

Abstract
The hypothalamic pituitary thyroid (HPT) axis plays a critical role in mediating changes in metabolism and thermogenesis. Thus, the central regulation of the thyroid axis by Thyrotropin Releasing Hormone (TRH) neurons in the paraventricular nucleus of the hypothalamus (PVN) is of key importance for the normal function of the axis under different physiological conditions including cold stress and changes in nutritional status. Before the TRH peptide becomes biologically active, a series of tightly regulated processes occur including the proper folding of the prohormone for targeting to the secretory pathway, its post-translational processing, and targeting of the processed peptides to the secretory granules near the plasma membrane of the cell ready for secretion. Multiple inputs coming from the periphery or from neurons present in different areas of the brain including the hypothalamus are responsible for the activation or inhibition of the TRH neuron and in turn affect the output of TRH and the set point of the axis.
Keywords: proTRH, TRH, neuroendocrine, peptides, HPT axis, prohormone processing, central nervous system, obesity, energy balance, catecholamines, prohormone convertases, thermoregulation, hypothalamus, STAT, CREB, NPY, MSH, leptin, melanocortin system
1. Introduction
The hypothalamic-pituitary-thyroid (HPT) axis plays an essential role in the maintenance of metabolic homeostasis in response to alterations in metabolism and external environment. Thyrotropin-releasing hormone (TRH) produced in the parvocellular division of the hypothalamus is the key peptide hormone responsible for HPT regulation. TRH is synthesized from a larger inactive precursor (proTRH) by a series of post-translational modifications while transported through the regulated secretory pathway (RSP). These processing events are conducted by two members of the family of prohormone convertases (PCs), PC1/3 and secondarily by PC2 [1; 2; 3]. The intermediate products of processing generated by these enzymatic activities are further subjected to additional modifications by exopeptidases, such as carboxyl peptidase E and D (CPE, CPD), to remove the C-terminal basic amino acids [4]. TRH-gly, the immediate precursor to TRH (pGlu-His-Pro-NH2; thyroliberin), is then amidated at its carboxyl terminus by the action of peptidylglycine α-amidating monooxygenase enzyme (PAM) [4; 5; 6] (see Fig. 1). Anatomically, a subgroup of TRH neurons (see section 2) originating in the paraventricular nucleus of the hypothalamus (PVN) project their axon terminals to the median eminence (ME) where they are in close proximity to the capillaries of the hypophysial-portal system. TRH released in these capillaries stimulate the biosynthesis and secretion of thyroid stimulating hormone (TSH) from the pituitary [7; 8], which in turn, stimulates biosynthesis of the thyroid hormones, thyroxine (T4), and it modified product triidothyronine (T3) by the action of deiodinases [9]. The thyroid hormone regulates the transcription of important genes by binding to a family of nuclear receptors throughout the body, and is of critical significance to sustain protein synthesis and metabolic activity in peripheral tissues. The thyroid hormone also plays a pivotal role in the regulation of thermogenesis. Roughly 30% of resting energy expenditure is dependent upon thyroid hormone, and it is critical in facultative thermogenesis during cold exposure [10; 11]. One mechanism by which thyroid hormone affects thermogenesis is by affecting uncoupling proteins (UCP-1, UCP-2 and UCP-3) in brown adipose tissue (BAT) and muscle, in combination with an activation of the sympathetic nervous system to accelerate ATP turnover [10; 11]. The thyroid gland provides an unchanging basal level of thyroid hormone to keep the basic metabolic rate of all cells at a constant level, and provides the overall set point of this endocrine axis. The maintenance of euthyroidism is dependent on TRH regulation of TSH [7; 8] as well as by feedback of thyroid hormones, which suppress preproTRH expression and TSH secretion [12; 13]. The significance of TRH in the regulation of thyroid axis was demonstrated in humans with a TRH receptor mutation and in mice lacking the TRH gene, which in both cases caused central hypothyroidism [14; 15]. In addition to the negative regulation to TRH by thyroid hormones, the TRH neurons in the PVN are also subjected to tight regulation by neuropeptide Y (NPY), the adipocyte hormone leptin, and two melanocortin peptides (melanocortin stimulating hormone (α-MSH) and agouti-related peptide (AgRP)) [6; 16; 17; 18]. Integration of all of these inputs provides an exact set point for the thyroid axis when changes in nutritional and external temperature occur. A better understanding of the mechanisms by which these inputs signal the TRH neurons is of paramount importance because of its impact of the thyroid metabolism at the central level.
Figure 1. Schematic representation of the biosynthesis and post-translational processing of rat proTRH.

A depicts the transcription and post-translational modifications in the rat proTRH composed of 231 amino acids. The signal sequence is cleaved from preproTRH upon delivery into the endoplasmic reticulum yielding proTRH. The conserved PGL sequence in the pYE26 peptide ensures the proper folding of proTRH within the lumen of the endoplasmic reticulum. The initial processing cleavage of proTRH by PC1/3 begins at the trans-Golgi network level generating an N-terminal and C-terminal intermediate forms. The intermediate forms of processed proTRH are then targeted to different secretory granules where processing continues by the action of the PCs, CPE/D and PAM until TRH and non TRH peptides are formed. The vertical bars on the right indicate in which intracellular compartment proTRH is cleaved and further procesed. Numbers indicates the positions of paired basic residues. Non TRH peptides are indicated in the proTRH molecule, and TRH is indicated by a black rectangle. Peptides are indicated as pXYZ nomenclature, where “p” means peptide, “X” is the first amino acid of each peptide, “Y” is the last one, and Z indicates the total number of amino acids in that given peptide. The nonTRH peptides are then targeted to different secretory granules ready for secretion. B depicts the proposed model of the unfolding process for proTRH after its initial cleavage by PC1/3, exposing potential sorting signals responsible for the targeting to different granules. Thus far a disulfide sequence has been identified in proTRH as an important sorting signal for the correct targeting of peptides to secretory granules. SS: signal sequence. ER: endoplasmic reticulum. TGN: trans Golgi network. ISGs: immature secretory granules. C-s-sC: disulfide bond. The reader should note that proTRH-derived peptides are named by “p” for peptide followed by the single letter amino acid designation for the first and last amino acid of the peptide, along with the peptide length in subscript. Where these peptides are first mentioned, they are followed by the longer prepro-TRH name that describes their amino acid residue positions within the precursor.
2. Hypothalamic TRH neuron and its neuronal and metabolic interactions
Hypophysiotropic TRH neurons have axon terminals present in high density in the external layer of the rat ME, in close apposition to capillaries of the hypophysial-portal system [19]. These axons originate from neuronal perikarya located in the PVN, of the hypothalamus. Destruction of this region results in the disappearance of up to 94% of TRH in the external layer of the ME, and the reduction of TSH secretion from the anterior pituitary gland [20]. After its biosynthesis in the hypothalamus through a post-translational processing mechanism, the TRH peptide is transported through axon terminals to the ME where is released into capillaries of the hypophysial-portal system for stimulating the synthesis and release of TSH from the pituitary [7; 8]. The TSH peptide is an important regulator of thyroid hormone levels and a major center of neural control on the endocrine system, and is thus responsible for controlling physiological responses to the external and internal environment [21; 22]. TRH neurons located in the “thyrotrophic area” (hypophysiotropic) receive inputs from other regions of the brain as well as from the circulation. The most important afferent connections to the TRH neurons in the PVN include catecholamine neurons from the brainstem [23] and neurons from the arcuate nucleus (ARC) of the hypothalmus. Neuropeptides derived from the ARC play a significant role in the response of thyroid axis to both starvation and illness, and catecholamines play a significant role in the up-regulation of TRH gene expression during cold-exposure [24]. In addition to the PVN, TRH neurons are present in other regions of the hypothalamus, including the preoptic area, anterior hypothalamus, supraoptic, arcuate, dorsomedial and premmamilary nuclei, as well as basolateral and prefornical hypothalmus [25; 26].
The rat PVN is composed of two major parts, the magnocellular and the parvocellular divisions. The magnocellular hypothalamic neurohypophysial tract carries vasopressin and oxytocin to the posterior pituitary. The parvocellular section is composed of a number of subcompartments including anterior, medial, periventricular, ventral, dorsal and lateral parvocellular subdivisions. A large population of TRH-immunoreactive neurons are located in medial and periventricular parvocellular subdivisions, organized in a triangular configuration, symmetric to the dorsal aspect of the third ventricle, whereas neurons expressing TRH in the anterior parvocellular subdivision are more disperse [26]. However, not all TRH containing neurons in the PVN project to the ME [27]. Only TRH neurons in the medial and periventricular parvocellular subdivisions project to the ME. Most likely the function of these neurons differ from those of the anterior parvocellular subdivision. Non-hypophysiotropic TRH neurons also present in the PVN has no known projections to the ME. It is presumed that they do not serve a direct hypophysiotropic function, but rather play a different role. Among other functions, they might act on the autonomic nervous system to induce thermoregulation. For example, after intra-cerebro-ventricular (ICV) administration of α-MSH [28], the peptide stimulated both hypohysiotrophic neurons and proTRH from the anterior and ventral parvocellular subdivisions of the PVN, a region associated with the stimulation of the autonomic regulation. This region sends neuronal projections to brain stem and spinal cord targets [29; 30; 31]. Given those facts, it is possible that non-hypophysiotropic TRH neurons might have an action in autonomic regulation by activating uncoupling protein-1 (UCP-1) in the brown adipose tissue [32] in concert with a simultaneous stimulation of the HPT axis by hypophysiotropic TRH neurons.
In the human PVN, the magnocellular and parvocellular neurons are not restricted to any particular subdivision of the nucleus. Although the literature on TRH neurons in the human PVN is limited, it was reported that most TRH neurons are present in its dorsomedial part [33]. Using in situ hybridization, TRH mRNA expressing neurons were reported not only in the human PVN, but also in the suprachiasmatic nucleus, perifornical area and lateral hypothalamus [34]. Therefore, the neuroanatomy of the TRH neuron is certainly not identical in all species and the rat hypothalamus cannot simply be taken as universal. On the other hand, NPY, AgRP and α-MSH containing axons are frequently present in close juxtaposition to TRH neurons in the human PVN, which is very similar to the rat PVN [35]. It is, therefore, important to point out that most data generated to date are derived from rat studies, and its extrapolation to humans may have its limitations in interpretation.
Although no definitive neuronal TRH projections from the PVN to the ventromedial hypothalamus (VMH, also know as the satiety center), or lateral hypothalamus (LH also known as the hunger center) were clearly defined, evidence suggests that TRH inhibits food and water intake. These results are consistent with high levels of TRH in the VMH [36; 37], and it was proposed that the LH is critical for TRH-induced anorexia [38]. What it is not clear, however, is whether those actions of TRH in these hypothalamic areas are derived from PVN fibers. TRH inhibits food [39; 40; 41] and water intake [26; 36]. For example, TRH can suppress eating without altering blood glucose levels [42] and without affecting TSH [43], suggesting that non-hypophysiotropic TRH neurons could regulate feeding behavior by another pathway aside from the HPT axis, perhaps by sending its axons to the VMH where no preproTRH mRNA was detected, but the presence of the TRH-1 receptor was identified [44]. The other possibility is that TRH produced in the LH [26; 44] or TRH derived from neurons located in the PVN and projecting their fibers to the LH will inhibit the secretion of the orexogenic peptides, MCH and orexin [45]. TRHR-1 and TRHR-2 are both present in the LH, and preproTRH mRNA is also abundant. Orexin/hypocretin neurons are critical for sustaining consciousness: their firing stimulates wakefulness and their destruction causes narcolepsy. In a recent study using whole-cell patch-clamp recordings, it was found that TRH robustly increased the action potential firing rate of these neurons. TRH-induced excitation persisted under conditions of synaptic isolation, and caused a Na (+)-dependent depolarization. The results of this study attributed a new role to TRH as a physiological modulator of orexin cell firing [46]. The largest concentrations of hypothalamic TRH neurons outside of the PVN are found in the dorsomedial nucleus, lateral hypothalamus and preoptic area, including medial, periventricular, suprachiasmatic and the sexual dimorphic nucleus of the preoptic area [25; 26]. In addition to the PVN, TRH neurons are present in many other regions of the CNS including regions in the diencephalon, telencephalon, mesencephalon, myelencephalon and spinal cord [26], and the majority of inputs to TRH neurons are derived from the diencephalon, telencephalon and brainstem [47]. The anatomic distribution of TRH neurons and their fibers in these tissues was described earlier [26]. As mentioned above, catecholaminergic neurons have one of the most prominent innervations to TRH neurons in the PVN arising primarily from A1/C1 groups in the medulla, and contributing approximately 20% of all synapses on these cells. The excitatory function of these neurons is suggested because the axons participate in asymmetric synapses with both perikarya and dendrites of TRH neurons. TRH neurons are densely innervated by norepinephrine (NE) containing axons that stimulate TRH secretion [19]. Their axon terminals are in close apposition to TRH axons in the external layer of the rat median eminence signifying that catecholamine inputs may influence TRH hypophysiotropic neurons directly on their perikarya, dendrites, but also on their axon terminals.
In addition to the catecholaminergic inputs to TRH neurons, four different peptides produced in the ARC are involved in regulating TRH by projecting axon terminals in synaptic contact with TRH hypophysiotropic neurons. These neuronal groups expressing the leptin receptor (ObRb) are considered as one of the main hypothalamic centers controlling food intake and energy homeostasis [48; 49]. One group of ARC neurons produces the α-MSH and cocaine and amphetamine regulated transcript (CART) [45; 50; 51]. Both are anorexic peptides but α-MSH is particularly potent and signals in the hypothalamus through both the melanocortin-3 and 4 receptors (MC3-R and MC4-R). TRH neurons in the PVN express MC4-R and are innervated by α-MSH nerve terminals [52; 53]. ICV administration of α-MSH can prevent the fasting-induced suppression of the thyroid axis [54; 55]. TRH neurons are also innervated by other ARC neurons involved in energy balance expressing ObRb receptor. They are the orexogenic AgRP and the neuropeptide Y (NPY). In the rat, α-MSH and CART are expressed in the same neurons situated in lateral portions of the ARC, while NPY and AGRP are coexpressed in a distinct population of neurons located in more medial portions of the ARC. This neuronal organization is somewhat similar in the human brain suggesting an evolutionary significance of this neuroendocrine system. However, unlike in rodents, CART in humans is absent from the perikarya and axons of α-MSH-synthesizing neurons, but is expressed in approximately one third of NPY/AgRP neurons in the human infundibular nucleus [56]. This anatomic distribution for CART seen in humans raises the question of whether CART is associated with an orexigenic role in the human brain. However, CART and α-MSH colocalize in the human lateral hypothalamus, which is similar to anatomy described in rodents. AgRP and NPY are down regulated by leptin and up-regulated during fasting [45; 50; 57; 58; 59]. When AgRP, an MC4-R antagonist and NPY are administered centrally they cause central hypothyroidism by downregulating preproTRH mRNA expression in the PVN [35; 60; 61]. Anatomically, NPY appears most prominent in its inputs to the periventricular and medial parvocellular divisions of the PVN [62]. NPY cell bodies principally reside in the medulla, often coexisting with NE and E [63].
