

Abstract
Arsenic is a widespread environmental toxicant with a diverse array of molecular targets and associated diseases, making the identification of the critical mechanisms and pathways of arsenic-induced cytotoxicity a challenge. In a variety of experimental models, over a range of arsenic exposure levels, apoptosis is a commonly identified arsenic-induced cytotoxic pathway. Human lymphoblastoid cell lines (LCL) have been used as a model system in arsenic toxicology for many years, but the exact mechanism of arsenic-induced cytotoxicity in LCL is still unknown. We investigated the cytotoxicity of sodium arsenite in LCL 18564 using a set of complementary markers for cell death pathways. Markers indicative of apoptosis (phosphatidylserine externalization, PARP cleavage, and sensitivity to caspase inhibition) were uniformly negative in arsenite exposed cells. Interestingly, electron microscopy, acidic vesicle fluorescence, and expression of LC3 in LCL 18564 identified autophagy as an arsenite-induced process that was associated with cytotoxicity. Autophagy, a cellular programmed response that is associated with both cellular stress adaptation as well as cell death appears to be the predominant process in LCL cytotoxicity induced by arsenite. It is unclear, however, whether LCL autophagy is an effector mechanism of arsenite cytotoxicity or alternatively a cellular compensatory mechanism. The ability of arsenite to induce autophagy in lymphoblastoid cell lines introduces a potentially novel mechanistic explanation of the well-characterized in vitro and in vivo toxicity of arsenic to lymphoid cells.
Keywords: Sodium arsenite, Autophagy, Cytotoxicity, Lymphoblastoid
Introduction
Human arsenic toxicology has been a scientific focus for centuries, but its mechanism of action continues to remain a contemporary conundrum. Arguably no other toxicant is associated with a comparably diverse collection of disrupted cellular pathways, tissue targets and associated diseases that produce degenerative and cancerous disorders. In the skin arsenic induces both non-cancerous pathology (palmoplantar keratosis, pigmentation loss and gain) as well as skin cancer (Guo et al., 2001). In peripheral nerve tissue arsenic induces a neuropathy characterized by paresthesia and impaired conduction velocity (Mukherjee et al., 2003). In the lung arsenic induces a non-cancerous severe obstructive condition called bronchiectasis as well as causing lung cancer (Smith et al., 2006). The immune system is also a target of arsenic toxicity, with evidence of lymphoid cell toxicity in epidemiological studies of humans as well as in ex vivo studies of lymphocytes (Gonsebatt et al., 1992; Raqib et al., 2009).
A similar level of diversity exists in the cellular processes and molecular targets that have been shown to be disrupted by arsenic. The molecular targets of arsenic include several proteins to which trivalent arsenic has been shown to bind, presumably via protein thiol groups (Zhang et al., 2007; Piatek et al., 2008). Bacterial and eukaryotic assays of mutagenesis have consistently failed to identify arsenic compounds as mutagenic, although arsenic does appear to induce clastogenic changes such as sister chromatid exchange, chromosomal aberrations and micronuclei formation (Hartmann and Speit, 1994). These observations are not entirely surprising given the broad spectrum of transcriptional activation pathways that arsenic has been shown to modulate, including NF-kappa B, AP-1, MAPK and p53 (Barchowsky et al., 1996; Salazar et al., 1997; Simeonova and Luster, 2000; Simeonova et al., 2000).
Despite this array of effects, some commonality exists in arsenic toxicology, including the ability of arsenic to induce apoptotic cell death under a variety of circumstances. At concentrations ranging from 0.1 uM to 100 uM, in cell lines of diverse lineage, arsenic has been demonstrated to be an apoptosis-inducing agent (Pulido and Parrish, 2003). This characteristic of arsenic has certainly had a role in the recent appearance of arsenic as an actively researched, clinically proven, anti-neoplastic compound. In HL-60, an acute promyelocytic leukemia cell line, several groups have established the induction of apoptosis by arsenic through mechanisms that may be initiated by reactive oxygen species (ROS) generation as well as direct arsenic-targeting of the mitochondrial permeability transition pore (Zhu et al., 1999; Sturlan et al., 2003). Charoensuk et al., documented morphological characteristics of apoptosis (membrane blebbing, chromatin condensation) in HL-60 cells exposed to several arsenic species, with particular potency in the trivalent arsenicals (Charoensuk et al., 2009). Glienke et al., also documented apoptosis in HL60 cells exposed to 1 uM arsenic trioxide for 48 hours (h) (Glienke et al., 2006). Notwithstanding these studies, conclusions regarding the mechanism of action of arsenic in HL-60 cells are not unanimous. Yang et al., provided evidence that arsenic trioxide-exposed HL-60 cells were undergoing cytotoxicity that was characterized by apoptosis, but also concurrently by another process, namely autophagy (Yang et al., 2008).
Autophagy is an organized cellular program that degrades superfluous or damaged organelles, portions of the cytosol, and a particular subset of cellular proteins. In autophagy the targets of degradation are enclosed within a vesicular structure called an autophagosome. The autophagosome then fuses with a lysosome, forming an autolysosome, in which hydrolytic lysosomal enzymes degrade the contents. The exception to this scheme occurs in chaperone-mediated autophagy, a process by which damaged proteins are directly delivered to autolysosomes. Even though the first report of autophagy was made over forty years ago, the process has only recently been an intense focus of scientific research (Yorimitsu and Klionsky, 2005). Autophagy is a highly conserved eukaryotic process, with many pathway members having been initially identified in Saccharomyces cerevisiae. In contrast to apoptosis, a process that launches a signaling cascade solely targeted to the ultimate death of the cell, understanding the functional context of autophagy is complicated in that autophagy can either be a constitutive homeostatic process, a stress-induced cell survival mechanism, or a stress-induced mechanism tied to the death of the cell, depending on the particular context in which the process occurs.
Our interest in arsenic-associate immunotoxicity in exposed human populations led to the study of lymphoblastoid cell lines (LCL) as an in vitro model system of normal human lymphocytes, an established arsenic target. While LCL have been studied for many years as models of arsenic toxicity, the specific mechanism or mechanisms of arsenic cytotoxicity in LCL has not yet been defined. Given the well-established role of autophagy within the immune system (Deretic and Levine, 2009), the possible involvement of autophagy in arsenic-induced cytotoxicity in LCL was of interest. Understanding the processes that underlie this cytotoxicity is key to understanding arsenic-induced immunopathology at the organism level.
