurode-generative disease. J Clin Invest. 2005;115:1449–1457. doi: 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- Blard O, Feuillette S, Bou J, Chaumette B, Frebourg T, Campion D, Lecourtois M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum Mol Genet. 2007;16:555–566. doi: 10.1093/hmg/ddm011. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brunkan AL, Goate AM. Presenilin function and gamma-secretase activity. J Neurochem. 2005;93:769–792. doi: 10.1111/j.1471-4159.2005.03099.x. [DOI] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Cao W, Song HJ, Gangi T, Kelkar A, Antani I, Garza D, Konsolaki M. Identification of novel genes that modify phenotypes induced by Alzheimer's beta-amyloid overexpression in Drosophila. Genetics. 2008;178:1457–1471. doi: 10.1534/genetics.107.078394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmine-Simmen K, Proctor T, Tschape J, Poeck B, Triphan T, Strauss R, Kretzschmar D. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol Dis. 2009;33:274–281. doi: 10.1016/j.nbd.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Sang TK, Lawless GM, Jackson GR. Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum Mol Genet. 2009;18:164–177. doi: 10.1093/hmg/ddn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee FC, Mudher A, Cuttle MF, Newman TA, MacKay D, Lovestone S, Shepherd D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol Dis. 2005;20:918–928. doi: 10.1016/j.nbd.2005.05.029. [DOI] [PubMed] [Google Scholar]
- Chen X, Li Y, Huang J, Cao D, Yang G, Liu W, Lu H, Guo A. Study of tauopathies by comparing Drosophila and human tau in Drosophila. Cell Tissue Res. 2007;329:169–178. doi: 10.1007/s00441-007-0401-y. [DOI] [PubMed] [Google Scholar]
- Cheng IH, Palop JJ, Esposito LA, Bien-Ly N, Yan F, Mucke L. Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat Med. 2004;10:1190–1192. doi: 10.1038/nm1123. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- Chiang HC, Iijima K, Hakker I, Zhong Y. Distinctive roles of different beta-amyloid 42 aggregates in modulation of synaptic functions. FASEB J. 2009;23:1969–1977. doi: 10.1096/fj.08-121152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M. Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci. 2004;5:677–685. doi: 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, Lomas DA. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Cummings JL. Cognitive and behavioral heterogeneity in Alzheimer's disease: seeking the neurobiological basis. Neurobiol Aging. 2000;21:845–861. doi: 10.1016/s0197-4580(00)00183-4. [DOI] [PubMed] [Google Scholar]
- Cummings JL. The neuropsychiatry of Alzheimer's disease and other dementias. Martin Dunitz; London: 2003. [Google Scholar]
- Deleault NR, Dolph PJ, Feany MB, Cook ME, Nishina K, Harris DA, Supattapone S. Post-transcriptional suppression of pathogenic prion protein expression in Drosophila neurons. J Neurochem. 2003;85:1614–1623. doi: 10.1046/j.1471-4159.2003.01819.x. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y, Callaerts P, Bellen HJ. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J Cell Biol. 2005;170:127–139. doi: 10.1083/jcb.200412001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117:236–245. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- Farris W, Schutz SG, Cirrito JR, Shankar GM, Sun X, George A, Leissring MA, Walsh DM, Qiu WQ, Holtzman DM, et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol. 2007;171:241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay DS, Fluet A, Johnson CJ, Link CD. In vivo aggregation of beta-amyloid peptide variants. J Neurochem. 1998;71:1616–1625. doi: 10.1046/j.1471-4159.1998.71041616.x. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer's Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Fortini ME, Bonini NM. Modeling human neurodegenerative diseases in Drosophila: on a wing and a prayer. Trends Genet. 2000;16:161–167. doi: 10.1016/s0168-9525(99)01939-3. [DOI] [PubMed] [Google Scholar]
- Fortini ME, Skupski MP, Boguski MS, Hariharan IK. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol. 2000;150:F23–F30. doi: 10.1083/jcb.150.2.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossgreen A, Bruckner B, Czech C, Masters CL, Beyreuther K, Paro R. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blisteredwing phenotype. Proc Natl Acad Sci USA. 1998;95:13703–13708. doi: 10.1073/pnas.95.23.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, Feldman RM, Guo M. ubiquilin antagonizes presenilin and promotes neurodegeneration in Drosophila. Hum Mol Genet. 2008;17:293–302. doi: 10.1093/hmg/ddm305. [DOI] [PubMed] [Google Scholar]
- Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, Mirkin CA. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer's disease. Proc Natl Acad Sci USA. 2005;102:2273–2276. doi: 10.1073/pnas.0409336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer's disease. J Neural Transm. 2005;112:813–838. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- Gotz J, Ittner LM. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve I, Kretzschmar D, Tschape JA, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 2004;24:3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GG, Feldman RM, Ganguly A, Wang J, Yu H, Guo M. Role of X11 and ubiquilin as in vivo regulators of the amyloid precursor protein in Drosophila. PLoS One. 2008;3:e2495. doi: 10.1371/journal.pone.0002495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986a;261:6084–6089. [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986b;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- Guo M, Hong EJ, Fernandes J, Zipursky SL, Hay BA. A reporter for amyloid precursor protein gamma-secretase activity in Drosophila. Hum Mol Genet. 2003;12:2669–2678. doi: 10.1093/hmg/ddg292. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurode-generation: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hama E, Shirotani K, Masumoto H, Sekine-Aizawa Y, Aizawa H, Saido TC. Clearance of extracellular and cell-associated amyloid beta peptide through viral expression of neprilysin in primary neurons. J Biochem. 2001;130:721–726. doi: 10.1093/oxfordjournals.jbchem.a003040. [DOI] [PubMed] [Google Scholar]
- Hamazaki H. Cathepsin D is involved in the clearance of Alzheimer's beta-amyloid protein. FEBS Lett. 1996;396:139–142. doi: 10.1016/0014-5793(96)01087-3. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- Hase M, Yagi Y, Taru H, Tomita S, Sumioka A, Hori K, Miyamoto K, Sasamura T, Nakamura M, Matsuno K, et al. Expression and characterization of the Drosophila X11-like/Mint protein during neural development. J Neurochem. 2002;81:1223–1232. doi: 10.1046/j.1471-4159.2002.00911.x. [DOI] [PubMed] [Google Scholar]
- Heidary G, Fortini ME. Identification and characterization of the Drosophila tau homolog. Mech Dev. 2001;108:171–178. doi: 10.1016/s0925-4773(01)00487-7. [DOI] [PubMed] [Google Scholar]
- Heisenberg M. Mushroom body memoir: from maps to models. Nat Rev Neurosci. 2003;4:266–275. doi: 10.1038/nrn1074. [DOI] [PubMed] [Google Scholar]
- Hemming ML, Patterson M, Reske-Nielsen C, Lin L, Isacson O, Selkoe DJ. Reducing amyloid plaque burden via ex vivo gene delivery of an Abeta-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Med. 2007;4:e262. doi: 10.1371/journal.pmed.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci. 2007;8:368–378. doi: 10.1038/nrn2124. [DOI] [PubMed] [Google Scholar]
- Hirano A. Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol. 1994;20:3–11. doi: 10.1111/j.1365-2990.1994.tb00951.x. [DOI] [PubMed] [Google Scholar]
- Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M, Staufenbiel M, Noda Y, Yamaguchi H, Nabeshima T, et al. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem. 2006;281:17941–17951. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- Iijima K, Iijima-Ando K. Drosophila models of Alzheimer's amyloidosis: the challenge of dissecting the complex mechanisms of toxicity of amyloid-beta 42. J Alzheimers Dis. 2008;15:523–540. doi: 10.3233/jad-2008-15402. [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS ONE. 2008;3:e1703. doi: 10.1371/journal.pone.0001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima-Ando K, Hearn SA, Granger L, Shenton C, Gatt A, Chiang HC, Hakker I, Zhong Y, Iijima K. Overexpression of neprilysin reduces alzheimer amyloid-beta42 (Abeta42)-induced neuron loss and intraneuronal Abeta42 deposits but causes a reduction in cAMP-responsive element-binding protein-mediated transcription, age-dependent axon pathology, and premature death in Drosophila. J Biol Chem. 2008;283:19066–19076. doi: 10.1074/jbc.M710509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Iwata N, Mizukami H, Shirotani K, Takaki Y, Muramatsu S, Lu B, Gerard NP, Gerard C, Ozawa K, Saido TC. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain. J Neurosci. 2004;24:991–998. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Higuchi M, Saido TC. Metabolism of amyloid-beta peptide and Alzheimer's disease. Pharmacol Ther. 2005;108:129–148. doi: 10.1016/j.pharmthera.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–642. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human Wild-Type Tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- Johansson AS, Berglind-Dehlin F, Karlsson G, Edwards K, Gellerfors P, Lannfelt L. Physiochemical characterization of the Alzheimer's disease-related peptides A beta 1-42Arctic and A beta 1-42wt. FEBS J. 2006;273:2618–2630. doi: 10.1111/j.1742-4658.2006.05263.x. [DOI] [PubMed] [Google Scholar]
- Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM, Sengupta S, Berry RW, Pomakian J, Oh HS, et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neuro-degeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Kauwe JS, Cruchaga C, Mayo K, Fenoglio C, Bertelsen S, Nowotny P, Galimberti D, Scarpini E, Morris JC, Fagan AM, et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci USA. 2008;105:8050–8054. doi: 10.1073/pnas.0801227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kelly JW. Attacking amyloid. N Engl J Med. 2005;352:722–723. doi: 10.1056/NEJMcibr044231. [DOI] [PubMed] [Google Scholar]
- Khurana V. Modeling Tauopathy in the fruit fly Drosophila melanogaster. J Alzheimers Dis. 2008;15:541–553. doi: 10.3233/jad-2008-15403. [DOI] [PubMed] [Google Scholar]
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16:230–241. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Klein JA, Ackerman SL. Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest. 2003;111:785–793. doi: 10.1172/JCI18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lee VM, Kenyon TK, Trojanowski JQ. Transgenic animal models of tauopathies. Biochim Biophys Acta. 2005;1739:251–259. doi: 10.1016/j.bbadis.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Leyssen M, Ayaz D, Hebert SS, Reeve S, De Strooper B, Hassan BA. Amyloid precursor protein promotes post-developmental neurite arborization in the Drosophila brain. EMBO J. 2005;24:2944–2955. doi: 10.1038/sj.emboj.7600757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Xie Z, Dong Y, McKay KM, McKee ML, Tanzi RE. Isolation and characterization of the Drosophila ubiquilin ortholog dUbqln: in vivo interaction with early-onset Alzheimer disease genes. Hum Mol Genet. 2007;16:2626–2639. doi: 10.1093/hmg/ddm219. [DOI] [PubMed] [Google Scholar]
- Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE. 2009;4:e4201. doi: 10.1371/journal.pone.0004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Lu B. Recent advances in using Drosophila to model neurodegenerative diseases. Apoptosis. 2009;14:1008–1020. doi: 10.1007/s10495-009-0347-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luheshi LM, Tartaglia GG, Brorsson AC, Pawar AP, Watson IE, Chiti F, Vendruscolo M, Lomas DA, Dobson CM, Crowther DC. Systematic in vivo analysis of the intrinsic determinants of amyloid Beta pathogenicity. PLoS Biol. 2007;5:e290. doi: 10.1371/journal.pbio.0050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Tully T, White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron. 1992;9:595–605. doi: 10.1016/0896-6273(92)90024-8. [DOI] [PubMed] [Google Scholar]
- Mah AL, Perry G, Smith MA, Monteiro MJ. Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol. 2000;151:847–862. doi: 10.1083/jcb.151.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney MB, Parks AL, Ruddy DA, Tiong SY, Esengil H, Phan AC, Philandrinos P, Winter CG, Chatterjee R, Huppert K, et al. Presenilin-based genetic screens in Drosophila melanogaster identify novel notch pathway modifiers. Genetics. 2006;172:2309–2324. doi: 10.1534/genetics.104.035170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Robakis NK. Genetic and molecular aspects of Alzheimer's disease shed light on new mechanisms of transcriptional regulation. Genes Brain Behav. 2005;4:134–146. doi: 10.1111/j.1601-183X.2005.00086.x. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, Verma IM, Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM. Can flies help humans treat neurodegenerative diseases? Bioessays. 2004;26:485–496. doi: 10.1002/bies.20029. [DOI] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM. Drosophila in the study of neurodegenerative disease. Neuron. 2006;52:169–178. doi: 10.1016/j.neuron.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Mershin A, Pavlopoulos E, Fitch O, Braden BC, Nanopoulos DV, Skoulakis EM. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem. 2004;11:277–287. doi: 10.1101/lm.70804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micchelli CA, Esler WP, Kimberly WT, Jack C, Berezovska O, Kornilova A, Hyman BT, Perrimon N, Wolfe MS. Gamma-secretase/presenilin inhibitors for Alzheimer's disease phenocopy Notch mutations in Drosophila. FASEB J. 2003;17:79–81. doi: 10.1096/fj.02-0394fje. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Santos MS, Oliveira CR, Shenk JC, Nunomura A, Smith MA, Zhu X, Perry G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol Disord Drug Targets. 2008;7:3–10. doi: 10.2174/187152708783885156. [DOI] [PubMed] [Google Scholar]
- Morimoto A, Irie K, Murakami K, Masuda Y, Ohigashi H, Nagao M, Fukuda H, Shimizu T, Shirasawa T. Analysis of the secondary structure of beta-amyloid (Abeta42) fibrils by systematic proline replacement. J Biol Chem. 2004;279:52781–52788. doi: 10.1074/jbc.M406262200. [DOI] [PubMed] [Google Scholar]
- Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, Herman M, Ho L, Kreber R, Honig LS, et al. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci USA. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Irie K, Morimoto A, Ohigashi H, Shindo M, Nagao M, Shimizu T, Shirasawa T. Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer's disease. J Biol Chem. 2003;278:46179–46187. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- Myers AJ, Kaleem M, Marlowe L, Pittman AM, Lees AJ, Fung HC, Duckworth J, Leung D, Gibson A, Morris CM, et al. The H1c haplotype at the MAPT locus is associated with Alzheimer's disease. Hum Mol Genet. 2005;14:2399–2404. doi: 10.1093/hmg/ddi241. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Nerelius C, Sandegren A, Sargsyan H, Raunak R, Leijonmarck H, Chatterjee U, Fisahn A, Imarisio S, Lomas DA, Crowther DC, et al. Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proc Natl Acad Sci USA. 2009;106:9191–9196. doi: 10.1073/pnas.0810364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Periz G, Fortini ME. Functional reconstitution of gamma-secretase through coordinated expression of presenilin, nicastrin, Aph-1, and Pen-2. J Neurosci Res. 2004;77:309–322. doi: 10.1002/jnr.20203. [DOI] [PubMed] [Google Scholar]
- Poirier R, Wolfer DP, Welzl H, Tracy J, Galsworthy MJ, Nitsch RM, Mohajeri MH. Neuronal neprilysin overexpression is associated with attenuation of Abeta-related spatial memory deficit. Neurobiol Dis. 2006;24:475–483. doi: 10.1016/j.nbd.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prion biology and diseases. 2nd. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2004. [Google Scholar]
- Qiu WQ, Borth W, Ye Z, Haass C, Teplow DB, Selkoe DJ. Degradation of amyloid beta-protein by a serine protease-alpha2-macroglobulin complex. J Biol Chem. 1996;271:8443–8451. doi: 10.1074/jbc.271.14.8443. [DOI] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- Quinn WG, Harris WA, Benzer S. Conditioned behavior in Drosophila melanogaster. Proc Natl Acad Sci USA. 1974;71:708–712. doi: 10.1073/pnas.71.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeber AJ, Muramoto T, Kornberg TB, Prusiner SB. Expression and targeting of Syrian hamster prion protein induced by heat shock in transgenic Drosophila melanogaster. Mech Dev. 1995;51:317–327. doi: 10.1016/0925-4773(95)00379-7. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, et al. Efficient inhibition of the Alzheimer's disease beta-secretase by membrane targeting. Science. 2008;320:520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- Ratnaparkhi A, Lawless GM, Schweizer FE, Golshani P, Jackson GR. A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE. 2008;3:e2334. doi: 10.1371/journal.pone.0002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, Liu B, Lewis H, Rosahl T, Hider R, Camargo LM, et al. Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer's disease. Eur J Neurosci. 2009;29:1335–1347. doi: 10.1111/j.1460-9568.2009.06701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochet JC, Lansbury PT., Jr Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- Rooke J, Pan D, Xu T, Rubin GM. KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science. 1996;273:1227–1231. doi: 10.1126/science.273.5279.1227. [DOI] [PubMed] [Google Scholar]
- Rorth P, Szabo K, Bailey A, Laverty T, Rehm J, Rubin GM, Weigmann K, Milan M, Benes V, Ansorge W, et al. Systematic gain-of-function genetics in Drosophila. Development. 1998;125:1049–1057. doi: 10.1242/dev.125.6.1049. [DOI] [PubMed] [Google Scholar]
- Sang TK, Jackson GR. Drosophila models of neurodegenerative disease. NeuroRx. 2005;2:438–446. doi: 10.1602/neurorx.2.3.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y, Syuzo-Takabatake A, Nakaya T, Saito Y, Tomita S, Itohara S, Suzuki T. Enhanced amyloidogenic metabolism of the amyloid beta-protein precursor in the X11L-deficient mouse brain. J Biol Chem. 2006;281:37853–37860. doi: 10.1074/jbc.M609312200. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura C, Choi SY, Beglopoulos V, Malkani S, Zhang D, Rao BSS, Chattarji S, Kelleher R, Kandel E, Duff K, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat Med. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- Seidner GA, Ye Y, Faraday MM, Alvord WG, Fortini ME. Modeling clinically heterogeneous presenilin mutations with transgenic Drosophila. Curr Biol. 2006;16:1026–1033. doi: 10.1016/j.cub.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, Kiryu-Seo S, Kiyama H, Iwata H, Tomita T, et al. Neprilysin degrades both amyloid beta peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem. 2001;276:21895–21901. doi: 10.1074/jbc.M008511200. [DOI] [PubMed] [Google Scholar]
- Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman JM, Shulman LM, Weiner WJ, Feany MB. From fruit fly to bedside: translating lessons from Drosophila models of neurodegenerative disease. Curr Opin Neurol. 2003;16:443–449. doi: 10.1097/01.wco.0000084220.82329.60. [DOI] [PubMed] [Google Scholar]
- Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C. Unfolding the role of protein misfolding in neurode-generative diseases. Nat Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- Spencer B, Marr RA, Rockenstein E, Crews L, Adame A, Potkar R, Patrick C, Gage FH, Verma IM, Masliah E. Long-term neprilysin gene transfer is associated with reduced levels of intracellular Abeta and behavioral improvement in APP transgenic mice. BMC Neurosci. 2008;9:109. doi: 10.1186/1471-2202-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell. 2007a;18:5060–5068. doi: 10.1091/mbc.E07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Mulkearns EE, Shulman JM, Biernat J, Mandelkow EM, Feany MB. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J Neurosci Res. 2007b;85:1271–1278. doi: 10.1002/jnr.21232. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- Tan L, Schedl P, Song HJ, Garza D, Konsolaki M. The Toll–>NFkappaB signaling pathway mediates the neuropathological effects of the human Alzheimer's Abeta42 polypeptide in Drosophila. PLoS One. 2008;3:e3966. doi: 10.1371/journal.pone.0003966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Tashiro K, Hasegawa M, Ihara Y, Iwatsubo T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport. 1997;8:2797–2801. doi: 10.1097/00001756-199708180-00029. [DOI] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ. 2006;2006:re1. doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol. 1999a;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V. The Drosophila beta-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci. 1999b;19:7793–7803. doi: 10.1523/JNEUROSCI.19-18-07793.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol [A] 1985;157:263–277. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- van de Hoef DL, Hughes J, Livne-Bar I, Garza D, Konsolaki M, Boulianne GL. Identifying genes that interact with Drosophila presenilin and amyloid precursor protein. Genesis. 2009;47:246–260. doi: 10.1002/dvg.20485. [DOI] [PubMed] [Google Scholar]
- Waddell S, Quinn WG. What can we teach Drosophila? What can they teach us? Trends Genet. 2001;17:719–726. doi: 10.1016/s0168-9525(01)02526-4. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci. 2007;27:574–581. doi: 10.1523/JNEUROSCI.5094-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- Watson MR, Lagow RD, Xu K, Zhang B, Bonini NM. A Drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem. 2008;283:24972–24981. doi: 10.1074/jbc.M804817200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen BM, Selkoe DJ, Hartley DM. Small non-fibrillar assemblies of amyloid beta-protein bearing the Arctic mutation induce rapid neuritic degeneration. Neurobiol Dis. 2005;20:254–266. doi: 10.1016/j.nbd.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Williams DW, Tyrer M, Shepherd D. Tau and tau reporters disrupt central projections of sensory neurons in Drosophila. J Comp Neurol. 2000;428:630–640. doi: 10.1002/1096-9861(20001225)428:4<630::aid-cne4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- Wirths O, Weis J, Szczygielski J, Multhaup G, Bayer TA. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer's disease. Acta Neuropathol (Berl) 2006;111:312–319. doi: 10.1007/s00401-006-0041-4. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurode-generation without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) Proc Natl Acad Sci USA. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff-Pak DS. Animal models of Alzheimer's disease: therapeutic implications. J Alzheimers Dis. 2008;15:507–521. doi: 10.3233/jad-2008-15401. [DOI] [PubMed] [Google Scholar]
- Yagi Y, Tomita S, Nakamura M, Suzuki T. Overexpression of human amyloid precursor protein in Drosophila. Mol Cell Biol Res Commun. 2000;4:43–49. doi: 10.1006/mcbr.2000.0248. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci Lett. 2001a;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- Yasojima K, McGeer EG, McGeer PL. Relationship between beta amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 2001b;919:115–121. doi: 10.1016/s0006-8993(01)03008-6. [DOI] [PubMed] [Google Scholar]
- Ye Y, Fortini ME. Apoptotic activities of wild-type and Alzheimer's disease-related mutant presenilins in Drosophila melanogaster. J Cell Biol. 1999;146:1351–1364. doi: 10.1083/jcb.146.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]