Other studies have suggested that the dorsomedial nucleus of the hypothalamus (DMN) could provide an inhibitory role to TRH neurons based on neuronal innervations to the PVN and TRH neurons [64], but no transmitter, catecholamine, or peptide was identified. However, it has been suggested that an important function of this nucleus is to mediate leptin action on the PVN. In support of this, a subset of leptin-responsive neurons in the DMN strongly innervates the PVN [65]. Moreover, the DMN may mediate the regulation of TRH neurons in the PVN by leptin via a multisynaptic ARC-DMN-PVN pathway. This idea is based on the fact that the DMN neurons, which innervate the PVN, are associated with terminal axons containing α-MSH likely from the ARC [66]. Therefore, leptin regulation of TRH neurons of the PVN is a complex system, which includes its direct action on these neurons, and several potential indirect pathways. In addition to the neuronal inputs to TRH neurons described above, other hormone and catecholamines also regulate TRH neurons in the PVN [26] including dopamine, serotonin, histamine, somatostatin, endogenous opiod peptides (such as beta endorphin, enkephalin, and dynorphin), vasoactive intestinal polypeptide, gamma-aminobutyric acid, cytoquines (such as interleukin 1 or interleukin 6), and recently adiponectin was shown to have a potential effect. Adiponectin is an adipocyte-derived hormone, which acts in the brain to modulate energy homeostasis and autonomic function including glucose regulation and fatty acid catabolism. Adiponectin exerts some of its weight reduction effects via the brain similar to leptin. A recent study investigated the role of adiponectin in controlling excitability in the magnocellular and parvocellular divisions of the PVN. Patch clamp recording identified a mix of PVN neurons with excitability. TRH neurons were depolarized in the presence of adiponectin, suggesting that adiponectin could play a specific role in the central nervous system to coordinate NE excitability of a certain group of PVN neurons [67].
3. Regulation of preproTRH gene and promoter
Hypophysiotropic preproTRH gene expression is continuously regulated in the medial and periventricular regions of the PVN to control the output of TSH secretion from the pituitary and subsequent thyroid hormone levels in response to physiological changes such as cold, illness, starvation, and pituitary or thyroid disease. The murine, rat and human TRH genomic structures contain the same 3 exons and 2 introns. Exon 1 encodes the 5′ untranslated region, while exon 2 and 3 contain the sequence that encodes the preproTRH message. Among the species, the two intron sequences are poorly conserved. The promoter region of the preproTRH gene is located immediately 5′ to exon 1, and it is believed that this is the major locus where regulation of the gene occurs. The preproTRH promoter region has a conserved TATAA box, required for transcriptional initiation by RNA polymerase II. Proximal to the TATAA box there is an important regulatory element termed Site 4 (TGACCTCA) that is conserved in the mouse, rat and human promoters [68; 69]. This is a critical thyroid hormone receptor-binding site. Site 4 serves as a multifunctional binding site for the transcription factor CREB, and thyroid hormone receptors (THR), including THR homodimers, and heterodimers of THR and the retinoic acid X receptor (RXR). However, a recent study challenges this view suggesting that site 4 does not bind pCREB. Using nuclear extracts from 8Br-cAMP-stimulated primary culture of hypothalamic cells, this study showed no binding to Site-4, but instead to cAMP response element (CRE)-2 (-101/-94). The authors suggest that pCREB binds to a response element in the TRH promoter (CRE-2) independently of Site-4 where TRbeta2 is bound. Therefore, they propose that pCREB and TRH do not have mutual interference on their binding sites [70]. The difference between the two studies is that the two research groups used different cell systems for their studies. It is proposed that Site 4 assists with a second weak THR binding site 11 bp 3′ to Site 4, that binds both THR homodimers and TR/RXR heterodimers. Therefore, Site 4 comes into view to be a critical sequence within the preproTRH promoter that is preserved across species and controls both basal and regulated expression of preproTRH gene expression.
T3 negatively feeds back on preproTRH gene expression and mediates its effects on gene expression via the three THR isoforms (THRa1, THRb1 and THRb2). Recent genetic studies have confirmed that TRb2 is critical for negative regulation of the TRH and the TSHa and b subunit genes by T3. However, the molecular mechanism controlling negative regulation of the preproTRH gene by T3 has not been completely determined. Site 4 and its surrounding region can bind THRb2, which appears to be essential for negative regulation. A second region within the TRH gene present within the first 55 bp of exon 1 may also be important in mediating the effects of T3. This region interacts with TRb monomers and its removal in functional studies causes a loss of negative regulation by T3 [71].
The canonical signal transducer of activated transcription (STAT) also activates preproTRH gene by having a binding site in the mouse and rat promoter 5′ to Site 4, which has a similar site in the human promoter located between −150 and −125. Interestingly, the leptin receptor activates transcription via the JAK-STAT signaling pathway and is able to significantly enhance preproTRH promoter activity in vitro [16]. Leptin binding to its receptor ObRb induces receptor-associated Janus tyrosine kinases (primarily JAK2) to phosphorylate both itself and the leptin receptor on specific tyrosine residues [72; 73; 74]. STAT isoforms then bind to these receptor phosphotyrosines, which allow STAT to be phosphorylated by JAK on tyrosine residue Y705. Phosphorylated STATs dimerize and enter the nucleus, where STAT regulates gene transcription by binding to STAT-responsive DNA elements [75]. In the hypothalamus, STAT3, but not STAT1, STAT5 or STAT6, was shown to be activated after in vivo leptin administration [76]. The short leptin receptor isoform, ObRa, does not contain intracellular tyrosine residues and cannot activate STAT3 signaling [74]. Although STATs are clearly important for leptin signaling through ObRb, like other members of this receptor class, ObRb can modulate other intracellular proteins and pathways, including insulin receptor substrate proteins and the RAS-MAPK pathway [74]. It is critical to emphasize that, for the biologically important actions of leptin regulation of appetite, energy expenditure, and neuroendocrine function, the relative roles of signaling via STATs and these or other pathways are unknown. The preproTRH promoter also contains binding sites for the glucocorticoid receptor and SP-1. The glucocorticoid receptor is a member of the nuclear receptor family, while SP-1 is a widely expressed transcription factor that binds to GC rich motifs in target promoters. Glucocorticoids appear to be inhibitory to preproTRH gene expression in the PVN. A more detailed revision of the regulation of the preproTRH promoter has been published recently elsewhere [77].
4. ProTRH biosynthesis, processing and trafficking to the regulated secretory pathway
One of the most well studied TRH precursors across species is the rat preproTRH, a 29kDa polypeptide composed of 255 amino acids. This precursor contains an N-terminal 25 amino acid leader sequence, five copies of the TRH progenitor sequence Gln-His-Pro-Gly flanked by paired basic amino acids (Lys-Arg or Arg-Arg), four non TRH peptides lying between the TRH progenitor sequences, an N-terminal flanking peptide, and a C-terminal flanking peptide [78; 79]. The N-terminal flanking peptide (preproTRH25-50-R-R- preproTRH53-74) is further cleaved at the C-terminal side of the arginine pair site to render preproTRH25-50 (pYE26) and preproTRH53-74 (pFT22), thus yielding a total of seven non-TRH peptides (Fig. 1). The rat preproTRH is approximately 88% homologous to the mouse preproTRH that contains 256 amino acids, also generates five copies of TRH by proteolytic processing. The human preproTRH contains 242 amino acids and generates six copies of the progenitor sequence for TRH. On the other hand, in the frog brain there are at least three different TRH preprohormones ranging between 224 and 227 amino acids and containing seven copies of the TRH progenitor sequence. Rat, mouse, and human proTRH contain multiple copies of the progenitor for TRH, Gln-His-Pro-Gly [80; 81], which could represent an evolutionary trend present in all species to ensure enough production of TRH molecules in response to signals that trigger its secretion. The complete sequence of proTRH for several species has been elucidated including salmon, frog, Zebrafish, chicken, Rhesus monkey, rat, mouse, and human. The more conserved prohormone sequence is found among the mammalian species, but common to all of them is the presence of multiple copies of a progenitor sequence for TRH, flanked on either side by paired basic amino acids, Lys-Arg or Arg-Arg.
Similarly other potent secretory molecules regulating key biological functions, the biosynthesis of TRH (pyroGlu-His-ProNH2, MW 362) and other non-TRH peptides begins with mRNA directed ribosomal translation of a larger inactive precursor called proTRH [26]. The maturation of TRH implicates many coordinated cellular and biochemical steps along the RSP (Fig. 1). First, The signal sequence, or presequence, directs cotranslational translocation of preproTRH into the lumen of the rough endoplasmic reticulum (ER) after which the signal sequence is removed. Once in the ER, the newly synthesized prohormone meets a unique environment containing a number of ER-specific chaperones involved in its folding pathway leading to its wild type conformation. In addition, proTRH, like all secretory proteins, encounter an exclusive set of post-translational modifications by specific peptide-bond cleavages provided by a network of processing peptidases. proTRH is transported from the ER to the Golgi Complex (GC) and then to the trans-Golgi network (TGN), where an initial proteolytic processing event occur. At the TGN, proTRH products are sorted to and stored into specialized secretory granules (SGs) that undergo secretion only after appropriate stimuli. In these SGs the final processing steps take place, which involves endoproteolysis by specific prohormone convertases at pair of basic residues [82; 83; 84], removal of the basic residues by a carboxypeptidase [85; 86; 87], and amidation [5; 88]. If one of these regulated steps is compromised, the biosynthesis of the prohormone derived peptides and their secretion may be affected.
Until recently, the initial maturation process of the proTRH precursor in the ER, and the structural features involved in intracellular trafficking from ER to the Golgi had to be elucidated. The primary sequence of proTRH has an N-terminal segment of 25 amino acid residues, pYE26, immediately after the signal sequence (Fig. 1). By expressing several deletions and point mutations cDNA constructs within the preproTRH31-52 sequence, and by monitoring the steady state production of proTRH in the ER, we identified a single tripeptide Pro-Gly-Leu (40PGL42) sequence, which is conserved in mammals and it turn out to be important for the stability of proTRH, in terms of resistance to protein degradation [84]. These studies revealed that PGL is primarily involved in the stability of proTRH in the early secretory pathway. Deletion of PGL destabilizes proTRH by targeting the protein to the proteasome for degradation. This was the first evidence showing that the structural role played by a short motif located in the N-terminal region of proTRH takes place early in the ER, and has important consequences on precursor stability rather than the sorting process to the SGs per se [84]. The processing modifications occur while proTRH is transported from the TGN to newly formed immature secretory granules [89]. The secretory granules then mature and are targeted to sites of secretion at the plasma membrane of the cell. Upon secretory granule maturation, electron-dense-secretory granules containing sorted products of proTRH fuse with the plasma membrane in response to an extracellular stimulation in a calcium-dependent manner, thereby releasing their contents into the external milieu [90].
Initial evidence supporting differential distribution in SGs for the N- and C-terminal proTRH peptides comes from immunocytochemical studies using transfected AtT20 cells with preproTRH cDNA, and in primary neuronal cultures. The TRH precursor and N-terminal intermediate forms are located in the GC and TGN (Fig. 2A). In contrast, end products including preproTRH25-50, preproTRH160-169, and TRH, are only present in SGs along the plasma membrane and in cell processes (Fig. 2F). C-terminally-directed antisera resulted in positive immunostaining in the GC, along the plasma membrane, and in cell processes (Fig. 2H). Thus, C-terminal intermediates appear to reach further along the secretory pathway before processing than their N-terminal counterparts. This differential processing event serves as a mechanism to regulate the timing of production of peptides such as preproTRH160-169, preproTRH178-199 and preproTRH53-75, and possibly TRH. Primary cultures of hypothalamic neurons, developed in our laboratory, provide a second model system to study proTRH processing. After 12-14 days in vitro [2], these hypothalamic neurons show growth of neurites similar in morphology to peptidergic neurons. Most neurons are bipolar with long axons containing varicosities, boutons and growth cones. Many of the growth cones are in contact with neurites of other neurons. Dendrite-like structures are also observed. N-terminal antiserum in these cultured hypothalamic neurons [2], stains the GC, ISGs budding from the GC, and terminal boutons (Fig. 2B and E). Immunoelectron microscopy confirms positive staining in the GC (Fig. 2E). Some ISGs budding from the TGN-like were also stained. Since positive staining was detected in boutons, it was proposed that these intermediates are processed to mature proTRH-derived peptides near the axon terminal, prior to secretion. ICC using anti-TRH, anti-preproTRH53-75, and anti-preproTRH160-169 showed positive staining only in the neuronal processes and axon terminals while the soma remained unstained (Fig. 2G). The positive staining observed in neuronal processes and axon terminals with these antibodies was confirmed by IEM (Fig. 2G (insert)). Immunostaining on transfected AtT20 cells using anti-TGN38, a TGN marker, indicated that the TGN could be projected away from the GC towards budding ISGs (Fig. 2C). Thus, in AtT20 cells processing of the N-terminal forms takes place somewhere between the TGN and ISGs, suggesting that preproTRH25-50, preproTRH53-75 and preproTRH83-106 are already formed by the time SGs mature. Using the C-terminal antiserum that recognizes C-terminal intermediate forms and end peptide products, positive staining was visualized in a patchy cytoplasmic distribution (Fig. 2I), often closely associated with the nucleus. Immunoreactivity was also observed in neuronal boutons, axon terminals and unbranched growth cones. IEM confirmed that the patchy cytoplasmic areas were within the ER and GC [2] (Fig. 2I insert); all layers of the GC (cis, medial and trans) were immunostained with this antibody [2]. The contrasting staining patterns for the two antisera (N- and C-terminal) (Fig. 2B,I) suggest the existence of a different peptide distribution for N-terminal versus C-terminal peptides, possibly, due to different intracellular routing of intermediates to SGs.
Fig.2.