Methods
Reagents
Sodium arsenite (dissolved in MilliQ H2O) and the proteasome inhibitor, epoxomicin (dissolved in dimethyl sulfoxide (DMSO)) were purchased from Sigma Aldrich (St. Louis, MO). The pan caspase inhibitor, quinolyl-valyl-O-methylaspartyl- [-2,6-difluorophenoxy]- methyl ketone (QVD) (dissolved in DMSO) was purchased from R&D Systems (Minneapolis, MN). The autophagy inhibitor, bafilomycin A1 (BafA1) was purchased from Enzo Life Sciences (Plymouth Meeting, PA).
Cell Culture and Exposure Conditions
The human Epstein-Barr virus (EBV) immortalized LCL GM18564, derived from a healthy female, was purchased from Coriell Cell Repositories (Camden, NJ). Cultures were maintained in RPMI media supplemented with L-glutamine, 15 % fetal bovine serum (FBS) and 1% antibiotic- antimycotic (ABAM) solution (Invitrogen, Carlsbad, CA). Cells were grown in T-25 culture flasks and the cultures were maintained between the cell concentrations of 350,000-2,000,000 cells/ml at 5 % CO2 and 37 °C. Cell cultures were seeded to 350,000-500,000 cells/ml of media and dosed with sodium arsenite, epoxomicin, or BafA1 for the indicated exposure length and then harvested by centrifugation and subsequently processed and analyzed by particular methods mentioned below.
Cytotoxicity Assay
Cells were exposed to sodium arsenite for 96 hours (h) and cell viability was analyzed using the Annexin V-FITC Apoptosis Detection Kit (Sigma Aldrich, St. Louis, MO) and flow cytometry following manufacturer's protocol. This assay uses fluorescein-labeled annexin-V (AV) to label phosphatidylserine and propidium iodide (PI) to label genomic DNA. Samples were analyzed using a LSR II Flow Cytometer (BD Biosciences, Sparks, MD). AV fluorescence was collected through the 525/50 bandpass filter and PI fluorescence was collected through the 660/20 bandpass filter. Data were analyzed using FacsDiva software (BD Biosciences, Sparks, MD), with fluorescence values plotted as dot plots comparing the level of AV fluorescence verses the level of PI fluorescence per cell in each sample. Quadrants were established in the dot plots based on the location of the live cell (AV-negative, PI-negative) cluster in control sample. Live cells for subsequent treatment groups for a given experiment were then defined as cells occurring in this lower-left quadrant in dot plots. To calculate arsenite-induced cytotoxicity, the percentage (of all cells measured) of live cells at each exposure condition was divided by the percentage of live cells in the vehicle control group for that experiment, with this ratio was expressed as percent cell survival.
Effect of caspase inhibition on arsenite and epoxomicin cytotoxicity
Cells were exposed to arsenite (6 uM for 96 h) or the apoptosis-inducing proteasome inhibitor epoxomicin (1 uM for 6 h) alone, or with a pre-treatment of the pan-caspase inhibitor, QVD (10 uM for 1 h prior to arsenic or epoxomicin exposure, continuing throughout exposure). Cells were then harvested and stained with AV/PI dyes and analyzed by flow cytometry as described above. To determine the extent to which QVD inhibited cell death in either the epoxomicin or sodium arsenite exposed cells, the percent of non-live cells (defined as all cells measured outside of the live quadrant) in the QVD pre-treatment group was divided by the percent of non-live cells in the group without QVD pre-treatment (perturbant alone). The resulting ratio was expressed as a percent. Separate calculations were made for epoxomicin-exposed and arsenite-exposed cells.
Western Blot for PARP
Cell pellets were lysed in RIPA #1 lysis buffer (5 M NaCl, 0.5 M Na2HPO4, 0.5 M NaH2PO4, 0.25 M EDTA, 10 % Sodium Deoxycholate, 20 % NP40, 10 % SDS, and protease inhibitors); samples were then sonicated and centrifuged. Supernatant was isolated and stored at -20 °C until use. Protein concentrations were determined using the Coomassie Protein Assay Kit (Thermo Scientific, Rockford, IL) and Ultraviolet Spectroscopy. Equal amounts of whole cell lysates were subjected to SDS-polyacrylamide gel (8 % acrylamide) electrophoresis, transferred to nitrocellulose membrane, and immunoblotted with antibodies. The following antibodies were used: rabbit anti-poly-ADP ribose polymerase (PARP) antibody, 1:1500 (Cell Signaling Technologies, Beverly, MA); mouse anti-actin antibody, 1:1500 (Millipore, Billerica, MA); goat anti-rabbit IgG-HRP antibody, 1:5000 (Santa Cruz Biotechnology, Santa Cruz, CA); and rabbit anti-mouse IgG, HRP, F(ab')2 antibody, 1:8000 (Millipore, Billerica, MA). The membrane was visualized by chemiluminescence detection (Thermo Scientific, Rockford, IL) and autoradiography.
Western Blot for LC3
Vehicle control and arsenite exposed cells were harvested with or without co-treatment with 100 nM BafA1 for the last 8 and 4 h of the exposure duration. Cell pellets were lysed in 1× Sample Buffer (50 mM Tris-HCl, 2 % Sodium Deoxycholate, 10 % glycerol, 100 mM DL-Dithiothreitol, and 0.1 % Bromophenol blue); samples were then sonicated and heated to 90 °C for 5 minutes (min). Cell lysates were stored at -20 °C until use. Equal amounts of whole cell lysates were subjected to SDS-polyacrylamide gel (10 % acrylamide (actin) and 12 % acrylamide (LC3)) electophoresis, transferred to nitrocellulose membranes, and immunoblotted with antibodies. The following antibodies were used: mouse anti-microtubule-associated protein 1 light chain 3B (LC3) antibody, 1:200 (Nano Tools, San Diego, CA); mouse anti-actin antibody, 1:8000 (Millipore, Billerica, MA); and rabbit anti-mouse IgG, HRP, F(ab')2 antibody, 1:8000 (LC3) and 1:20000 (actin) (Millipore, Billerica, MA). The membranes were visualized by chemiluminescence detection (Thermo Scientific, Rockford, IL) and autoradiography.
Electron Microscopy
Cells (5 × 106) were harvested following exposure to sodium arsenite (6 uM for 96 h) or vehicle control. Cells were fixed in 2.5 % glutaraldehyde in 0.1 M cacodylate buffer, postfixed in 1 % osmium tetroxide, washed and pelleted. Cells were then stained in 4 % aqueous uranyl acetate, dehydrated with ethanol infiltrated with Spurr's Resin, and allowed to polymerize overnight at 60 °C. Silver-gold sections (70-100 nm) were cut, mounted on 150 mesh copper grids, and stained with 2 % lead acetate for 3 min. Sections were examined using a FEI (Philips) CM12 Transmission Electron Microscope (TEM) operated at 80 kv and images collected on an Optronics Macrofire AMT 542 digital camera.
Lysotracker Red Staining
Cells (1 × 106) were harvested, centrifuged, and resuspended in 20 nM Lysotracker Red dye (LRD) (Invitrogen, Carlsbad, CA) and incubated at 37 °C for 30 min. Cells were then washed once in phosphate buffered saline (PBS) (Invitrogen, Carlsbad, CA) and resuspended in 500 ul PBS at a final cell concentration of 2 × 106 cells/ml. Samples were analyzed by flow cytometry on a LSR II Flow Cytometer (BD Biosciences, Sparks, MD). Fluorescence of LRD was collected through the 610/20 nm bandpass filter. Data were analyzed using FacsDiva (BD Biosciences, Sparks, MD) and FlowJo (Tree Star, Inc. Ashland, OR) software. In order to measure the effect of the autophagy inhibitor BafA1 on LRD fluorescence, 100 nM BafA1 was added to arsenite exposed cells for the last 4 h of the exposure length. The co-exposed cells were harvested and analyzed in the same way as the single-exposure samples mentioned above.
Fluorescence Microscopy
Cells (1 × 106) were harvested after exposure to 6 uM sodium arsenite for 96 h, centrifuged and stained with LRD as described above. After the incubation period, cells were centrifuged, washed once in PBS and resuspended in 500 ul PBS. PBS-suspended, stained cells were placed on glass slides and examined using a Bio-Rad MRC 1024 confocal system (Hercules, CA), which is mounted on an Olympus BX50 fluorescence microscope (Center Valley, PA). An argon laser was excited to 568 nm and LRD fluorescence was collected using a 585 long pass filter. All images were taken with a 60× objective lens with oil immersion using Laser Sharp 2000 software (Bio-Rad, Hercules, CA).
Statistical analysis
Comparisons of LCL18564 under control and treated conditions was evaluated by paired Student's t-test using SPSS version 15.0 (SPSS/IBM, Chicago, Illinois, USA).
Results
Cytotoxicity Assay
A two-color fluorescence cytotoxicity assay that labeled phosphatidylserine with FITC-tagged annexin-V (AV) and labeled cell membrane-permeable cells with propidium iodide (PI) was used to evaluate cytotoxicity. Cells undergoing canonical apoptosis display high AV fluorescence, due to externalized phosphatidylserine, and low PI fluorescence reflecting an intact plasma membrane that excludes PI. Cells with compromised plasma membrane structure display high PI fluorescence. Thus, high PI fluorescence is not specific for a particular cell death pathway.
Evaluation of apoptosis in LCL cytotoxicity from arsenite
Using a 96 h arsenite toxicity dose-response relationship (Figure 1), an arsenite exposure level of 6 uM was selected for subsequent studies. This concentration resulted in an average cell survival of 38% (S.D. = 9.7%), an intermediate level of cytotoxicity allowing an investigation of the underlying pathways. This cytotoxicity evolved gradually, with average survival of 81% at 24 h, 65% at 48 h, and 55% at 72 h.
Figure 1.
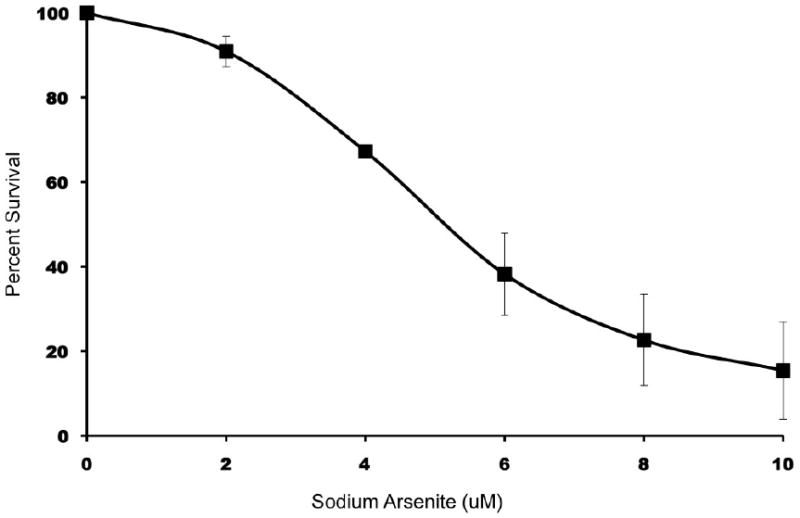
Cytotoxicity analysis of LCL 18564 exposed to increasing concentrations of sodium arsenite. Cells were stained with AV/PI and analyzed by flow cytometry. The data were normalized to the percent of live cells verses control for each concentration of arsenite. Data represents the mean ± S.D. for three separate experiments.
When LCL 18564 cells were exposed to 6 uM arsenite for 96 h the predominant population of high AV staining cells also stains positively for PI, suggesting that apoptosis is not induced by arsenite under these conditions (Figure 2A). To ensure that the 96 h time point had not missed an earlier execution of apoptosis, similar cytotoxicity experiments using arsenite exposure lengths of 6, 12, 24, 48 and 72 h (data not shown) likewise failed to demonstrate the emergence of a high AV, low PI population, supporting the conclusion that 6 uM arsenite does not induce apoptosis in this LCL. In order to verify that LCL18564 is indeed capable of apoptosis, the AV/PI assay was used to evaluate cells exposed to epoxomicin, a proteasome inhibitor reported to induce apoptosis (Concannon et al. 2007). The presence of a large population of apoptotic cells (Figure 2B) is in contrast to the arsenite-exposed cells shown in Figure 2A, indicating that LCL18564 has an intact apoptotic pathway that is not activated by cytotoxic concentrations of arsenite.
Figure 2.