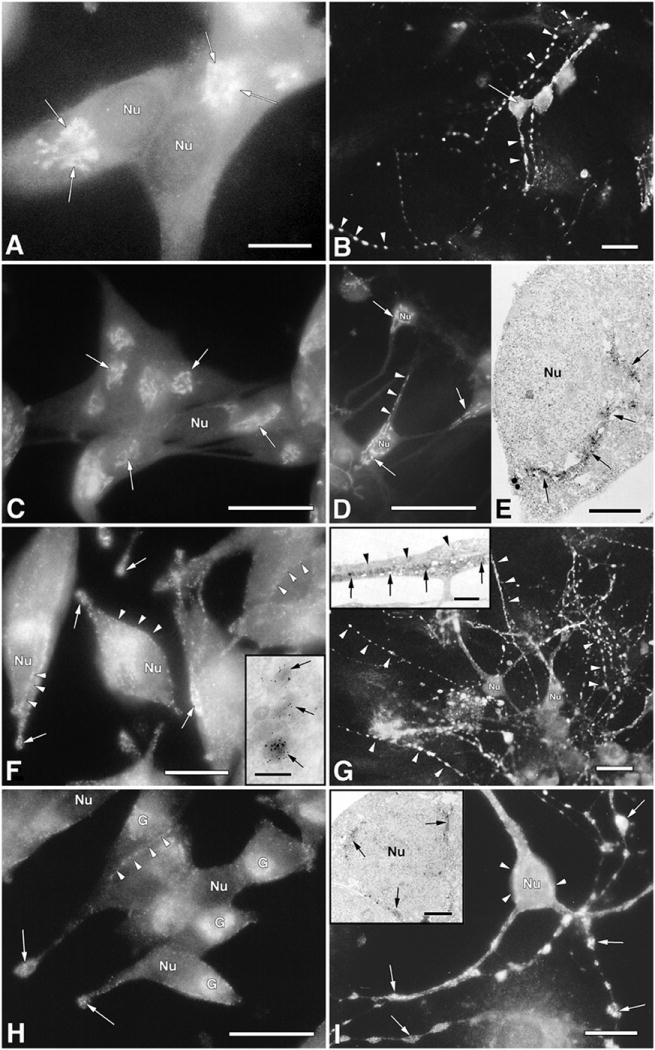
Subcellular distribution of proTRH intermediate and end products of processing, in transfected AtT20 cells encoding the TRH gene, and in primary cultures of hypothalamic neurons. The cells were fixed with 4% paraformaldehyde followed by immunostaining with different antibodies against the proTRH sequence. Fluorescein isothiocyanate conjugated to goat antirabbit globulin was used as a probe. Panel A, AtT20 cells: positive staining in the GC and TGN (arrows) using an antibody against proTRH and N-terminal intermediate forms (anti-pCC10). Bar =25μm. Panel C, AtT20 cells: cells immunostained with anti-TGN38 (arrows), a TGN marker. Bar = 50μm. Panel F, AtT20 cells: a typical positive staining along the plasma membrane (arrow heads), a common granule distribution of corticotropic cells, and processes (arrows) using anti-non-TRH peptides and anti-TRH. Bar = 25μm. Panel F, AtT20 cells; inset: typical positive staining of SGs by IEM using anti-pST10 antibodies (5 nm gold particles). Bar = 200μm. Panel H, AtT20 cells: positive staining in the GC and processes using an antibody that recognizes pro-TRH and C-terminal intermediate forms (anti-pYE17). Bar = 50μm. Panel B, Hypothalamic neurons: positive staining in the GC (arrow) and TGN using an antibody against proTRH and N-terminal intermediate forms, and in all boutons distributed along the neuronal processes (arrowheads) Bar = 50μm. Panel D, hypothalamic neurons: cells immunostained with anti-TGN38, a TGN marker. Bar = 50μm. Panel E, hypothalamic neurons, a higher magnification of panel B showing positive staining in stacked Golgi cisternae and in some forming granules (arrows) using the peroxidase-DAB reaction (arrows) Bar = 5μm. Panel G, hypothalamic neurons: positive fluorescence is observed only in neurites (arrowheads) and axon terminals, while the cell body remain unstained. Bar = 50μm. Panel G, hypothalamic neurons; inset: an IEM of neurites using peroxidase-DAB staining reaction. Of the two adjacent neurites shown, the lower one is positively stained (large arrows), whereas the upper one is negative (small arrow). Bar = 1μm. Panel I, hypothalamic neurons: positive staining in several areas of the cell body (arrowheads) and in all boutons distributed along the neuronal processes (arrowheads) using an antibody against pro-TRH and C-terminal intermediate forms. Bar = 25μm. Panel I, hypothalamic neurons, inset: a higher magnification of cytoplasmic areas from panel I showing positive staining in the endoplasmic reticulum and GC (arrows) as well as in SG near the plasma membrane (arrows). Bar = 2μm. nu, Nucleus; G, Golgi complex. The polyclonal antibodies used in this ICC are as follow: Anti-pCC10 [made against a synthetic decapeptide (Cys-Lys-Arg-Gln-His-Pro-Gly-Lys-Arg-Cys)], which recognizes prepro-TRH25–255 (26 kDa) prepro-TRH25–151 (15 kDa) prepro-TRH25–112 (9.5 kDa) prepro-TRH25–74 (6 kDa). Anti-pYE17 (made against prepro-TRH240–255), which recognizes prepro-TRH25–255 (26 kDa) prepro-TRH115–255 (16.5 kDa), prepro-TRH160–255 (10 kDa), prepro-TRH208–255 (5.4 kDa), anti-pST10 (made against preproTRH160–169), and anti-TRH. [Panels B, D, G, and I were reproduced with permission from E. A. Nillni et al.: Endocrinology 137:5651–5661, 1996 (36). Panels were reproduced with permission from E. A. Nillni and K. Sevarino: Endocr Rev. 1999 Oct;20(5):599-648. © The Endocrine Society.
How do we explain these early findings? Synthesis of prohormone polypeptides and their processing to end products occur while they are sorted to the SGs [91; 92; 93; 94]. Several structural elements have been related to the sorting mechanism within the RSP. The N-terminal hydrophobic domain in certain prohormones could represent a sorting signal [95]. Also, a disulfide loop in the N-terminal region found in certain prohormones seems to be essential for targeting to the RSP [96; 97; 98]. Many prohormones contain pairs of basic residues, which could also act as a sorting signal [99; 100]. Additionally, the RGD (Arg-Gly-Asp) motif present in processing enzymes is also implicated in trafficking within the RSP [101]. In the ProTRH polypeptide there are 11 pairs of basic residues, one disulfide loop and two RGD motifs (Fig. 1). While evidence showed that the RGD motif did not play a role in sorting, the disulfide loop present in the C-terminal side of proTRH was involved in the sorting of proTRH-derived peptides and in their retention in the SGs [102]. In separate study, the data demonstrated that the initial processing of proTRH by PC1/3 in the TGN, and not a cargo-receptor relationship, is important for the downstream sorting events that result in the storage of proTRH-derived peptides in mature SGs [103]. The same initial cleavage by PC1/3 produced the unfolding of a partially folded proTRH, and it was a key determinant mechanism for the delivering of N- and C-terminal proTRH-derived peptides to different SGs of the RSP [104] (Fig.1B). This could be a common mechanism used by neuroendocrine cells to independently regulate the secretion of different bioactive peptides derived from the same gene product [104]. The important evolutionary concept that arises from these studies is that post-translational processing of hormone precursor proteins is a critical mechanism by which cells increase their biological and functional diversity, such that two or more peptides with different biological functions originate from the same precursor. It is through differential post-translational processing mechanisms that cells selectively produce specific peptides for secretion [26; 105; 106]. The correct sorting events of proteins and peptides to their target organelles are of fundamental importance in cell biology. However, identification of the signals or physical properties that ensure proper intracellular sorting of peptides to dense core SGs is not yet fully established. In a recent review article, Dikeakos and Reudelhuber [107] subdivided the targeting function of proteins that are sorted to SGs into three groups: membrane-associated (or traversing) tethers, tether-associated cargo, and aggregation. The enzyme PC1/3 is a granule-tethered protein that may act as a sorting chaperone for its substrates in addition to being a processing enzyme. In the case of proTRH, as described above, initial processing action of PC1/3 on proTRH, and not a cargo-receptor relationship, is important for the sorting of the proTRH-derived peptides. Thus, it is possible that key paired basic amino acids of proTRH (residues 107, 108, 113,114, 152, 153, and 159), which constitute a cleavage site for PC1/3, would also function as granule sorting domains [103; 107]. For proTRH thus far, N- and C-terminal proTRH derived fragments behave independently after the prohormone is cleaved (Fig. 3). It is possible that the initial cleavage changes the folding conformation and/or exposes different motifs that allow N- and C-terminal proTRH fragments to aggregate independently or to interact with different components of the RSP. Interestingly, the differential sorting is independent of the cell type [104]. This suggests that the N- and C-terminal ends of proTRH have conserved properties, which are sufficient for the sorting events. This finding also suggests that the cellular environment is not a critical factor, although the presence of RSP machinery should be necessary. Peptides derived from the same precursor, which are differentially packaged, have been shown for the egg-laying hormone (ELH) precursor in both Aplysia californica and Lymnaea stagnalis (37–39), but this phenomenon had never been demonstrated in mammalian prohormones, and proTRH represents the first mammalian case in which differential sorting of its processing products is demonstrated.
Fig. 3.
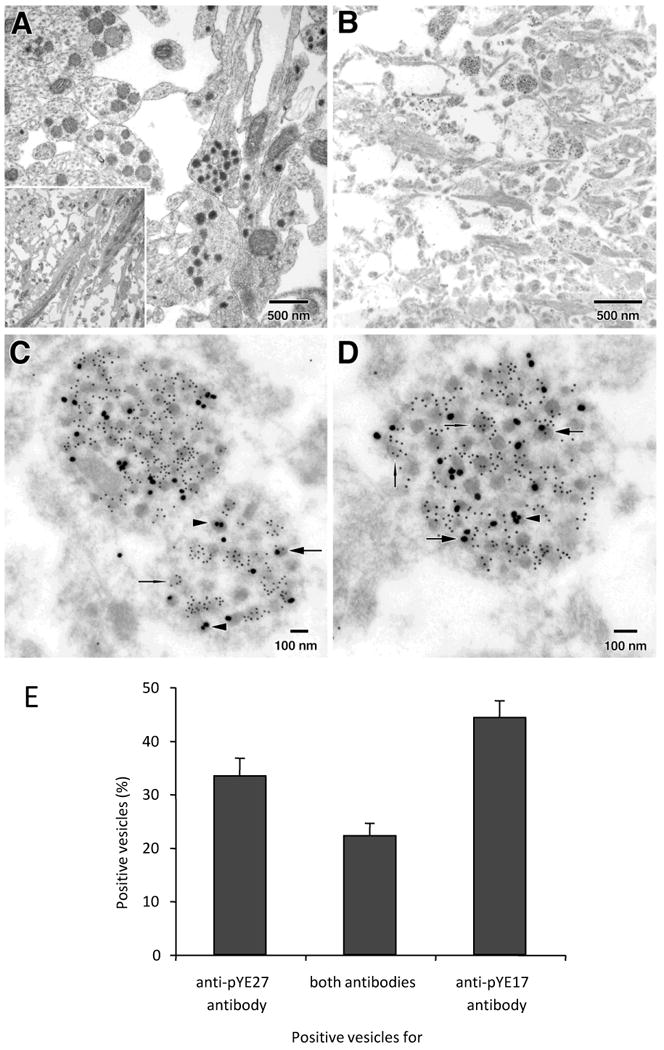
N- and C-terminal end products of proTRH are partially located in different vesicles within the ME fibers. Sprague-Dawley rats were perfused with Karnovsky's fixative (A, inlet shows a panoramic micrograph of this region) or with 4% paraformaldehyde/0.15% glutaraldehyde in PBS (B-D). Panels C-D show an increased magnification of the panel B. Images show three classes of positive vesicles for either anti-pYE17 antibody alone (arrows-head), anti-pYE17 antibody alone (needles), or a combination of both antibodies (arrows). Panel 3E shows quantitative results obtained from sections labels first with anti-pYE17 (10 nm gold-particles), and then with anti-pYE27 (25nm gold-particles). Mean percentages represent the proportion of a particular positive vesicle compared to the total number of positivevesicles observed per micrograph. This Figure was reproduced with permission of Perello et al. J Biol Chem. 2008 Jul 18;283(29):19936-47.
Among the seven members of the PC family cloned thus far, PC1/3 and PC2 are specifically found in neuronal and endocrine cells containing secretory granules [108; 109; 110]. Their involvement in the processing of neuropeptide precursors has been suggested by the finding that PC1/3 and PC2 transcripts and protein products are widely distributed in different areas of the brain, including the cerebral cortex, hippocampus, and hypothalamus [111; 112]. Within the hypothalamus, these enzymes display an extensive overlapping pattern of expression [111; 112]. The critical role of PC1/3 and PC2 in prohormone processing is underscored by studies from animals lacking the genes encoding PC1/3 [113] and PC2 [114], as well as 7B2 [115; 116], a neuropeptide essential for the maturation of PC2. Disruption of the gene encoding mouse PC1/3 results in a syndrome of severe postnatal growth impairment and multiple defects in processing for many hormone precursors, including hypothalamic progrowth hormone releasing hormone (proGHRH) to mature GHRH, pituitary proopiomelanocortin hormone (POMC) to adrenocorticotropic hormone, islet proinsulin to insulin, and intestinal proglucagon to glucagon-like peptide-1 and -2. PC1/3-/- mice are normal at birth, but display impaired postnatal growth and are about 60% of normal size at 10 weeks. They lack mature GHRH, have low pituitary growth hormone and hepatic insulin-like growth factor-1 mRNA levels, and phenotypically are smaller than a normal mouse (34). Mice with disruption of the gene-encoding PC2 appear to be normal at birth. However, they exhibit a small decrease in rate of growth. They also have chronic fasting hypoglycemia and a reduced blood glucose level during an intraperitoneal glucose tolerance test, both of which are consistent with a deficiency of circulating glucagons [117]. The processing of proglucagon, prosomatostatin, and proinsulin in the alpha, delta, and beta cells of the pancreatic islets is severely impaired in PC2 null mice [118].
An original work published in 1997 [3] and prior supporting data provided unequivocal evidence for the role of PC1/3 and PC2 in the processing of proTRH [1; 2; 3; 119]. Initial processing of proTRH occurs in the TGN at the prepro152-153-TRH-158-159 site to generate the 15 and 10kDa peptides (Fig. 1). In subsequent steps, the 15 kDa N-terminal intermediate moiety of proTRH is processed to a 9.5kDa peptide followed by continuous processing until the end products are generated in secretory granules. It is proposed that the processing of the remaining 10 kDa C-terminal fragment produces the 5.4 kDa C-terminal peptide preproTRH208-255 early in the secretory pathway [89] and the remaining intermediate form is further processed to end products in secretory granules [26; 105; 120; 121]. Thus far, only two sites processed by PC2 have been identified. The pFE22 peptide is further cleaved to two smaller forms of about 1.6 (preproTRH178-184 pFQ7) and 0.84 (preproTRH186-199, pSE14) kDa [121]. The extended form of pEH24, TRH-pEH24 is processed to generate TRH and pEH24 [3] (Fig. 1). Preliminary studies using the PC1/3-/- mice confirmed the initial in vitro finding that attributes a primary role of PC1/3 in the processing of proTRH [3], while PC2 has a specific role in cleaving TRH from its extended forms [121]. PC1/3 null mice show a dramatic decrease in the biosynthesis of all proTRH-derived peptides analyzed, including TRH and its proform, TRHGly. However, proTRH is still processed to its end products suggesting, as demonstrated by us in earlier studies, that PC2 and potentially furin can compensate for the lack of PC1/3 in the processing of proTRH [3] (unpublished results). In the PC2 null mice, although TRH and TRHGly showed some decrease as compared with the wild type animals, the concentrations of N-terminal peptides preproTRH25-50 and preproTRH83-106 did not change. This indicates that PC1/3 is the primary enzyme involved in the processing of proTRH that occurs at the pairs of basic residues flanking the TRH sequence including the sequence Arg51-Arg52, which does not contain TRH.