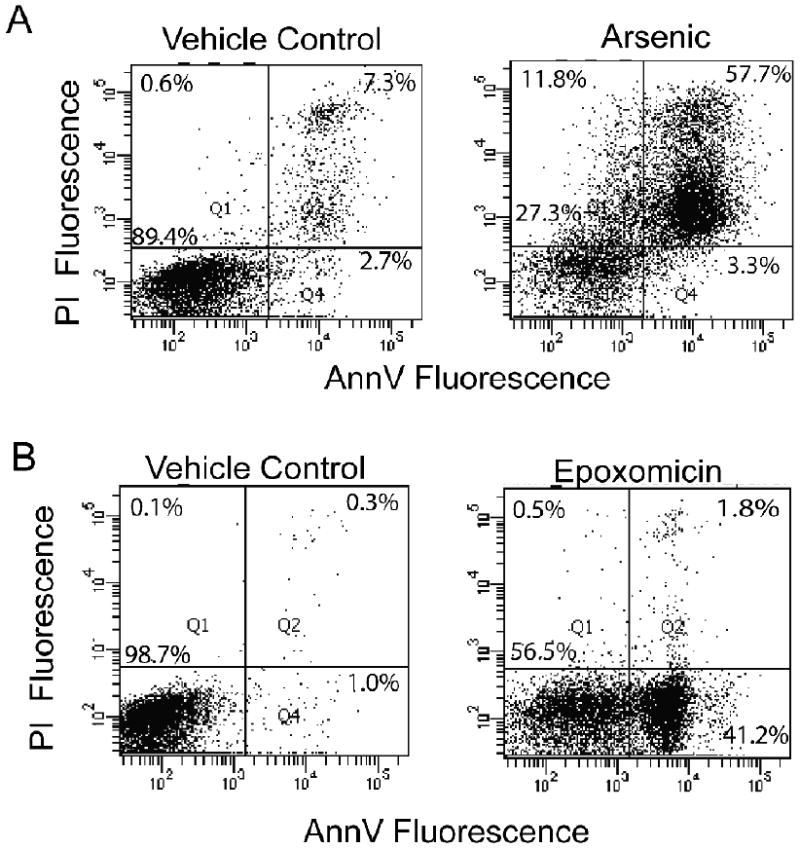
Representative (three independent experiments) AV/PI dot plots of arsenite and epoxomicin exposed LCL 18564. A) Cells were cultured with or without 6 uM sodium arsenite for 96 h; B) Cells were cultured with or without 1 uM epoxomicin for 6 h. Cells were stained with AV/PI and analyzed by flow cytometry.
To corroborate the data suggesting that apoptosis was not activated by arsenite exposure, the effect of an apoptosis inhibitor was tested. LCL18564 cells were exposed to arsenite or epoxomicin under similar conditions as in the previous experiments, however with or without pretreatment with 10 uM QVD, a pan-caspase inhibitor (Figure 3). In the face of ongoing apoptosis, QVD pre-treatment should have the effect of increasing the number of cells in the live quadrant (low AV, low PI) and reducing the cell number in the remaining quadrants. QVD pre-treatment of epoxomicin-treated cells had that effect (Figure 3), reducing the fraction of cells in the three non-live quadrants by 80%, from 100% of the cell total (set as the sum of cells in the three non-live quadrants in the epoxomicin only group) to 20 % of the cell total. This reversal was statistically significant. In contrast, no such significant redistribution of cell fluorescence after QVD pre-treatment was observed in arsenite-exposed cells (Figure 3), supporting the existence of non-apoptotic processes induced by arsenite. The contrasting effects of QVD on arsenite-exposed and epoxomicin-exposed cells were also evaluated using a complementary marker of apoptosis, namely the cleavage of poly-ADP ribose polymerase (PARP) from its native state of 116 kDa to a cleaved fragment of 89 kDa (Figure 4). Consistent with previous experiments, arsenite exposure is not associated with PARP cleavage, while epoxomicin exposure does result in cleaved PARP, in a QVD-reversible manner.
Figure 3.
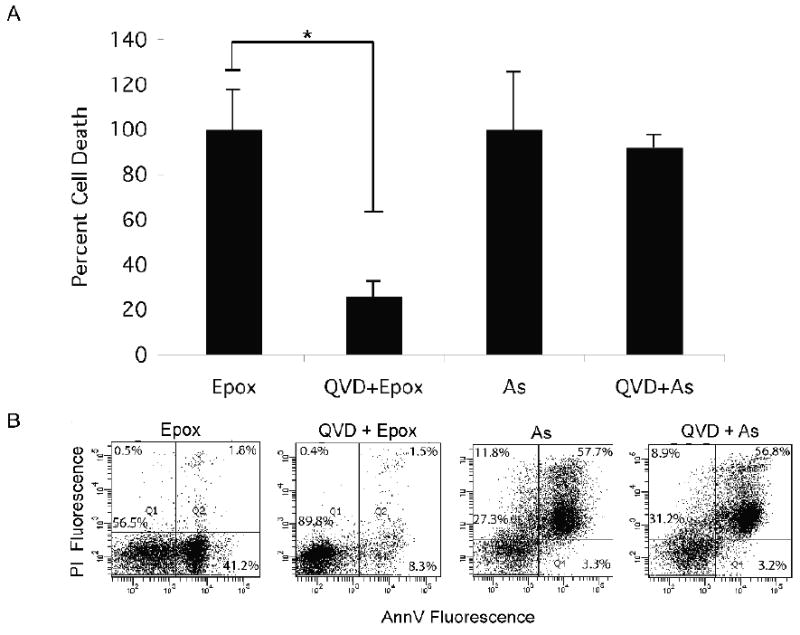
The effect of QVD pretreatment on epoxomicin and arsenite induced cytotoxicity. A) Summary graph of the effect of QVD pretreatment on the cytotoxicity of epoxomicin (Epox, 1 uM for 6 h) or arsenite (As, 6 uM for 96 h) exposed LCL 18564. The graph represents the mean percent cell death ± S.D. for three separate experiments. * Paired t-test p=0.012. B) Representative flow cytometry AV/PI dot plots for the four exposure groups.
Figure 4.
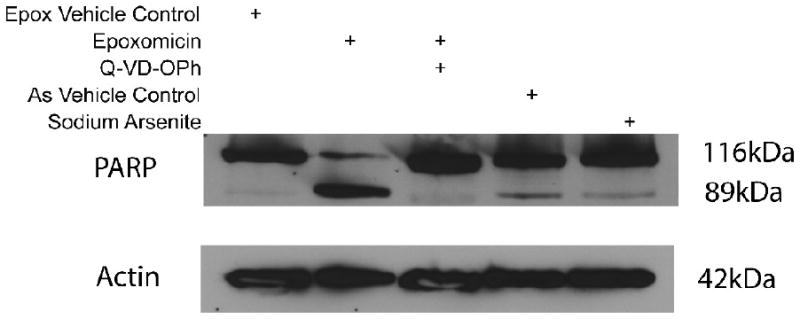
Western blot analysis of PARP cleavage in epoxomicin and arsenite exposed 18564 LCL. Lanes: 1) epoxomicin vehicle control (DMSO); 2) epoxomicin exposed cells (1 uM, 6 h); 3) pretreatment with QVD (50 uM, 1 h), and treated with epoxomicin (1 uM, 6 h); 4) arsenite vehicle control (media); 5) arsenite exposed cells (6 uM, 96 h). Full length PARP is the 116 kDa band and cleaved PARP is the 89 kDa band, β-actin served as loading control for this experiment.
Evaluation of autophagy in LCL cytotoxicity from arsenite
Since microscopic cellular morphology represents a key discriminant of different cell death pathways, transmission electron microscopy (TEM) was used to evaluate LCL18564 (Figure 5). A key difference between microscopic morphology in arsenite-exposed and control cells was the existence of increased numbers of membrane bound vesicles in arsenite-exposed cells (black arrows, Figures 5B, 5C). These vesicles appeared to contain cell structures that were at various stages of degradation, characteristic of cells undergoing autophagy. Co-localizing vesicular structures, consistent with an autophagosomal cluster, were also evident (Figure 5B, 5C) (Calle et al. 1999). Noteworthy is a mitochondrion in an apparently damaged condition with incomplete cristae (open arrow, Figure 5C), which appears to be partially surrounded by a double-membrane structure, suggestive of early engulfment during formation of nascent autophagosomes. Arsenite exposure also increased the presence of cells containing late-stage autophagic vacuoles. (Figure 5D).
Figure 5.
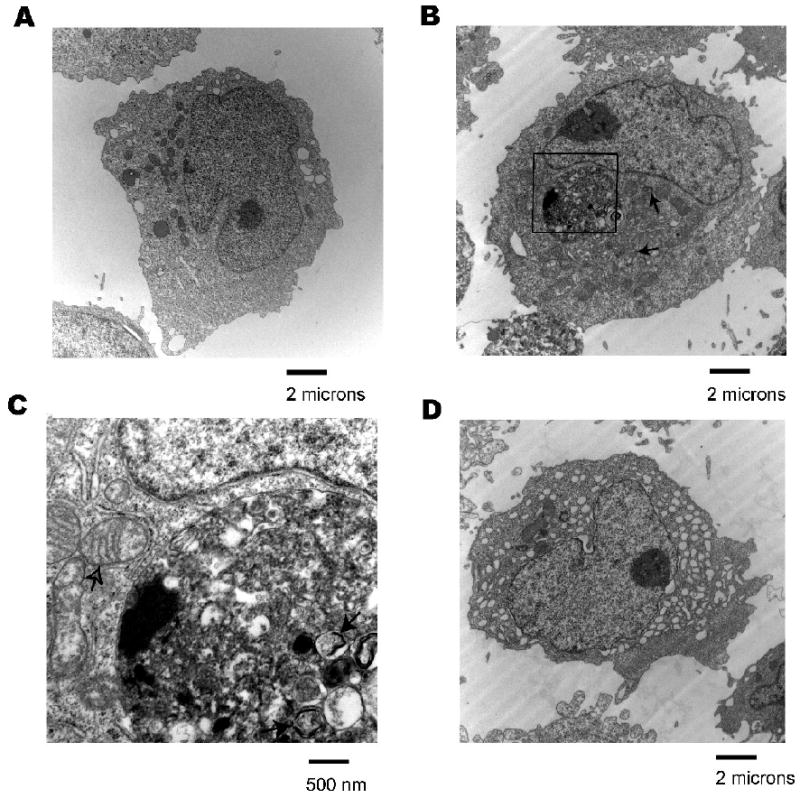
Transmission electron micrographs of arsenite exposed 18564 LCL. A) Control cells, 5600× magnification, B) LCL treated with 6 uM sodium arsenite for 96 h, 5600× magnification. Black arrows indicate examples of autophagosomes, C) 25000× magnification image of boxed area from image B. Black arrows indicate examples of autophagosomes. Open arrow indicates a damaged mitochondrion partially enveloped by a double-membrane structure, D) LCL treated with 6 uM sodium arsenite for 96 h, 5600× magnification.
Because the formation of autolysosomes containing acidic hydrolases increases the total volume of membrane-bound acidic compartments within cells undergoing autophagy, acidophilic fluorescent dyes that label these compartments are commonly used as markers of autophagy. One such dye, Lysotracker Red (LRD), was used to monitor the acidic compartments in LCL18564 following arsenite exposure (Figure 6A). Flow cytometry analysis of cells exposed to 6 uM arsenite for 96 h demonstrated a four-fold increase in fluorescence, indicating an expansion of the cells' acidic compartment. Experiments aimed at inhibiting autophagy used BafA1, a specific inhibitor of vacuolar proton ATPase, a protein essential for maintenance of internal acidity in autolysosomes (Yoshimori et al., 1991). The BafA1-induced de-acidification of lysosomes inhibits their fusion to autophagosomes, in turn inhibiting autophagy (Yamamoto et al., 1998). Cells that had been exposed to 6 uM arsenite for 96 hours were exposed to BafA1 or vehicle control for 4 hours, then stained with LRD. As expected, BafA1 exposure reduced LRD fluorescence, further confirming that the arsenic-induced, enhanced LRD fluorescence was due to an expansion of lysosomes. Efforts to determine the effect of BafA1 on arsenite-induced cytotoxicity were unsuccessful because 96 h exposures to BafA1 at concentrations that inhibit autophagy are extremely cytotoxic to LCL (data not shown). To confirm that the LRD fluorescence measured by flow cytometry was distributed in a punctate pattern consistent with autolysosomal vesicles we examined arsenite-exposed, LRD-stained LCL using fluorescence microscopy (Figure 6B), and found the staining pattern to be consistent with the punctate distribution of autolysosomes.
Figure 6.
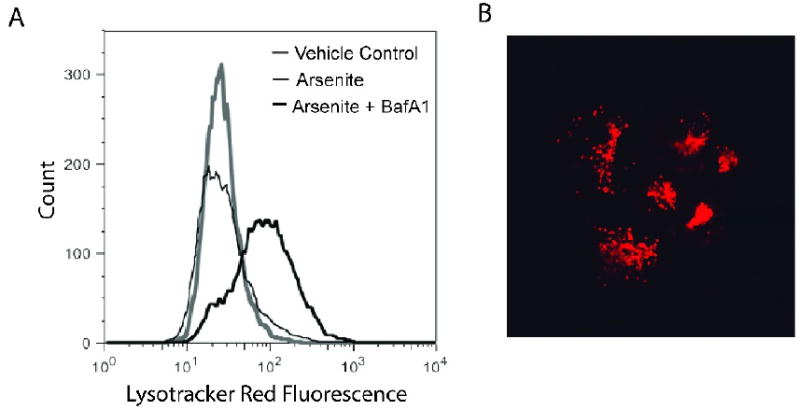
Change in LRD fluorescence after exposure to arsenite. 6A) Flow cytometry histograms of LRD-stained LCL 18564 after exposure to vehicle control (gray histogram) or to 6 uM sodium arsenite (thick black histogram) for 96 h, or arsenite for 96 h followed by BafA1 for 4 h (thin black histogram). 6B) Fluorescence micrograph of LRD-stained, arsenite-exposed cells. Image acquired with a 60× objective lens.