A second mouse model of PC1/3 deficiency generated by random mutagenesis was recently reported [122]. This mouse has a missense mutation in the PC1/3 catalytic domain (N222D) that leads to obesity with abnormal proinsulin processing and multiple endocrine deficiencies. Although there was defective proinsulin processing leading to glucose intolerance, neither insulin resistance nor diabetes developed despite obesity. The apparent key factor in the induction of obesity was impaired autocatalytic activation of mature PC1/3 causing reduced production of hypothalamic α-MSH. This is the first published characterization of a Pc1 mutation in a model organism that mimics human PC1 deficiency [122]. For example, a patient with a compound heterozogous mutation in the PC1/3 gene resulting in production of nonfunctional PC1/3 had severe childhood obesity [123]. An analogous obese condition was found in a patient with a defect in POMC processing [124]. It remains to be studied what effect the mouse Pc1 (N222D) with an obese phenotype will have on the processing of proTRH. However, the real challenge for the thyroid axis occurs when animals are physiologically perturbed such as during cold stress or during changes in the nutritional status, situations in which more TRH peptide is necessary to increase the output of the thyroid axis. For example, the role of CPE in processing has been elucidated in CPEfat/fat mice, which lack functional CPE resulting from a naturally occurring mutation [125]. This mouse is obese, diabetic, and infertile [4]. Using this mouse, it was found that hypothalamic TRH was depressed by at least 75% compared to wild type controls [4], and that CPD was probably responsible for the generation of 21% of the TRH produced in the hypothalamus of these animals [4]. Their body temperatures had declined by an additional 2.1°C as compared with wild type controls. Furthermore, these animals cannot maintain a cold challenge for 2 hours at 4°C because of alterations in the HPT axis represented by deficits in proTRH processing, which resulted in hypothalamic TRH depletion and a reduction in thyroid hormone levels [4]. More insight can be obtained from other genetic models as useful tools to understand the role of leptin in the thyroid axis, but not without simultaneously raising puzzling questions. One important point to be considered is that the status of the thyroid axis for each genetic model is different, which makes the interpretation of the results obtained from different animals difficult to compare. For example, to understand the implications of TRH in the physiology of the HPT axis, previous studies generated mice that lack TRH. Although the TRH-/- mice showed signs of hypothyroidism with characteristic elevation of serum TSH level and diminished TSH biological activity, they were viable through adulthood and showed no gross anatomic abnormalities, and exhibited normal development. The decrease in TSH immunopositive cells could be reversed by TRH [15]. This TRH deficient mouse also exhibited hyperglycemia, which was accompanied by impaired insulin secretion in response to glucose, providing a good model of tertiary hypothyroidism. This hyperglycemia seen in the TRH-/- mice indicates that TRH is involved in the regulation of glucose homeostasis. Supporting this hypothesis, a recent study showed that TRH was able to reverse STZ-induced hyperglycemia by increasing pancreatic islet insulin content, preventing apoptosis, and potentially inducing islet regeneration [126]. Also, perinatal TRH treatment enhanced basal insulin secretion in STZ-induced animals of both sexes and partially restored the insulin response to glucose stimulation in females [127].
These observations suggested that, similar to other genetic models, there must be some compensatory responses during ontogeny to compensate for the lack of TRH. In the ob/ob mouse, which could be important for the understanding of the direct pathway, there is a compensatory response by other unknown mechanisms during development that keeps T3/4 almost at normal levels. However, ob/ob mice are inefficient in their response to cold stress, indicating a deficiency in HPT axis regulation [128; 129]. A similar response was observed in CPEfat/fat mice where the level of T3/4 was normal but the animals failed to maintain their body temperature during cold stress [4]. In both cases the thyroid axis was relatively normal under basal conditions, but unable to respond to cold stress. The other interesting and highly relevant genetic model is the mouse lacking the MC4 receptor (MC4-/-), which is essential for α-MSH activity in TRH neurons. Utilizing the MC4-/- mouse model, it was found that TRH peptide levels fluctuate during fasting and respond to leptin treatment in a similar fashion to wild-type controls. Additional roles for the two pathways of leptin action on TRH neurons [18] (see section 7. The direct and indirect pathways of leptin action on TRH neurons) were uncovered in studies related to the role of the melanocortin system and adaptive thermogenesis of brown adipocyte tissue [130]. In rodents, thyroid hormones act synergistically with the SNS to regulate UCP1 expression [131]. UCP1 expression relies on functional T3 response elements and cAMP response element binding protein motifs in the UCP1 gene upstream enhancer region [10]. BAT contains abundant type 2 deiodinase (D2), which catalyzes the conversion of T4 to the more biologically active and potent T3 and which is activated by the SNS [10]. Since the melanocortin system can modulate both sympathetic outflow to the BAT [32] and the function of the HPT axis [132], the question was whether the MC4R-mediated up-regulation of UCP1 expression in response to a high fat diet involves the HPT axis. These studies led to the hypothesis that perhaps there may be a defect in the MC4R-HPT axis that contributes to the inability of MC4R-/- mice to up-regulate UCP1 expression in BAT in response to a HF diet. However, MC4R-/- and AgRP-treated mice exhibit the same increase in total T3 hormone levels as wild type controls when switched from a low fat to a high fat diet. The total T4 levels in high-fat-fed MC4R-/- and wild-type mice are decreased, which is likely related to the observed increases in T3, because T4 is converted to T3. Altogether, these results suggest that the central melanocortin-mediated regulation of the HPT axis may not be involved in the diet induce thermogenesis provoked up-regulation of UCP1, although the melanocortin system may mediate part of the leptin-induced secretion of TRH from hypothalamic explants [18].
At the physiological level, the PCs are regulated by states of hyperglycemia [133], inflammation [134], suckling [121], starvation [135], cold stress [136] and morphine withdrawal [137]. In the case of morphine withdrawal, for example, it was previously demonstrated that during opiate withdrawal, preproTRH mRNA increased in neurons of the midbrain periaqueductal gray matter (PAG), caused a significant change in the level of some post-translational processing products derived from the TRH precursor, and the mature form of PC2 increased only in PAG as compared with their respective controls demonstrating a region-specific regulation of proTRH processing in the brain, which may engage PC2 [137] [138]. Another interesting example was provided in alterations seen in proTRH processing in the PVN during lactation where PC2 was involved [121]. Other examples of the regulation of PCs include rats exposed to streptozotocin, where it was demonstrated that the diabetic state altered alpha-cell processing of proglucagon to give increased levels of glucagon-like peptide 1 [133]. Li et al also identified regions in PC1/3 and PC2 human promoters that contain putative negative thyroid hormone response elements and has shown that T3 negatively regulates PC1/3 and/or PC2 expression in rat GH3 cells, rat anterior pituitary, hypothalamus and cerebral cortex [139; 140; 141; 142]. In addition to the PCs, other enzymes involved in the maturation of peptides could be affected under different physiological conditions including cold exposure, fasting, or changes in thyroid status [6]. For example, the amidating enzyme PAM activity on TRH neurons is regulated by the thyroid status. TRH and TRH-Gly levels increase under low iodine/PTU diet-induced hypothyroidism and decrease under TH-induced hyperthyroidism [6; 143]. However, the ratio TRH/TRH-Gly, which depends on PAM enzymatic activity, is different for each condition. In hypothyroidism, the ratio TRH/TRHGly increases suggesting an increase in PAM activity, and in hyperthyroidism the ratio decreases suggesting a decrease in PAM activity. Since PAM activity is responsive to changes in thyroid hormone levels, this could represent another level of control in the final production of mature TRH.
To make matters more complex in the relationship between prohormones and processing enzymes, a major breakthrough study in the biology of leptin added an extra level of complexity in the maturation of TRH, that is, in addition to regulating peptide hormone expression, leptin also controls prohormone processing by regulating PC1/3 and PC2 [135]. Since leptin regulation of energy balance works through the activity of several neuropeptides, it was hypothesized that leptin might regulate processing enzymes in addition to regulating peptide production. These studies clearly supported the hypothesis that the regulation of hypophysiotropic TRH biosynthesis by leptin, occurs not only at the transcriptional level, but also at the post-translational level through changes in proTRH processing by the action of PC1/3 and PC2 [135] (Fig. 2). A more comprehensive review on this subject was recently published elsewhere [144]. Utilizing in vitro and in vivo approaches, strong evidence was shown implicating leptin in the regulation of PC1/3 and PC2 expression/protein biosynthesis, and as a consequence of those regulatory changes, the post-translational processing of proTRH is affected [135]. Thus, the results from this study demonstrate that leptin couples the up-regulation of preproTRH expression and its protein biosynthesis with the up-regulation of the processing enzymes in a coordinated fashion. Such regulation ultimately leads to more effective processing of leptin-regulated pro-neuropeptides into mature peptides, such as TRH and α-MSH, which are critical for leptin action. Therefore, transcriptional control of PC1/3 and PC2 gene expression by leptin is another level at which this hormone regulates energy homeostasis. This adds a novel key checkpoint that is tightly regulated in the control of energy consumption. Since leptin-induced increase in the PC levels in vivo is partially dependent on the activation of the melanocortin system [18], and since PC1/3 promoter contains two CREB response elements, which are transactivated by CREB-1 [145], the P-CREB transcription factor could activate in a coordinated fashion the synthesis of preproTRH and PCs. Therefore, leptin can act on the TRH neurons of the PVN directly (P-STAT) and indirectly via the melanocortin system (P-CREB) regulating in a synchronized manner the biosynthesis of proTRH and the PCs through both pathways (see Figs. 2 and 3).
5. Regulation of hypophysiotropic TRH neurons by cold exposure
Exposure of animals to cold stress strongly stimulates the HPT axis through adrenergic inputs from the medulla [24], and the catecholamine NE has long been proposed to be the main regulator. As mentioned above, the periventricular and medial parvocellular divisions of the PVN are densely innervated by NE-containing and epinephrine (E)-containing inputs from the medulla and pons [23]. Further, NE-containing neurons densely innervate the midregion of the external layer of the ME. ICV injections of NE, E, and α2-adrenergic agonists stimulate basal TSH secretion [146; 147], and NE/E treatment of hypothalamic preparations stimulates TRH release [148]. Inhibitors of catecholamine biosynthesis or α2-adrenergic antagonists lead to a fall in basal TSH secretion. Thus, NE and E exert a tonic, stimulatory regulation on TSH secretion principally through α2-adrenergic receptors. Stimulated release from the ME appears to be mediated via α1-adrenergic receptors [149]. In contrast, locus coeruleus afferents are inhibitory when activated during stress [150]. NE/E excitation of PVN TRH neurons mediates the rise in TSH in response to acute cold exposure or hypovolemia [149; 151; 152]. Rats subjected to cold temperatures increase preproTRH mRNA levels in the hypothalamic paraventricular nucleus within 30 to 60 min of the stimulus [153], augment TRH release from the median eminence [154], stimulate TSH secretion from pituitary, and potentiate T4 and T3 contents in blood [155]. Similarly to the situation seen during starvation, where the HPT axis set point is altered to reduced energy expenditure [135; 156], acute cold-induced stimulation of the HPT axis by increasing preproTRH mRNA when at the same time circulating levels of thyroid hormone are elevated. Consequently, cold exposure and starvation can shift the set point for feedback regulation of the preproTRH gene.
Although early studies showed that the activation of α1-adrenergic receptors in the ME increased the release of TRH [149] and cold exposure increased preproTRH mRNA in the PVN [157], the specific role of NE on the biosynthesis of proTRH at this level was not fully studied. Recent new studies showed that NE stimulates preproTRH gene expression and proTRH biosynthesis in cultures of hypothalamic neurons [136], and in an in vivo model the β-adrenergic receptor mediated the cold-induced increase in the biosynthesis of proTRH in the PVN [136] through P-CREB signaling [136]. The cold exposure-induced increase of proTRH levels in the PVN was also coupled to an up-regulation in the biosynthesis of PC1/3 and PC2. The up-regulation of these enzymes is mediated by the activation of β-adrenergic receptors [136]. Similar to the melanocortin system, this activation may occur via the CREB response elements contained in the PC1/3 promoter [158]. Specific regulation of prohormone processing is likely another key step where final amounts of bioactive peptides can be tightly regulated. Different factors, such as thyroid hormones, leptin, the melanocortin system and NE, potently stimulate proTRH biosynthesis in concert with an increase in the PC enzymes in the hypothalamic PVN [18; 135; 143]. In another example, it was recently showed that regulation of POMC biosynthesis also occurs at the post-translational level through coordinated changes in the PC enzymes in hypothalamus and medulla [159], and pro-vasopressin prohormones as well PCs [160]. Altogether these collective findings seen for several prohormones suggest the existence of a common mechanism used by different types of cells to generate bioactive peptides in a more efficient way. Thus, the coordinated regulation of PCs may play an important role in neuroendocrine cells in maintaining a proper enzyme-substrate homeostasis, and ensuring adequate processing of newly synthesized prohormones (Fig. 4).
Fig. 4. Schematic representation of the TRH neuron with the most relevant inputs controlling its gene expression.

The illustration shows the multiple signals affecting the TRH neuron coming from the peripheral circulation including leptin and T3, and from the ARC, α-MSH, NPY, and AgRP. The TRH promoter integrates each of these inputs to determine the set point of the HPT axis. The different hormonal inputs regulating the PCs are also represented.
6. Thyroid hormones selectively regulate proTRH processing in the PVN by affecting the PCs
At the cellular level, changes in the thyroid status also have an effect on the processing of proTRH by altering the level of PCs [143; 161]. These changes in proTRH processing were only observed in hypophysiotropic TRH neurons. During PTU (6-n-propyl-2-thiouracil) diet-induced hypothyroidism, in addition to increasing hypophysiotropic preproTRH mRNA and proTRH biosynthesis [162], increases the processing of proTRH prohormone also occurred combined with an increase in PC1/3, PC2 [143]. In the PVN of hypothyroid rats, an increase for the end products of proTRH processing analyzed peptides was found [143]. Hyperthyroidism induced by T4 treatment decreased preproTRH mRNA and TRH levels in the PVN [162], decreased the post-translational processing of proTRH rendering an accumulation of unprocessed intermediate forms combined with a downregulation of the PCs [143]. This finding was further supported by studies demonstrating that the PC promoters contain thyroid receptors, which can be negatively regulated by thyroid hormone [134; 140; 142]. Thus, regulation of prohormone and enzyme by changes in thyroid status may lead to altered hormonal biosynthesis. The selective co-regulation of proTRH processing by thyroid status in the PVN is a novel aspect of the regulation of the HPT axis. Further support for these findings includes the regions in the PC1/3 and PC2 human promoters containing putative negative thyroid response elements, and the fact that T3 negatively regulates PC1/3 and/or PC2 expression in rat GH3 cells, rat anterior pituitary, hypothalamus and cerebral cortex [139; 140; 141; 142]. These findings suggested the possibility that thyroid hormones could regulate PC1/3 and PC2 expression in specific hypothalamic nuclei by directly regulating proTRH processing. Experiments altering the thyroid hormone status in rats and examining the effects on proTRH processing in the PVN and in other extra-hypophysiotropic areas clearly showed that a high level of thyroid hormone down-regulates TRH biosynthesis, and that this was coupled with the down-regulation of PC1/3 and PC2 in the PVN. Conversely, low levels of thyroid hormone up-regulated TRH biosynthesis and PC1/3 levels [143; 161]. This regulation represents a novel aspect in the way the HPT axis is integrated between the central and peripheral inputs, which may have important implications in the pathophysiology of hypo- and hyperthyroidism (Figs. 2 and 4).
7. The direct and indirect pathways of leptin action on TRH neurons
At the central level, the hypothalamus is the primary component of the nervous system in interpreting adiposity or nutrient related inputs; it delivers hormonal and behavioral responses with the ultimate purpose of regulating energy intake and consumption. At the molecular level, enzymes called nutrient or energy sensors mimic the role of the tissues involved in energy balance [163]. Two key energy/nutrient sensors, mTOR (mammalian target of rapamycin) and AMPK (AMP activated kinase), in addition to their extensive metabolic roles in the peripheral tissues, are involved in the control of food intake in the hypothalamus [164; 165]. Recently, the enzyme Sirt1, an evolutionarily conserved NAD+-dependent deacetylase involved in many biological processes including cellular differentiation, apoptosis, metabolism, and aging, was showed to regulate the central melanocortin system in a FoxO1 dependent manner [Cakir, 2009 #5369]. Within the hypothalamus, neurons in the ARC receive circulating adiposity signals and relay their responses to “second-order” neurons in the PVN and lateral hypothalamus to mediate effects on food intake and energy homeostasis. TRH neurons represent the second order of neurons when stimulated by the melanocortin system. The fall in circulating levels of the hormone leptin during starvation is the signal to the brain that suppresses preproTRH expression in the PVN. Serum leptin decreases considerably during fasting, and leptin replacement during a 48-hour fast partly prevents the fasting-induced decrease in serum T4 and increase in serum corticosterone in mice. In turn, leptin was proposed as a vital signal that initiates the neuroendocrine response to fasting [166]. The hormone leptin (16kDa) is expressed predominantly in adipose cells, and the lack of it, as in the ob/ob mouse produces severe obesity [167]. Initially considered as a hormone to prevent obesity, it was later showed that the major role of leptin is to signal the switch from the fed to the starved state at the hypothalamic level [166; 168; 169]. The fall in circulating levels of leptin is perceived in the hypothalamus to increase appetite, decrease energy expenditure, and change neuroendocrine function in a direction that favors survival. The major consequences of falling leptin include activation of the stress axis, and suppression of reproduction, linear growth, and the thyroid axis [166]. In summary, leptin has a central physiologic role in providing information on energy stores and energy balance to brain centers that regulate appetite, energy expenditure and neuroendocrine function [166; 167; 170; 171; 172]. When leptin signaling is deficient, due either to mutation of leptin peptide or leptin receptor genes, severe obesity results in both rodents and humans [167; 173; 174; 175; 176], underscoring the fundamental role of leptin in the physiology of energy balance. Leptin induces STAT3 phosphorylation in nuclei of POMC neurons in the ARC of rats, and can directly activate the proximal pomc promoter in cells expressing the leptin receptor [177]. Leptin also induces STAT3 phosphorylation, mediated by Y1138 on ObRb, in nuclei of TRH neurons in the hypothalamus of rats [178]. This suggests that STAT3 is critical for mediating leptin-induced regulation of preproTRH gene-expression in the hypothalamus.
During fasting AgRP and NPY mRNAs increase and POMC mRNA decreases in the ARC. Although insulin increases POMC mRNA expression and both insulin and glucose reduce NPY mRNA expression in the ARC, the fasting induced a decrease in TRH mRNA expression and thyroid hormone concentrations, which is not prevented by glucose or insulin administration [179] supporting an exclusive role for leptin. Although the hypothalamic effects of leptin are mediated via the hypothalamic melanocortin system, which plays an essential role in feeding behavior, two landmark studies demonstrated the existence of a direct effect of leptin on TRH neurons [16; 17]. These studies showed that leptin dose-dependently stimulated the increase in proTRH biosynthesis and TRH release in primary cultures of hypothalamic neurons [17]. In the same primary cultures ObRb and proTRH showed to co-localize, which was consistent with the finding of the leptin receptor ObRb in the PVN [48]. In addition, leptin stimulates preproTRH gene expression via a STAT3 response element located in the preproTRH promoter in isolated cells [16], and induce STAT3 phosphorylation in TRH neurons in the PVN of leptin-treated rats [178]. It was also demonstrated that STAT3 mediates the transcriptional effects of leptin in vivo suggesting that the TRH promoter is directly regulated by leptin [71]. Leptin rapidly regulates polarization of neurons in isolated brain slices of the PVN and can stimulate TRH peptide release from dispersed hypothalamic cultures and hypothalamic tissue ex vivo [17; 55]. Leptin produced a five-fold induction of luciferase activity in CV-1 cells transfected with a TRH promoter-luciferase reporter and the long form of the leptin receptor cDNA [16]. Additionally, peripheral administration of leptin to rodents activates SOCS-3 (suppressor-of-cytokine-signaling) mRNA (which is the sensitive marker of direct leptin action) in neurons located in the PVN [16; 180]. Since ObRb [181] is present in TRH neurons directly activated by leptin (via STAT3) and indirectly (via CREB) by the melanocortin system [18] (Fig. 4), one may ask whether the increase in preproTRH gene expression and proTRH biosynthesis in response to leptin is mediated by both pathways, each one individually or a combination of both. This question was recently clarified where it was determined that leptin affects both pathways [16; 18; 120; 178; 182], providing compelling evidence in support of the direct action of leptin on TRH neurons. Two additional findings support the existence of the direct pathway, 1) MC4-R KO mouse and patients with MC4-R mutations have normal thyroid hormone levels [183; 184], 2). While the MC4-R is expressed in the PVN [53], it was found that only approximately 48% of TRH neurons in the PVN express this receptor [16]. It is interesting to note that only 30% of the POMC neurons derived from the ARC have synapses with 34% of TRH neurons in the medial parvocellular subdivision [179]. The majority of TRH neurons in the medial parvocellular subdivision of the PVN where most of the hypophysiotropic TRH neurons reside [27], are contacted by AGRP and not by α-MSH-containing axon terminals [185]. In the same studies, 300ng of α-MSH (physiological levels are 2.5 ng) administrated to fasting rats every 6 hours for 64 hours by ICV injections were needed to raise the level of thyroid hormone to only ∼50% of the levels observed in the fed animals [185]. In contrast systemic leptin administration to fasting animals not only increases preproTRH mRNA in the PVN but also fully restores thyroid hormone levels and TRH peptide to normal or higher levels [135; 156]. Therefore, α-MSH appears to affect only part of the full regulatory effects of leptin on the thyroid axis, primarily via actions directed on the regulation of preproTRH gene expression.
Although the above data are consistent with a direct ability of leptin to promote TRH biosynthesis through actions on TRH neurons, the ARC-derived neuropeptides play the most significant role in the regulation of proTRH neurons, in the normal but not in the obese condition as its was shown that pretreatment of fasted rats with a melanocortin antagonist fully blocks the leptin-induced effect on the HPT axis [18] (see below section 10 Obesity and the Thyroid axis), via synaptic input from leptin-responsive POMC and NPY/AgRP neurons located in the ARC [179; 186]. Using nuclear P-CREB staining and a pharmacological antagonist of MC4-R, it was shown that the melanocortin system has a primary role in the leptin-mediated activation of hypophysiotropic proTRH neurons [18]. ObRb mRNA is also highly expressed in the ARC. Specifically, ObRb is present in neurons expressing NPY, AgRP, POMC, and CART [50; 51; 57; 187; 188; 189]. Leptin resistance suppresses coexpression of NPY and AgRP in one population of arcuate neurons. Since NPY, AgRP and POMC expression in the ARC are regulated by leptin in vivo [50; 51; 57; 187; 189] growing evidence shows that these neuropeptides play an indirect role in the regulation of TRH neurons by leptin. These include: a) an inhibitory action of leptin on NPY and AgRP release from the ARC, leading to a reduced inhibitory effect of these peptides on TRH expression in the PVN; and/or b) a stimulatory action of leptin on α-MSH release resulting in stimulation of TRH release originally shown for the first time using primary hypothalamic cultures [17] (Fig. 4 and 5)
Fig. 5. Proposed model of leptin and NPY action on TRH neurons through the direct and indirect pathways.

This diagram shows the two subgroups of TRH neurons recently identified based on their signaling modalities. Leptin acts directly on TRH neurons expressing ObRB through P-STAT3 signaling, and indirectly by acting on POMC neurons expressing ObRB that in turn release α-MSH and stimulate TRH neurons expressing the MC4 receptor through P-CREB signaling. A third unidentified group of TRH neurons is proposed to be potentially present in the PVN carrying both ObRb and MC4R. The TRH neurons carrying the MC4R are mainly involved in the regulation of the HPT axis, whereas the direct pathway could be essential in the obese condition. Other neurons stimulated by the melanocortin pathway might be involved in sympathetic activity or food intake regulation. NPY may regulate TRH neurons through mechanism 1 or 2 depending upon the status of the melanocortin system.
Previous data suggest that the primary action of leptin on the HPT axis is exclusively mediated by hypothalamic ARC neurons through direct axonal projections to the PVN. It was proposed that if the ARC is ablated by neonatal treatment with mono-sodium glutamate (MSG), not only is the response of the thyroid axis to fasting abolished, but its response to the exogenous administration of leptin is lost as well [182]. However, this is not the ideal model to test this hypothesis. Rats subjected to neonatal MSG are less sensitive to peripherally administered leptin [190; 191]. Moreover, since MSG treatment causes lesions to blood-brain barrier-free areas, it is possible that the leptin insensitivity is due to an impaired leptin transport system rather than lesions of specific leptin-sensitive cells. The complex metabolic disturbances of MSG rats (hyperinsulinaemia, hypercorticosteronaemia, hyperleptinaemia, hyperglycaemia) could contribute significantly to the peripheral leptin resistance observed by Dawson et al. [191]. Hyperleptinemia observed in MSG treated rats was sustained for at least 150 days of age [192] indicating that the administration of leptin to MSG treated rats will not have an effect on the HPT axis because the animals are already leptin resistant. ICV administration of α-MSH can fully restore the fasting-induced decrease of preproTRH mRNA in the PVN of starved rats, however, it only partially restores the fasting-induced decrease of thyroid hormones [54]. Similarly, CART ICV administration completely restores the fasting-induced decrease of proTRH mRNA in the PVN in fasted rats; however, it does not restore the fasting-induced decrease of thyroid hormones [179]. Conversely, AgRP and NPY ICV administered in fed rats induce a state of central hypothyroidism similar to the fasting-induced state [60; 185]. Therefore, these ARC-derived peptides play a significant role in the indirect regulation of TRH neurons by leptin in vivo [51; 57; 187], and the P-CREB signaling is critical during the indirect leptin actions on proTRH neurons. α-MSH binds to the Gs-protein coupled melanocortin 4 receptor (MC4R), which signals by increasing P-CREB levels subsequently activating different genes, including preproTRH [16; 193]. AgRP acts as a competitive antagonist or inverse agonist on the melanocortin receptors [194; 195], and blocks α-MSH-induced effects on TRH release [55]. NPY action on TRH neurons is mediated by Y1 and Y5 receptors [61], which couple to Gi-protein and, when activated, decrease P-CREB levels [196]. Anatomical analysis shows that 48% of leptin-responsive TRH neurons are concentrated in the caudal region of the medial and periventricular parvocellular subnucleus of the PVN [178]. In another study using double in situ hybridization for TRH mRNA and MC4-R mRNA it was reported that approximately 50 to 60% of the TRH neurons located at the medial parvocelluar division of the PVN coexpress MC4R mRNA [197]. On the other hand, α-MSH activates the proTRH gene, at least in part, by increasing the phosphorylation of CREB in the nucleus of these neurons [16]. Therefore, observing the distribution of P-STAT3 and P-CREB expression in different subsets of hypophysiotrophic TRH neurons carrying ObRb and/or MC4 receptors in the PVN has been of key importance to identify the anatomical locations of proTRH neurons stimulated by both pathways [18] (Figs. 2 and 3).
8. Regulation of hypophysiotropic TRH neurons by NPY
NPY negatively regulates TRH production in the PVN, and acts by binding to NPY receptors on TRH neurons [60; 179; 186]. It was early suggested that NPY, in parallel with triggering feeding behavior, interrupts normal thyroid feedback during food deprivation. An early study showed that NPY and catecholaminergic inputs on TRH neurons are of mixed origin, and those cell bodies and proximal dendrites of TRH neurons receive a robust, putative inhibitory NPY input from the hypothalamus [198]. NPY action on TRH neurons leads to activation of an inhibitory G-protein that blocks phosphorylation of the CREB transcription factor downstream of the MC4R receptor. NPY neurons in the ARC also inhibit POMC neurons (Fig. 5) in the ARC by release of NPY and GABA [199]. α-MSH production is inhibited by both activation of the NPY1 receptor and GABA induced reduction of action potentials [199]. There are at least 6 NPY receptors subtypes, but Y1 and Y5 have received specific attention because of their ability to increase food intake when specifically stimulated [132]. These two receptors are present in cell bodies in overlapping regions of the rat PVN and ARC [200]. It is unknown the degree that Y1 and Y5 are present within the same neuron in the PVN or ARC, but in other regions these receptors have been seen individually in separate neurons or colocalized [200]. Y1 has been shown to colocalize with many but not all TRH neurons in the PVN [201]. The NPY innervation of TRH neurons has been seen throughout all subdivisions of the PVN [186; 202]. There is a great deal of evidence that these two receptors mediate different effects of NPY on the HPT axis, suggesting that they may be located on physiologically different subsets of neurons in the PVN and ARC. Y1 specific agonists stimulate the appetite phase of food intake, while Y5 specific agonists stimulate the meal size and duration during food intake [203]. Y1 but not Y5 antagonists block the effects of NPY on food intake in rats. Fasting-induced food intake was reduced in Y1 but not Y5 knockout mice [204]. We already know that there are at least two subsets of TRH neurons in the PVN, one directly responsive to leptin (P-STAT3) and located in the medial and posterior part of the PVN, and a second group indirectly responsive to leptin (P-CREB) located mainly in the anterior, medial and periventricular part of the PVN [18]. Therefore, it is tempting to speculate that NPY downregulation on TRH neurons will occur by two different pathways or mechanisms, similar to what it is was shown for leptin. The first one will involve NPY neurons sending axonal projections to TRH neurons where the NPY peptide may bind to either Y1 or Y5 receptor. In the second pathway NPY neurons will send its axonal projections to POMC neurons in the arcuate nucleus affecting the release of α-MSH that in turn will regulate the release of TRH through the MC4R. In a recent study conducted in our laboratory, we showed that the NPY1R colocalizes with TRH neurons mostly located in the anterior part of the PVN. These results are consistent with a pCREB activation seen previously in the TRH neurons located in the anterior part of the PVN [18]. In addition, with a combination of NPY plus leptin and NPY plus α-MSH ICV treatments in fasted animals we were able to show the possibility of two mechanisms (Fig. 5) by which NPY could regulate TRH neurons (unpublished results).