To corroborate data suggesting that arsenite induces autophagy in LCL we measured levels of microtubule-associated protein 1 light chain 3-B (LC3) using Western immunoblot analysis (Figure 7). LC3 is cleaved by the gene ATG4 to form LC3-I which is distributed throughout the cytoplasm. During autophagy, LC3-I is cleaved and conjugated to phosphatidylethanolamine, producing LC3-II, a lipophilic protein that localizes to the membranes of autophagosomes and autolysosomes. For this reason LC3-II is a specific marker for the quantity of autophagosomes and autolysosomes (Rubinsztein et al., 2009). Figure 7 shows that LCL exposed to arsenite (alone) have a higher level of LC3-II protein than do control cells, consistent with an expansion of autophagosomes and autolysosomes. LC3-II exists on both the luminal and the cytosolic surfaces of autophagosomes. Luminal LC3-II is degraded in the autolysosome, while LC3-II on the cytosolic surface can be delipidated and recycled. Because of this, analysis of LC3-II levels at a single point in time reflects the balance of synthesis of LC3-II and its degradation, complicating the interpretation of the differences in expression measured at a single time-point. Insight into the flux of LC3-II can be gained by blocking its degradation using BafA1. In addition to evaluating single time-point levels of LC3, we also evaluated time-dependent flux of LC3 using 4 h and 8 h exposures to BafA1. Inhibition of autolysosomal degradation by BafA1 resulted in the accumulation of LC3-II at 4h and at 8h to a level which was similar between arsenite-exposed and control cells.
Figure 7.
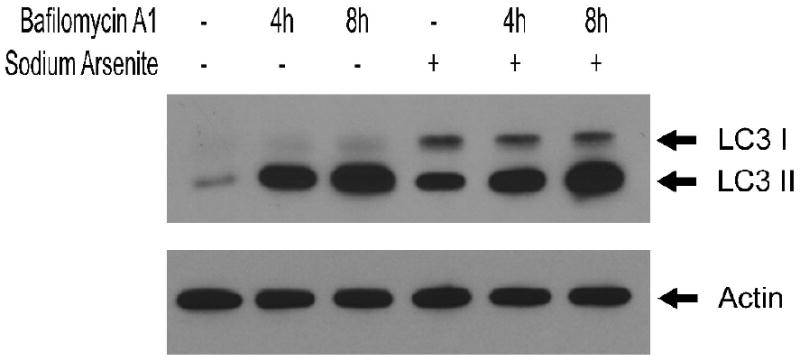
Western blot analysis of LC3 expression in LCL 18564 in the absence (lanes 1,2,3) or presence (lanes 4,5,6) of 6 uM sodium arsenite for 96 h, ending with 100 nM BafA1 exposure for 0 h (lanes 1,4), 4 h (lanes 2,5) or 8 h (lanes 3,6).
Discussion
The data presented here strongly suggest that in human lymphoblastoid cells autophagy is a key component in the cytotoxicity resulting from arsenite exposure. Microscopic cellular morphology revealed an increase in both early and late-stage autophagic structures in arsenite-exposed LCL. Flow cytometry and fluorescence microscopy using an acidophilic dye documented a significant expansion of acidic vesicular structures following arsenite exposure. Concurrent with these changes, arsenite-exposed LCL have increased levels of LC3-II, a specific marker for autophagic vesicles. This arsenite-induced accumulation of autophagosomes and autolysosomes in LCL 18564 appears to be a general phenomenon, as we have observed cellular morphology consistent with autophagy by TEM and increased LRD fluorescence by flow cytometry in three other human LCL that were exposed to arsenite at concentrations ranging from 0.75 uM to 6 uM (data not shown).
In contrast to reports describing trivalent arsenic cytotoxicity in many other cell types, arsenite cytotoxicity in LCL does not proceed through apoptosis as measured by cellular morphology, phosphatidylserine externalization, PARP cleavage, or by functional blockage of apoptosis via caspase inhibition. Importantly, LCL 18564 cells were demonstrated to be capable of executing apoptosis under different perturbation (epoxomicin).
Yet a third possible pathway of cell death that should be considered is necrotic cell death. While experiments specifically targeting an evaluation of necrosis were not conducted, the lack of cytoplasmic swelling in electron microscopy analysis, and the maintenance of lysosomal integrity seen in fluorescence microscopy using LRD argue against necrosis as the primary cytotoxic pathway.
While relatively few reports describe the induction of autophagy by trivalent arsenic, this effect of arsenic seems plausible. Using selected, model proteins and three chemical forms of arsenic, Ramadan et al. demonstrated that arsenic compounds are capable of inhibiting oxidative protein folding, a process that occurs in the physiologic maturation of proteins within the endoplasmic reticulum (ER) (Ramadan et al., 2009). Using a combined proteomic and genomic approach, Zhang et al. concluded that arsenic trioxide exposure in K562 (myeloid leukemia) cells promoted development of the unfolded protein response (UPR), a stress response triggered by detection of failed oxidative protein folding in the ER (Zhang et al., 2009). Coupled with the suggestion from these studies, that arsenic can induce the UPR, are studies that establish the UPR as a process that can lead to the induction of autophagy (Rouschop and Wouters, 2009; Szegezdi et al., 2009). Taken together, the possibility that arsenic induces autophagy is mechanistically reasonable. In fact, some evidence currently exists to support this effect of trivalent arsenic in cell culture models of urothelium, glioblastoma and mononuclear leukocytes (Qian et al., 2007; Aoki et al., 2008; Huang et al., 2009).
More challenging, however, is understanding the functional context and consequences of arsenic-associated autophagy. Unlike the singular death-focused context of apoptosis, autophagy can occur within different contexts that include constitutive function, inducible function, cell survival, cell death, oncogenesis and anti-oncogenesis. In the case of a low-level UPR, the induction of autophagy appears to be a compensation mechanism that can accelerate protein disposal and return the cell to homeostasis. However, there are also examples in which experimental up-regulation of autophagy appears to result in cell death that can be blocked by the silencing of autophagy pathway members (Pattingre et al., 2005). Nevertheless, a clear consensus supporting autophagy as a cell death pathway has not emerged, and even the idea of considering autophagy as a cell death pathway is currently debated in the literature (Kroemer and Levine, 2008).