9. The TRH neuron, thyroid hormones and nutrition
The physiological role of the thyroid goes beyond maintaining the basic metabolic rate in all cells. A common situation in which thyroid hormone levels are subject to major physiological changes is during the transition from the fed to the starved state. Starvation is a severe threat to survival, and in rodents, the capacity to survive without food can be measured in a matter of days. Given that thyroid hormone has the ability to set the basal metabolic rate, a suppression of thyroid hormone during starvation reduces metabolic rate and preserves energy stores. Since animals in the wild commonly experience periods of starvation, the thyroid response to starvation should be considered as a major evolutionary mechanism designed to ensure survival when food is scarce. Through evolution, primitive humans faced long periods of food scarcity and starvation, and those who survived these conditions adapted their evolutionary traits to provide a greater capacity for conserving energy in fat stores until food became available again. However, these properties that in the past constituted an evolutionary advantage, today make humans more susceptible to become obese during times of large quantities of available food. Undeniably, this prosperous genotype in modern and industrialized societies, characterized by food abundance and reduced physical activity, culminated in an obesity of gigantic proportions, mostly in developed countries [205; 206; 207]. How does this nutritional adaptation take place? The thyroid system has several levels of regulation beginning at the level of the hypothalamus. There is a tightly regulated relationship to maintain homeostatic concentrations of thyroid hormones, by activating the feedback mechanisms on the biosynthesis of proTRH in the PVN followed by the pulsatile secretion of TRH from the ME [26; 162]. However, during starvation this dynamic is completely altered. Starvation suppresses hypophysiotropic preproTRH gene expression, while other TRH producing neurons inside or outside the hypothalamus are not affected [135]. Such a decrease in circulating thyroid hormone levels is associated with a reduction in the biosynthesis of TRH and the secretion of TSH [135; 208; 209]. By creating this state of central hypothyroidism, the resulting reduction in thyroid stimulating thermogenesis may serve as an important energy conservation mechanism until re-feeding occurs. How and where does the signal to the brain coordinate this adaptation? It was suggested that reduced TRH secretion might be due to the fasting-induced increase of corticosterone [210; 211]. The reason for this alteration is that TRH neurons are strongly affected by the nutritional status of the animal, which is similar in humans. This discovery adds a new level of understanding of the evolutionary role of the HPT axis, which involves an environmental adaptation to preserve energy during periods of malnourishment. We now know that the response to starvation not only causes changes in carbohydrate and fat metabolism, but also affects several neuroendocrine axes [26; 166].
How do thyroid hormones produced in the thyroid gland regulate these processes throughout the body [77; 212: Nillni, 1999 #4678] to maintain homeostasis at a constant rate? It does it largely by increasing basal metabolic rate in cells through the action of T3 on the uncoupling proteins (UCP-1, UCP-2 and UCP-3) [213] [9] [9; 10; 11; 214]. Performing work or generating heat in different ways can be the source for spending energy in the form of thermogenesis. Adaptive thermogenesis or regulated production of heat is affected by environmental temperature and diet. The mitochondria organelles that convert food to carbon dioxide, water and ATP, are fundamental in mediating effects on energy dissipation. Thyroid hormone-dependent energy expenditure is related to adaptive or facultative thermogenesis, which is characterized by an uncoupling of oxidative phosphorylation in cold-exposed brown adipose tissue (BAT) dependent on locally generated thyroid hormone. For example, shivering reaction during cold exposure uses energy and this is an example of adaptive thermogenesis. In small mammals, sympathetic adrenergic stimulation of BAT induces uncoupling protein-1 (UCP- 1) that uncouples the mitochondrial proton gradient from ATP production promoting the generation of heat. A major contributor in this reaction is D2, which increases local, intracellular T3 production from T4 [215] (Fig.4), while serum T3 levels do not appear to be affected in this condition [215]. The important central role of D2 in the generation of T3 in cold-induced BAT thermogenesis was demonstrated by a blocked in non-shivering thermogenesis in response to cold exposure [216] giving an important role to the interaction between the adrenergic sympathetic innervation cascade and thyroid hormone action [10; 11]. In humans, skeletal muscle plays the thermogenic role accomplished by brown adipose tissue in small mammals, a tissue that contains sufficient mass, innervation, and metabolic activity. Therefore, a prominent portion of the basal metabolic rate activity among normal humans could be assigned to this tissue alone [217]. The family of deiodinases converts less active T4 to the more active T3 or inactive reverse T3 in various tissue sites. It has been recently hypothesized that leptin and/or glucocorticoids play a role in these mechanisms and their interactions with D2 may be an important regulator of the HPT axis [218]. Since the hypothalamic TRH expression is regulated by T3 via negative feedback inhibition the availability of T3 converted from T4 is of key importance in this regulation. Several deiodinases are involved in the production of T3 in a tissue-specific manner. D2 is expressed in the central nervous system, the anterior pituitary, and brown adipose tissue (Fig. 4). Indeed, the thyroid regulation of TRH also involves a monosynaptic pathway between the ARC that contains the highest enzyme activity of D2, and those TRH neurons of the PVN that are projected to the external layer of the ME. This unique signaling modality plays an important role in thyroid feedback on TRH cells [219].
Hypothyroid and fasted rats show elevated hypothalamic D2 mRNA and D2 activity, and while T4 administration restores D2 mRNA expression in hypothyroid rats, it does not restore the fasting-induced increase of D2 expression, suggesting that the elevation of D2 production and activity during fasting is not related to the thyroid [220]. In another set of studies, the same group showed that hypothalamic D2 activity was not affected by either leptin or corticosterone administration in the fed state, but it was restored by leptin in the fasting state. They concluded that a decrease in serum leptin seen during fasting plays an important role in the increased serum corticosterone concentrations to elevate D2 activity [218]. Inhibition of D2 activity by ICV infusion of the deiodinase inhibitor iopanoic prevented the fasting-induced hypothalamic TRH decrease consistent with the observation that increased hypothalamic D2 activity during starvation may cause a higher production of local T3 that in turn decreases the TRH expression in the PVN [221]. In a further study, the local increase of bioactive T3 in tanycytes fasted animals was associated with an activation of neuronal uncoupling protein 2 (UCP2), and mitochondrial proliferation in the orexigenic AgRP/NPY neurons [222]. There is also a possibility that leptin may regulate deiodinases in other tissues to alter the set point of the thyroid axis. Supporting this hypothesis, a previous study showed that central administration of leptin affected D2 activity in BAT through a possible activation of the sympathetic nervous system [223]. This reaction resulted in a significant increase in plasma T3 levels, a situation that favors the activity of hepatic D1, given the high sensitivity of this enzyme to T3 [224]. However, when these studies were performed there was no information about the role of leptin regulating the TRH neuron. Therefore, it remains to be confirmed what contribution each of these leptin targets have in controlling the synthesis of T3 during changes in nutritional status. When leptin levels are high, does T3 increase because of a leptin action on TRH neurons stimulating the HPT axis that, in turn, provides more T4 available for conversion by deiodinases to T3, or does leptin increase deiodinase activity in specific tissues to convert the existing T4 to T3, or indeed are both mechanisms involved? Aside from the role of thyroid hormone regulating TRH in the PVN at the level of gene expression and post-ranslational processing, new studies [225] showed that T4 also controls TRH degradation by regulating the enzyme pyroglutamyl peptidase II (PPII). The PPII enzyme is commonly expressed in tanycytes and glial cells lining the third ventricle in the hypothalamus. The cytoplasmic processes of these cells reach the median eminence suggesting that PPII regulates the output of TRH peptide from the ME before its reaches the pituitary.
Long-term hypocaloric dietary regimens resulted in alterations similar to those observed during fasting. Although serum T3 and rT3 tended to return to basal concentrations during the last period of hypocaloric regime, TSH release upon TRH infusion remained low during the whole period, indicating that central and peripheral thyroid hormone metabolisms are affected via separate process [226]. In an effort to understand whether the decreased in hypothalamic TRH secretion during fasting is responsible for the decrease in TSH, the effect of 48-hour TRH infusion during a 6-day fast was investigated. On the second day of fasting, serum TSH decreased, while a rise in FT4 concentrations was observed suggesting that the transient suppression of serum TSH was not TRH mediated but due to the elevation of FT4 [227]. Another study determined the effect of fasting on circadian and pulsatile TSH secretion in eight healthy humans by measuring serum TSH every 10 minutes during the last 24 hours of a 60-hour fast. The decreased nocturnal TSH surge during fasting was associated with a significantly decreased TSH pulse amplitude, but with an unaltered number of TSH pulses between 2000-0400 h. These results seem to indicate that fasting decreases 24-h TSH secretion and the nocturnal TSH surge in the absence of a change in plasma T4 concentration [228]. The discovery of leptin as an important regulator of the thyroid axis provided an additional insight about the multiple mechanisms regulating the thyroid axis [135; 156; 166; 229]. In humans, Chan et al. studied the effects of recombinant human leptin on 72-hour fasting in healthy men. Fasting decreased serum T3, TSH and increased serum rT3. The administration of recombinant human leptin, prevented the fall in TSH secretion, suggesting that leptin regulates the alterations in TSH levels and TSH pulsatility during fasting [230]. In a follow up study, the effects of a 72-hour fast in healthy women resulted in a similar relative decrease of serum leptin, but exogenous leptin did not affect TSH pulsatility [231], although leptin replacement during the 4-day fast did result in higher 24-hour mean TSH concentrations compared to placebo [232]. In another study, healthy subjects on restricted caloric intake who lost 10% of body weight showed a restoration of serum T3 and T4 after leptin treatment suggesting that differences in the experimental protocol of these various studies, acute fasting during a short period versus a longer-term hypocaloric diet may account for the different outcomes [233].
The thyroid axis responds at multiple levels to illness rendering inappropriate secretion of TSH, which is seen in non-thyroidal illness. Non-thyroidal illness is generally associated with anorexia, suggesting that the leptin and melanocortin signaling pathways may play a key role in this process. A previous study suggested that central hypothyroidism associated with infection might be due to up-regulation of D2 activity in tanycytes present in the walls and floor of the third ventricle. These cells could lead to inhibition of TRH gene expression by increasing local T3 production [234].
In the human hypothalamus (postmortem), the HPT axis response to illness produced extensive changes. For example, there was a decrease in serum concentrations of T3 and TSH during illness, which were paralleled by decreased preproTRH mRNA. These results suggest that the T4 is taken up in glial cells that convert T4 into the biologically active T3 via the enzyme D2, which in turn binds to THRs and/or may be degraded into inactive iodothyronines by D3 [235]. Cancer anorexia-cachexia syndrome observed in 80% of patients in the advanced stages of cancer alters a multifactorial process involving many mediators, including hormones like leptin, neuropeptides such as α-MSH, and cytokines. It is likely that close interrelation among these mediators exists in the hypothalamus, decreasing food intake and leading to cachexia by the robust increase of α-MSH that in turn stimulate the TRH neuron and activete the thyroid axis and increase in energy expenditure [236].
Initial observations showed that leptin can prevent the fall of T4 and T3 in starved rodents, and the expression of preproTRH mRNA in the PVN [156; 166]. Consistent with this hypothesis, it was recently demonstrated that the biosynthesis of the TRH peptide in the PVN and its release from the ME is restored after the administration of leptin to fasted animals, independent of calorie intake [135]. After leptin treatment in fasted animals, the T4 values exceeded the normal values seen in fed control animals [135]. The TSH levels, consistent with previous studies [237], showed a small, but significant, decrease in male rats during fasting. It has been suggested previously that most of the TSH detected during fasting, in the absence of TRH, may not be functional. TRH plays a key role in the glycosylation of TSH in thyrotrophs by regulating the posttranslational maturation of TSH oligosaccharide chains to promote full biological activity, which means that such a reduction in TRH leads to TSH with less complex carbohydrate structures and reduced bioactivity that can be restored by TRH administration [237]. Humans with central hypothyroidism hypothalamic disease frequently have normal, or even slightly higher, serum TSH concentrations. These patients with hypothalamic diseases showed poor glycosylation of TSH, which is mannose-rich compared to control and primary hypothyroid patients. Again, consistent with this fall in pituitary and thyroid hormones, a reduction in preproTRH mRNA in the rat PVN was observed. The preproTRH mRNA expression was restored after leptin treatment. Similar studies done in humans were consistent with these findings. Rosenboun et al [233] found that in clinical studies maintenance of reduced body weight was associated with decreased 24-hour energy expenditure, a decrease in circulating plasma concentration of leptin, and thyroid hormones. All these endocrine changes were reversed by administration of replacement doses of leptin. The conclusion from studies done in animals and humans support the hypothesis that the neuroendocrine changes associated with constant reduced body weight are due, partially, to the lower circulating levels of leptin secondary to reduced fat mass. It is clear now that nutritional regulation of the thyroid axis through TRH in rodents and humans is important to both food deprivation and to illness. Humans with mutations of the ObRb have central hypothyroidism [174]. Also, controlled caloric restriction in humans, which leads to weight loss, results in a decline in serum T3 levels, which can be reversed by leptin [233]. On the other hand, acute fasting in humans leads to a loss in the normal pulsatile release of TSH, a situation that can be reversed by leptin replacement [228; 230]. Humans with MC4-R mutations have an obese phenotype, however, they have a normal thyroid axis [184; 238; 239; 240]. Some patients with central hypothyroidism of hypothalamic origin are associated with the secretion of TSH molecules, but decreased bioactivity because of changes in the oligosaccharide chains [241].
10. Obesity and the thyroid axis
Obesity and its related medical complications including type 2 diabetes, cardiovascular disease, dyslipidemia, and cancer account for more than 300,000 deaths per year in the United States. Obesity treatment strategies often do not result in adequate sustained weight loss, and the prevalence and severity of obesity in the U.S. and many developed countries is progressively increasing. Recent surveys classify roughly one third of all Americans as obese [242]. The complexity of the obesity condition results from the interaction between environmental and predisposed genetic factors [243]. A more thorough understanding of the molecular mechanisms underlying the pathogenesis of obesity and regulation of energy metabolism is essential for the development of effective therapies. Obesity occurs as a result of a longstanding imbalance between energy intake and energy expenditure, which is influenced by a very complex set of biological pathway systems regulating appetite [168; 212; 244; 245]. A better understanding of the causes of obesity lies in uncovering the molecular and physiologic mechanisms that regulate appetite, satiety, and energy balance [166; 167; 168; 170; 171; 172; 246] and will further improve the possibility to develop new anti-obesity drugs.
The interaction between the thyroid axis and development of obesity in humans is characterized by a complex relationship between leptin and hypothalamic TRH with important connotations in the thyroid status [247]. Different from insulin or cortisol, which fluctuate in response to nutritional changes and stress, thyroid hormones are typically maintained at a constant level, keeping the metabolic machinery running at a proper metabolic rate [212: Nillni, 1999 #4678]. Thyroid hormones are crucial for the survival of both rodents and humans by adjusting its levels from fed to starved state where in the case of starvation a rapid suppression of T4 and T3 levels occurs to preserve energy stores. The secretion of T3 and T4 is controlled by a feedback system involving the pituitary gland and hypothalamus that produces hormones that regulate thirst, hunger, body temperature, sleeps, moods and sex drive. When plasma levels of thyroid hormone fall, the biosynthesis and secretion of hypophysiotropic TRH increase, raising the threshold for feedback inhibition by thyroid hormone on anterior pituitary thyrotrophs, and thus increasing TSH secretion. Conversely, elevations in plasma concentrations of thyroid hormone suppress the biosynthesis and secretion of TRH, causing a reduced threshold for feedback regulation by thyroid hormone on thyrotrophs resulting in suppression of TSH secretion, thus leading to a reduced release of thyroid hormones [19]. Changes of thyroid function are associated with changes in both body weight and energy expenditure, but despite intensive research in this area, results are still not very clear on the role of leptin in thyroid pathophysiology [229]. Patients with thyroid disease usually have secondary disturbances of body weight, food intake and thermogenesis [248]. In healthy adults, plasma TSH and leptin concentrations are synchronized, suggesting that leptin regulates the HPT axis in humans [249; 250]. Patients with leptin receptor mutations or leptin deficiency show features of hypothalamic hypothyroidism [174; 176], and leptin treatment increases plasma thyroid hormones in leptin deficient humans [129]. Also, leptin administration reverses the low thyroid hormones in subjects on hypocaloric diets [233]. Based on these observations, one would expect to find high activity of the HPT axis in hyperleptinemic obese subjects. Although this has not been consistently observed, several studies reported that obese individuals have a higher activity of thyroid axis within the normal range [251; 252; 253; 254; 255]. Patients with morbid obesity have higher levels of thyroid hormones compared to normal weight subjects [256]. Therefore, it is possible that this model of leptin-induced resetting of the hypothalamic thyrostat may explain the alteration of the thyroid axis found in, at least, a subpopulation of obese individuals. Intriguingly, most obese humans and rodents have high levels of plasma leptin, which neither reduces appetite nor increases energy expenditure [257; 258]. This condition of hyperleptinemia is known as leptin resistance. Many aspects of this “leptin resistant” state are still poorly understood.
While the neuronal circuits controlling food intake and energy expenditure in lean and obese animals have been extensively investigated [259; 260], the neuronal circuits controlling the HPT axis in obese animals are poorly understood. In rodents, the ARC neurons were defined as the major site of the hypothalamic “leptin resistance” induced by high fat diet (HFD) [261; 262] affecting the melanocortin system, which is essential to activate the HPT axis [18]. Even with high leptin levels, most obese humans and rodents lack responsiveness to its appetite-suppressing effects. Our laboratory in collaboration with Michael Cowley's laboratory has shown that leptin modulates NPY/AgRP and α-MSH secretion from the ARC of lean mice, and diet-induced obesity (DIO) mice have normal ObRb levels and increased SOCS-3 levels, but leptin fails to increase α-MSH secretion. We also showed that by decreasing the fat content of the mouse's diet, leptin responsiveness of NPY/AgRP and POMC neurons recovered simultaneously, with mice regaining normal leptin sensitivity and glycemic control. These observations underscore the physiological significance of leptin sensing in the melanocortin circuits and show that loss of leptin sensitivity contributes to the pathology of leptin resistance. Therefore, it appears that leptin resistant is only a temporary phenomena. So even when overweight humans are likely to be leptin resistant, this condition can improve by losing weight. This circumstance is quite different to type 2 diabetes and insulin resistance, which is very hard to reverse, while leptin resistance is fairly correctable with a normal, healthy diet and exercise.
Previous studies have shown that pretreatment of fasted rats with a melanocortin antagonist fully blocks the leptin-induced effect on the HPT axis [18] demonstrating that this neuronal pathway is heavily implicated in the activation of the HPT axis. Since “leptin resistance” does not appear to affect other hypothalamic nuclei [261], we recently hypothesized that TRH neurons in the PVN of obese animals could remain sensitive to the direct action of leptin. Experiments conducted in vivo demonstrated that hyperleptinemic DIO rats have higher plasma thyroid hormones levels than controls, as well as increased preproTRH mRNA, TRH peptide, and STAT3 signaling increased in TRH neurons (unpublished results). This intriguing observation suggests that the direct pathway of leptin action on TRH neurons could play a significant role during the stage of leptin resistance to maintain the overall homeostasis. Confirming these initial observations, DIO rats, similarly to lean animals, down-regulated the HPT axis during fasting. In addition, TRH synthesis of MC4KO mice during fasting or administration of leptin is similar to wild type animals. In the absence of a functional melanocortin system, which is the case of the DIO rat, the TRH synthesis in the MC4KO mice during fasting or administration of leptin was similar to wild type animals [130]. To further test the hypothesis that the higher plasma T3 levels seen in DIO rats occur because of direct leptin action, fed Zucker rats (Zucker rats bear a mutation in the leptin receptor gene and lacks leptin response [263; 264]) with regular chow or HFD were unable to increase the levels of T3 seen in regular DIO rats. This finding confirms the notion that higher levels of plasma T3 seen in DIO rats depends, at least in part, on leptin signaling (unpublished results). Consequently, these preliminary studies could provide the first demonstration that the TRH neurons directly regulated by leptin play a significant role in the DIO condition by allowing the HPT axis to continue to be functional when leptin resistance is established at the level of the ARC. Although these preliminary data are compelling, these neurons also contain classical transmitters like GABA, glutamate and acetyl-choline that could influence TRH neurons even when the melanocortin receptors are not functional. Further studies are needed to demonstrate this possibility.
Altogether, these results suggest that leptin can affect hypothalamic functions of DIO rats in the presence of “resistance” to its anorexic activity. According to this idea, Halaas et al. showed that the leptin-mediated anorexic response attenuated earlier than the leptin-mediated increase of energy expenditure after central infusion of leptin in lean mice [265]. Scarpace et al. also showed that resistance to leptin-induced anorexogenic actions develops earlier than the resistance to thermogenic effects using rats centrally treated with virus encoding leptin cDNA [266]. Thus, it seems that the direct action of leptin on TRH neurons is one of the potential mechanisms to increase energy expenditure independently of the anorectic action coming from the ARC. As described earlier in this review, thyroid hormones could also increase energy expenditure in DIO rats either directly, via up regulation of UCPs and basal body temperature, or indirectly via an increase in the sensitivity to the sympathetic activity [214; 267]. However, it is important to mention that changes in the activity of the autonomic system, independent of the HPT axis and leptin, may also contribute to increase the energy expenditure of DIO rats [268]. Direct leptin action on TRH neurons may explain other side effects of obesity such as hypertension. It has been shown that diencephalic TRH mediates the central leptin-induced increase of blood pressure in rats [269]. Moreover, Landa et al. recently reported that obesity-induced hypertension decreases when diencephalic TRH is knocked down [270]. These findings suggest that the selective leptin action on TRH neurons could contribute to the overall phenotype of obese individuals. In conclusion, the possibility of the existence of a group of hypothalamic TRH neurons sensitive to the leptin-induced increase of pSTAT3 signaling in obese animals is a novel aspect in the interaction between the HPT axis and leptin that may have important implications in the future development of anti-obesity drugs. A recent study examined the effect of thyrotoxicosis and hypothyroidism on whole body energy metabolism and spontaneous physical activity in rats. Thyrotoxicosis showed to induce hyper-metabolism, increase food intake, and favoring lipid replenishment by increasing triglyceride derived fatty acid uptake in oxidative tissues. This finding suggests that thyroid hormone might regulate fatty acid uptake from triglycerides rich lipoproteins in a tissue specific-manner [271].
11. Summary of regulatory check points on the TRH neuron biology
This current review is intended to describe the latest knowledge on the TRH neuron research by providing new insights into the cellular and molecular mechanisms controlling preproTRH gene expression, proTRH biosynthesis, the molecular determinants involved in the proper folding of the polypeptide, post-translational processing, and the differential sorting of the processing products to mature secretory granules (Fig.1). Since the discovery of the PCs in the late 1980s a new frontier on the propeptide hormone biosynthesis and processing research has come to fruition. It is particularly relevant to emphasize the concept that secretion of any particular peptide or neuropeptide hormone depends on a series of post-translational modification carried out by a group of different processing enzymes located in a unique environment within the secretory system. These enzymes, as we recently learned, are also regulated by important hormones involved thyroid physiology such as leptin and T3 itself (Figs. 4 and 6). The hormone leptin, like NE and T3, regulates the biosynthesis, processing and secretion of proTRH while simultaneously regulated PC1/3 and 2, which are responsible for the maturation proTRH. These sequences of events further support the existence of a coupled regulation of proneuropeptide/processing enzymes as a common process by which cells generate more effective processing of prohormones into mature peptides ready for release. It is also described to some extend the biological action of TRH in the thyroid axis during metabolic perturbations including cold stress, and the thyroid response in starvation and obesity. The recent view on this endocrine axis is that in order to maintain the homeostasis of the whole body metabolism and temperature it requires the participation of multiple factors coming from different brain nuclei, including the hypothalamus, and hormones from the periphery acting in a very coordinated and tightly regulated fashion. Although there are other factors affecting the regulation of the hypophysiotropic TRH neurons, emphasis was placed in the review on those related to nutritional control of the HPT axis and cold stress (Fig. 4). The review describes the way Leptin activates different subgroups of TRH neurons directly via the leptin receptor, inducing activation of the TRH promoter, and indirectly via effects on the stimulation of α-MSH release from POMC neurons and acting through neuronal synapsis on TRH neurons, while inhibiting AgRP/NPY neurons. The review also proposes a new modality for the inhibitory action of NPY on TRH neurons (Fig. 5). At the hypothalamic level, evidence shows that fasting causes the increase of D2 in tanycytes promoting a conversion of T4 to T3 (Fig. 4) representing another point of regulation of the TRH expression in the PVN by acting on THRb2 [272]. While the preproTRH expression remains higher in THRb2-null mice compared to wild type, leptin restores preproTRH expression in TRb2-null and wild-type mice as well, suggesting that responsiveness of TRH neurons to leptin might be independent of the THRb2 [273]. The information on human preproTRH expression is limited however, a recent study had showed that the participating proteins in the regulation of hypothalamic TRH during fasting in rodents are also expressed in the human hypothalamus [274]. D2 is expressed in glial cells of the infundibular nucleus, which is the human homologue of the rat ARC [274]. The recently identified thyroid hormone transporter MCT8 is expressed in neurons of the infundibular nucleus and PVN and plays a role in the access of T3 to the preproTRH gene in the nucleus (Fig. 6) [275; 276]. T3 also plays a down regulatory role on the PCs and PAM to prevent further processing of proTRH. The physiological role of MCT8 was confirmed in mutations found in humans and mouse genetic models showing impairment in the regulation of preproTRH mRNA expression by thyroid hormone [235; 277; 278; 279]. In summary, the hypophysiotropic TRH neurons in the PVN are sensitive to T3 levels and other modulators of energy balance including leptin, α-MSH, AgRP, and NPY. The TRH neuron integrates these inputs to determine the set point of preproTRH expression and biosynthesis of TRH as seen during fasting. These mechanisms of gene regulation are accompanied by a tight regulation of proTRH processing by PC1/3, PC2, CPE/D and PAM. Based on the new knowledge on the TRH neuron biology, it has been recently suggested to operate as a metabolic sensor since it is regulated at multiple levels to control thyroid function and metabolism [280].
Fig. 6. Integrated regulatory mechanisms controlling TRH transcription, posttranslational processing, and degradation by leptin and thyroid hormone.
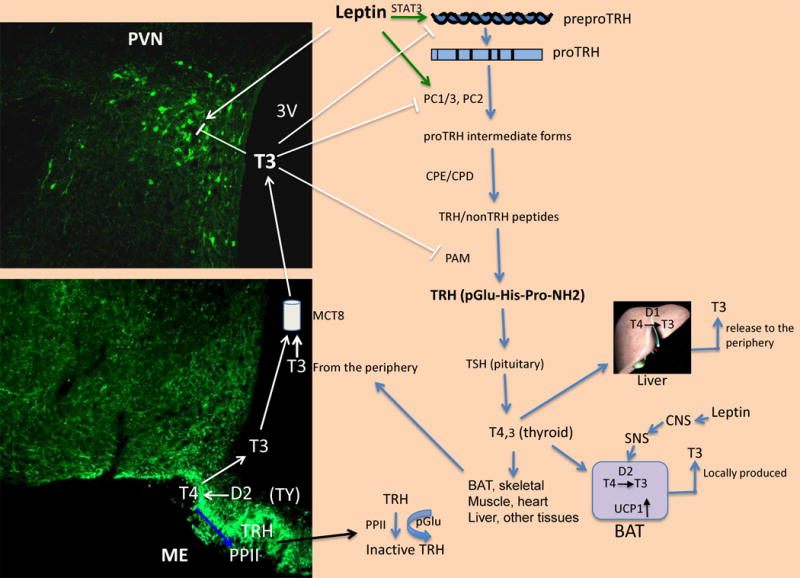
This illustration shows the multiple targets that leptin and T3 have in regulating the output of TRH. Tanycytes incorporate T4 and converted it to T3 by D2, and together with T3 coming from the periphery reach TRH neurons via specific transporters such as the monocarboxylate transporter (MCT8) inhibiting gene expression by affecting the TRH promoter through binding to the THRb2 isoform leading to the recruitment of cofactors such as SRC-1. PreproTRH, PC1/3, PC2, and PAM are inversely regulated by T3. Post-translational processing of proTRH occurs during the transport of the prohormone in the axons reaching the median eminence. The amount of TRH released from the axon terminals in the median eminence to the defenestrated capillaries can be controled by the degrading tanycyte-bound enzyme PPII, which is positively regulated by thyroid hormone. The potential effect of leptin via an activation of the sympathetic nervous system has been suggested, and in turn, increases the expression of BAT-UCP1 mRNA by the conversion of T4 to T3 facilitated by D2 increased activity. The staining in the PVN and ME was performed using an antibody against the proTRH sequence.