While there is a clear arsenite-induced expansion of autophagosomes and autolysosomes in LCL, we were unable to determine whether autophagy in this scenario was compensatory or a primary cell death mechanism. This was complicated by our inability to inhibit autophagy over the entire course of the arsenite exposures due to the cytotoxicity of BafA1 exposures lasting longer than 8 h. This cytotoxicity appears to be common to compounds that de-acidify lysosomes, as we observed a similar level of cytotoxicity in LCL exposed to chloroquine at 50 uM for 96 h (data not shown). One possible explanation for the cytotoxicity of autophagy inhibitors is that LCL have a constitutive level of autophagy that is required for survival (Lee and Sugden, 2008). Our studies of LC3-II expression support this idea. Inhibition of autophagy-mediated degradation of LC3-II by BafA1 in control LCL resulted in a substantial increase in LC3-II, suggesting that autophagy is constitutively active in LCL. If this constitutive autophagy is required for LCL survival, then extended exposures to autophagy inhibitors would be expected to be cytotoxic, an effect we observe in LCL. Interesting in this context is our data suggesting that arsenite may also be inhibiting autophagic degradation. Arsenite treatment (no BafA1) elevates LC3-II levels, however the rate of LC3-II accumulation when degradation is blocked by BafA1 is similar regardless of arsenite exposure. This suggests that while arsenite exposure may be expanding the number of autolysosomes, protein degradation within them is inhibited. We conclude this because the higher levels of LC3-II observed in arsenite-only exposed cells would result in higher LC3-II levels when degradation is blocked by BafA1 if degradation rates were identical in control and arsenite-exposed cells. Since LC3-I is not degraded by autophagy, we did not expect BafA1 treatment to modulate levels and this was confirmed in our study. Arsenite exposure moderately elevated LC3-I levels, possibly reflecting increased LC3 gene expression associated with autophagy, or alternatively increased delipidation and recycling of LC3-II from the outer autolysosomal membrane resulting from arsenite-induced inhibition of the normal progression of autophagic degradation and removal of the autolysosomes. Thus it is possible that the inhibition of autophagic degradation within an expanded population of autolysosomes may contribute to arsenite cytotoxicity in LCL.
There is an intriguing overlap in the disease spectra associated with defects in autophagy and with diseases associated with arsenic exposure, including neurodegeneration, atherosclerosis, skin pigmentation changes, diabetes, and cancer (Martinet and De Meyer, 2008; Apel et al., 2009; Hartley et al., 2009; Lee, 2009; Marks, 2009; Rahman et al., 2009; States et al., 2009). Autophagy plays a prominent role in the human immune system, including the processing of internalized microbes and loading of MHC-II. Establishing that arsenite perturbs autophagy in LCL offers an additional mechanism by which arsenic exposure could result in immunosuppresion. The observation that arsenite exposure results in the induction of a relatively novel cell damage-related pathway opens new possibilities in the area of arsenic toxicology, not the least of which is a new set of potential biological targets that may explain at least part of the arsenic story.
Acknowledgments
Funding for this project included a pilot project from the NIEHS Center Grant (ES 006694) and the NIEHS Superfund Basic Research Program Grant (ES 04940), and a NIEHS Human Gene by Environment Interaction Training Grant (ES 16652).
Abbreviations Used
- LCL
Lymphoblastoid cell line
- LRD
Lysotracker red dye
- UPR
Unfolded protein response
- PARP
Poly-ADP ribose polymerase
- QVD
Quinolyl-valyl-O-methylaspartyl- [-2,6-difluorophenoxy] –methyl ketone
- AV
Annexin V
- PI
Propidium Iodide
- TEM
Transmission electron microscopy
- EBV
Epstein-Barr virus
- BafA1
Bafilomycin A1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki H, Kondo Y, Aldape K, Yamamoto A, Iwado E, Yokoyama T, Hollingsworth EF, Kobayashi R, Hess K, Shinojima N, Shingu T, Tamada Y, Zhang L, Conrad C, Bogler O, Mills G, Sawaya R, Kondo S. Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy. 2008;4:467–475. doi: 10.4161/auto.5668. [DOI] [PubMed] [Google Scholar]
- Apel A, Zentgraf H, Buchler MW, Herr I. Autophagy-A double-edged sword in oncology. Int J Cancer. 2009;125:991–995. doi: 10.1002/ijc.24500. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Dudek EJ, Treadwell MD, Wetterhahn KE. Arsenic induces oxidant stress and NF-kappa B activation in cultured aortic endothelial cells. Free radical biology & medicine. 1996;21:783–790. doi: 10.1016/0891-5849(96)00174-8. [DOI] [PubMed] [Google Scholar]
- Calle E, Berciano MT, et al. Activation of the autophagy, c-FOS and ubiquitin expression, and nucleolar alterations in Schwann cells precede demyelination in tellurium-induced neuropathy. Acta Neuropathol. 1999;97(2):143–155. doi: 10.1007/s004010050967. [DOI] [PubMed] [Google Scholar]
- Charoensuk V, Gati WP, Weinfeld M, Le XC. Differential cytotoxic effects of arsenic compounds in human acute promyelocytic leukemia cells. Toxicology and applied pharmacology. 2009 doi: 10.1016/j.taap.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Concannon CG, Koehler BF. Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene. 2007;26(12):1681–1692. doi: 10.1038/sj.onc.1209974. [DOI] [PubMed] [Google Scholar]
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glienke W, Chow KU, Bauer N, Bergmann L. Down-regulation of wt1 expression in leukemia cell lines as part of apoptotic effect in arsenic treatment using two compounds. Leuk Lymphoma. 2006;47:1629–1638. doi: 10.1080/10428190600625398. [DOI] [PubMed] [Google Scholar]
- Gonsebatt ME, Vega L, Herrera LA, Montero R, Rojas E, Cebrian ME, Ostrosky-Wegman P. Inorganic arsenic effects on human lymphocyte stimulation and proliferation. Mutation research. 1992;283:91–95. doi: 10.1016/0165-7992(92)90139-9. [DOI] [PubMed] [Google Scholar]
- Guo HR, Yu HS, Hu H, Monson RR. Arsenic in drinking water and skin cancers: cell-type specificity (Taiwan, ROC) Cancer Causes Control. 2001;12:909–916. doi: 10.1023/a:1013712203455. [DOI] [PubMed] [Google Scholar]
- Hartley T, Brumell J, Volchuk A. Emerging roles for the ubiquitin-proteasome system and autophagy in pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2009;296:E1–10. doi: 10.1152/ajpendo.90538.2008. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Speit G. Comparative investigations of the genotoxic effects of metals in the single cells gel (SCG) assay and the sister chromatid exchange (SCE) test. Environmental and molecular mutagenesis. 1994;23:299–305. doi: 10.1002/em.2850230407. [DOI] [PubMed] [Google Scholar]
- Huang YC, Hung WC, Chen WT, Yu HS, Chai CY. Sodium arsenite-induced DAPK promoter hypermethylation and autophagy via ERK1/2 phosphorylation in human uroepithelial cells. Chem Biol Interact. 2009;181:254–262. doi: 10.1016/j.cbi.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Sugden B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene. 2008;27:2833–2842. doi: 10.1038/sj.onc.1210946. [DOI] [PubMed] [Google Scholar]
- Lee JA. Autophagy in neurodegeneration: two sides of the same coin. BMB Rep. 2009;42:324–330. doi: 10.5483/bmbrep.2009.42.6.324. [DOI] [PubMed] [Google Scholar]
- Marks MS. Eating thyself toward the dark side? Pigment Cell Melanoma Res. 2009;22:251–252. doi: 10.1111/j.1755-148X.2009.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, De Meyer GR. Autophagy in atherosclerosis. Curr Atheroscler Rep. 2008;10:216–223. doi: 10.1007/s11883-008-0034-y. [DOI] [PubMed] [Google Scholar]
- Mukherjee SC, Rahman MM, Chowdhury UK, Sengupta MK, Lodh D, Chanda CR, Saha KC, Chakraborti D. Neuropathy in arsenic toxicity from groundwater arsenic contamination in West Bengal, India. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2003;38:165–183. doi: 10.1081/ese-120016887. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Piatek K, Schwerdtle T, Hartwig A, Bal W. Monomethylarsonous acid destroys a tetrathiolate zinc finger much more efficiently than inorganic arsenite: mechanistic considerations and consequences for DNA repair inhibition. Chemical research in toxicology. 2008;21:600–606. doi: 10.1021/tx7003135. [DOI] [PubMed] [Google Scholar]
- Pulido MD, Parrish AR. Metal-induced apoptosis: mechanisms. Mutat Res. 2003;533:227–241. doi: 10.1016/j.mrfmmm.2003.07.015. [DOI] [PubMed] [Google Scholar]
- Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res. 2007;31:329–339. doi: 10.1016/j.leukres.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Rahman MM, Ng JC, Naidu R. Chronic exposure of arsenic via drinking water and its adverse health impacts on humans. Environ Geochem Health. 2009;31 1:189–200. doi: 10.1007/s10653-008-9235-0. [DOI] [PubMed] [Google Scholar]
- Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C. Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry. 2009;48:424–432. doi: 10.1021/bi801988x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raqib R, Ahmed S, Sultana R, Wagatsuma Y, Mondal D, Hoque AM, Nermell B, Yunus M, Roy S, Persson LA, Arifeen SE, Moore S, Vahter M. Effects of in utero arsenic exposure on child immunity and morbidity in rural Bangladesh. Toxicology letters. 2009 doi: 10.1016/j.toxlet.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Rouschop KM, Wouters BG. Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med. 2009;9:417–424. doi: 10.2174/156652409788167131. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, Klionsky DJ. In search of an “autophagomometer”. Autophagy. 2009;5:585–589. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- Salazar AM, Ostrosky-Wegman P, Menendez D, Miranda E, Garcia-Carranca A, Rojas E. Induction of p53 protein expression by sodium arsenite. Mutation research. 1997;381:259–265. doi: 10.1016/s0027-5107(97)00207-8. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Luster MI. Mechanisms of arsenic carcinogenicity: genetic or epigenetic mechanisms? J Environ Pathol Toxicol Oncol. 2000;19:281–286. [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Toriuma W, Kommineni V, Matheson J, Unimye N, Kayama F, Harki D, Ding M, Vallyathan V, Luster MI. Arsenic mediates cell proliferation and gene expression in the bladder epithelium: association with activating protein-1 transactivation. Cancer research. 2000;60:3445–3453. [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yuan Y, Ferreccio C, Liaw J, von Ehrenstein O, Steinmaus C, Bates MN, Selvin S. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ Health Perspect. 2006;114:1293–1296. doi: 10.1289/ehp.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- States JC, Srivastava S, Chen Y, Barchowsky A. Arsenic and cardiovascular disease. Toxicol Sci. 2009;107:312–323. doi: 10.1093/toxsci/kfn236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturlan S, Baumgartner M, Roth E, Bachleitner-Hofmann T. Docosahexaenoic acid enhances arsenic trioxide-mediated apoptosis in arsenic trioxide-resistant HL-60 cells. Blood. 2003;101:4990–4997. doi: 10.1182/blood-2002-08-2391. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. American journal of physiology. 2009;296:C941–953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Yang YP, Liang ZQ, Gao B, Jia YL, Qin ZH. Dynamic effects of autophagy on arsenic trioxide-induced death of human leukemia cell line HL60 cells. Acta Pharmacol Sin. 2008;29:123–134. doi: 10.1111/j.1745-7254.2008.00732.x. [DOI] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell death and differentiation. 2005;12 2:1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. The Journal of biological chemistry. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Zhang QY, Mao JH, Liu P, Huang QH, Lu J, Xie YY, Weng L, Zhang Y, Chen Q, Chen SJ, Chen Z. A systems biology understanding of the synergistic effects of arsenic sulfide and Imatinib in BCR/ABL-associated leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3378–3383. doi: 10.1073/pnas.0813142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yang F, Shim JY, Kirk KL, Anderson DE, Chen X. Identification of arsenic-binding proteins in human breast cancer cells. Cancer letters. 2007;255:95–106. doi: 10.1016/j.canlet.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XH, Shen YL, Jing YK, Cai X, Jia PM, Huang Y, Tang W, Shi GY, Sun YP, Dai J, Wang ZY, Chen SJ, Zhang TD, Waxman S, Chen Z, Chen GQ. Apoptosis and growth inhibition in malignant lymphocytes after treatment with arsenic trioxide at clinically achievable concentrations. Journal of the National Cancer Institute. 1999;91:772–778. doi: 10.1093/jnci/91.9.772. [DOI] [PubMed] [Google Scholar]