Acknowledgments
This work was supported by NIDDK/NIH R01 DK58148
Abbreviations
- TRH
Thyrotropin Releasing Hormone immunoreactive
- PTU
propylthiouracil
- HPT
hypothalamic-pituitary-thyroid
- DVC
dorsal vagal complex
- DMN
dorsal motor nucleus of the vagus
- NTS
nucleus tractus solitaris
- NPY
neuropeptide Y
- NE
norepinephrine
- GABA
g-aminobutyric acid
- CPE/D
carboxypeptidase E/D
- CSF
cerebrospinal fluid
- PVN
periventricular nucleus of the hypothalamus
- ICV
intracerebroventricular
- SC
subcutaneous, IC: intracerebral
- CNS
central nervous system
- ME
median eminence
- RER
rough endoplasmic reticulum
- PCs
proconverting enzymes
- RSP
regulated secretory pathway
- GC
Golgi complex
- SG
secretory granules
- ISG
immature secretory granules
- TGN
trans-Golgi network
- ICC
immunocytochemistry
- TRE
thyroid response element
- CRE
cAMP response element
- PAM
peptidylglycine α-amidating monooxygenase enzyme
- TSH
thyroid stimulating hormone
- T4
thyroxine
- T3
triidothyronine
- UCPs
uncoupling proteins
- BAT
brown adipose tissue
- α-MSH
melanocortin stimulating hormone
- AgRP
agouti-related peptide
- ARC
arcuate nucleus of the hypothalamus
- VMH
ventromedial hypothalamus
- LH
lateral hypothalamus
- ObRb
leptin receptor
- MC3-R and MC4-R
melanocortin-3 and 4 receptors
- STAT
signal transducer of activated transcription
- JAK
Janus tyrosine kinases
- 40PGL42
Pro-Gly-Leu
- ICC
immunocytochemistry
Footnotes
Disclosure statement: The author has nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nillni EA, Friedman TC, Todd RB, Birch NP, Loh YP, Jackson IM. Pro-thyrotropin-releasing hormone processing by recombinant PC1. J Neurochem. 1995;65:2462–72. doi: 10.1046/j.1471-4159.1995.65062462.x. [DOI] [PubMed] [Google Scholar]
- 2.Nillni EA, Luo LG, Jackson IM, McMillan P. Identification of the thyrotropin-releasing hormone precursor, its processing products, and its coexpression with convertase 1 in primary cultures of hypothalamic neurons: anatomic distribution of PC1 and PC2. Endocrinology. 1996;137:5651–61. doi: 10.1210/endo.137.12.8940396. [DOI] [PubMed] [Google Scholar]
- 3.Schaner P, Todd RB, Seidah NG, Nillni EA. Processing of prothyrotropin-releasing hormone by the family of prohormone convertases. J Biol Chem. 1997;272:19958–68. doi: 10.1074/jbc.272.32.19958. [DOI] [PubMed] [Google Scholar]
- 4.Nillni EA, Xie W, Mulcahy L, Sanchez VC, Wetsel WC. Deficiencies in pro-thyrotropin-releasing hormone processing and abnormalities in thermoregulation in Cpefat/fat mice. J Biol Chem. 2002;277:48587–95. doi: 10.1074/jbc.M206702200. [DOI] [PubMed] [Google Scholar]
- 5.Eipper BA, Stoffers DA, Mains RE. The biosynthesis of neuropeptides: peptide α-amidation. Annu Rev Neurosci. 1992;15:57–85. doi: 10.1146/annurev.ne.15.030192.000421. [DOI] [PubMed] [Google Scholar]
- 6.Perello M, Nillni EA. The biosynthesis and processing of neuropeptides: lessons from prothyrotropin releasing hormone (proTRH) Front Biosci. 2007;12:3554–65. doi: 10.2741/2334. [DOI] [PubMed] [Google Scholar]
- 7.Hall R, Amos J, Garry R, Buxton RL. Thyroid-stimulating hormone response to synthetic thyrotropin releasing hormone in man. Br Med J. 1970;2:274–277. doi: 10.1136/bmj.2.5704.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris AR, Christianson D, Smith M, Fang SL, Braverman S, Vagenakis A. The physiological role of thyrotropin-releasing hormone in the regulation of thyroid-stimulating hormone and prolactin secretion in the rat. J Clin Invest. 1978;61:441–448. doi: 10.1172/JCI108955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianco AC, Kim BW. Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest. 2006;116:2571–9. doi: 10.1172/JCI29812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva JE. Thyroid hormone and the energetic cost of keeping body temperature. Biosci Rep. 2005;25:129–48. doi: 10.1007/s10540-005-2882-9. [DOI] [PubMed] [Google Scholar]
- 11.Silva JE. Thermogenic mechanisms and their hormonal regulation. Physiol Rev. 2006;86:435–64. doi: 10.1152/physrev.00009.2005. [DOI] [PubMed] [Google Scholar]
- 12.Morley JE. Extrahypothalamic thyrotropin-releasing hormone (TRH) -- its distribution and its functions. Life Sci. 1979;25:1539–1550. doi: 10.1016/0024-3205(79)90435-1. [DOI] [PubMed] [Google Scholar]
- 13.Yarbrough GG. On the neuropharmacology of thyrotropin-releasing hormone (TRH) Prog Neurobiol. 1979;12:291–312. doi: 10.1016/0301-0082(79)90012-1. [DOI] [PubMed] [Google Scholar]
- 14.Collu R, Tang J, Castagne J, Lagace G, Masson N, Huot C, Deal C, Delvin E, Faccenda E, Eidne KA, Van Vliet G. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab. 1997;82:1561–5. doi: 10.1210/jcem.82.5.3918. [DOI] [PubMed] [Google Scholar]
- 15.Yamada M, Saga Y, Shibusawa N, Hirato J, Murakami M, Iwasaki T, Hashimoto K, Satoh T, Wakabayashi K, Taketo MM, Mori M. Tertiary hypothyroidism and hyperglycemia in mice with targeted disruption of the thyrotropin-releasing hormone gene. Proc Natl Acad Sci U S A. 1997;94:10862–7. doi: 10.1073/pnas.94.20.10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris M, Aschkenasi C, Elias CF, Chandrankunnel A, Nillni EA, Bjoorbaek C, Elmquist JK, Flier JS, Hollenberg AN. Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J Clin Invest. 2001;107:111–20. doi: 10.1172/JCI10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nillni EA, Vaslet C, Harris M, Hollenberg A, Bjorbak C, Flier JS. Leptin regulates prothyrotropin-releasing hormone biosynthesis. Evidence for direct and indirect pathways. J Biol Chem. 2000;275:36124–33. doi: 10.1074/jbc.M003549200. [DOI] [PubMed] [Google Scholar]
- 18.Perello M, Stuart RC, Nillni EA. The role of intracerebroventricular administration of leptin in the stimulation of prothyrotropin releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2006;147:3296–306. doi: 10.1210/en.2005-1533. [DOI] [PubMed] [Google Scholar]
- 19.Toni R, Lechan RM. Neuroendocrine regulation of thyrotropin-releasing hormone (TRH) in the tuberoinfundibular system. J Endocrinol Invest. 1993;16:715–53. doi: 10.1007/BF03348918. [DOI] [PubMed] [Google Scholar]
- 20.Brownstein MJ, Eskay RL, Palkovits M. Thyrotropin releasing hormone in the median eminence is in processes of paraventricular nucleus neurons. Neuropeptides. 1982;2:197. [Google Scholar]
- 21.Fliers E, Guldenaar SE, Wiersinga WM, Swaab DF. Decreased hypothalamic thyrotropin-releasing hormone gene expression in patients with nonthyroidal illness. J Clin Endocrinol Metab. 1997;82:4032–4036. doi: 10.1210/jcem.82.12.4404. [DOI] [PubMed] [Google Scholar]
- 22.Boelen A, Wiersinga WM, Fliers E. Fasting-induced changes in the hypothalamus-pituitary-thyroid axis. Thyroid. 2008;18:123–9. doi: 10.1089/thy.2007.0253. [DOI] [PubMed] [Google Scholar]
- 23.Sawchenko PE, Swanson LW. The organization of noradrenergic pathways from the brainstem to the paraventricular and supraoptic nuclei in the rat. Brain Res Rev. 1982;4:275. doi: 10.1016/0165-0173(82)90010-8. [DOI] [PubMed] [Google Scholar]
- 24.Arancibia S, Rage F, Astier H, Tapia-Arancibia L. Neuroendocrine and autonomous mechanisms underlying thermoregulation in cold environment. Neuroendocrinol. 1996;64:257–267. doi: 10.1159/000127126. [DOI] [PubMed] [Google Scholar]
- 25.Lechan RM, Toni R. Thyrotropin-releasing hormone neuronal systems in the central nervous system. In: Nemeroff CB, editor. Neuroendocrinology. CRC; Boca Raton: 1992. pp. 279–330. [Google Scholar]
- 26.Nillni EA, Sevarino KA. The biology of pro-thyrotropin-releasing hormone-derived peptides. Endocr Rev. 1999;20:599–648. doi: 10.1210/edrv.20.5.0379. [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa K, Taniguchi Y, Inoue K, Kurosumi K, Suzuki M. Immunocytochemical delineation of thyrotropic area: origin of thyrotropin-releasing hormone in the median eminence. Neuroendocrinol. 1988;47:384. doi: 10.1159/000124943. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar S, Legradi G, Lechan RM. Intracerebroventricular administration of alpha-melanocyte stimulating hormone increases phosphorylation of CREB in TRH- and CRH-producing neurons of the hypothalamic paraventricular nucleus. Brain Res. 2002;945:50–9. doi: 10.1016/s0006-8993(02)02619-7. [DOI] [PubMed] [Google Scholar]
- 29.Roeling TA, Veening JG, Peters JP, Vermelis ME, Nieuwenhuys R. Efferent connections of the hypothalamic “grooming area” in the rat. Neuroscience. 1993;56:199–225. doi: 10.1016/0306-4522(93)90574-y. [DOI] [PubMed] [Google Scholar]
- 30.Swanson LW, Sawchenko PE, Wiegand SJ, Price JL. Separate neurons in the paraventricular nucleus project to the median eminence and to the medulla or spinal cord. Brain Res. 1980;198:190–5. doi: 10.1016/0006-8993(80)90354-6. [DOI] [PubMed] [Google Scholar]
- 31.Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology. 1980;31:410–7. doi: 10.1159/000123111. [DOI] [PubMed] [Google Scholar]
- 32.Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33:542–7. doi: 10.1161/01.hyp.33.1.542. [DOI] [PubMed] [Google Scholar]
- 33.Fliers E, Noppen NW, Wiersinga WM, Visser TJ, Swaab DF. Distribution of thyrotropin-releasing hormone (TRH)-containing cells and fibers in the human hypothalamus. J Comp Neurol. 1994;350:311–23. doi: 10.1002/cne.903500213. [DOI] [PubMed] [Google Scholar]
- 34.Guldenaar SE, Veldkamp B, Bakker O, Wiersinga WM, Swaab DF, Fliers E. Thyrotropin-releasing hormone gene expression in the human hypothalamus. Brain Res. 1996;743:93–101. doi: 10.1016/s0006-8993(96)01024-4. [DOI] [PubMed] [Google Scholar]
- 35.Mihaly E, Fekete C, Tatro JB, Liposits Z, Stopa EG, Lechan RM. Hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in the human hypothalamus are innervated by neuropeptide Y, agouti-related protein, and alpha-melanocyte-stimulating hormone. J Clin Endocrinol Metab. 2000;85:2596–603. doi: 10.1210/jcem.85.7.6662. [DOI] [PubMed] [Google Scholar]
- 36.Winokur A, Utiger R. Thyrotropin-releasing hormone: regional distribution in rat brain. Science. 1974;185:265–266. doi: 10.1126/science.185.4147.265. [DOI] [PubMed] [Google Scholar]
- 37.Browstein MJ, Palkovits M, Saavedra JM, Bassiri RM, Utiger RD. Thyrotropin-releasing hormone in specific nuclei of rat brain. Science. 1974;185:267–269. doi: 10.1126/science.185.4147.267. [DOI] [PubMed] [Google Scholar]
- 38.Shian LR, Wu MH, Lin MT, Ho LT. Hypothalamic involvement in the locomotor stimulant or satiety action of thyrotropin-releasing hormone and amphetamine. Pharmacology. 1985;30:259–265. doi: 10.1159/000138076. [DOI] [PubMed] [Google Scholar]
- 39.Kow LM, Pfaff DW. The effects of the TRH metabolite cyclo (His-Pro) and its analogs on feeding. Pharmacol Biochem Behav. 1991;38:359–364. doi: 10.1016/0091-3057(91)90291-9. [DOI] [PubMed] [Google Scholar]
- 40.Choi YH, Hartzell D, Azain MJ, Baile CA. TRH decreases food intake and increases water intake and body temperature in rats. Physiol Behavior. 2002;77:1–4. doi: 10.1016/s0031-9384(02)00784-9. [DOI] [PubMed] [Google Scholar]
- 41.Steward CA, Horan TL, Schuhler S, Bennett GW, Ebling FJ. Central administration of thyrotropin releasing hormone (TRH) and related peptides inhibits feeding behavior in the Siberian hamster. Neuroreport. 2003;14:687–691. doi: 10.1097/00001756-200304150-00006. [DOI] [PubMed] [Google Scholar]
- 42.Woods SC, Porte D., Jr The central nervous system, pancreatic hormones, feeding, and obesity. Adv Metab Disord. 1978;9:283–312. doi: 10.1016/b978-0-12-027309-6.50020-3. [DOI] [PubMed] [Google Scholar]
- 43.Vogel RA, Cooper BR, Barlow TS, Prange AJ, Jr, Mueller RA, Breese GR. Effects of thyrotropin-releasing hormone on locomotor activity, operant performance and ingestive behavior. J Pharmacol Exp Ther. 1979;208:161–168. [PubMed] [Google Scholar]
- 44.Heuer H, Schafer MK, O'Donnell D, Walker P, Bauer K. Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J Comp Neurology. 2000;428:319–336. [PubMed] [Google Scholar]
- 45.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez JA, Horjales-Araujo E, Fugger L, Broberger C, Burdakov D. Stimulation of orexin/hypocretin neurones by thyrotropin-releasing hormone. J Physiol. 2009;587:1179–86. doi: 10.1113/jphysiol.2008.167940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawchenko PE, Swanson LW. The organization of forebrian afferents to the paraventricular and supraoptic nuclei of the rat. J Comp Neurol. 1983;218:121–132. doi: 10.1002/cne.902180202. [DOI] [PubMed] [Google Scholar]
- 48.Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–47. [PubMed] [Google Scholar]
- 49.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–86. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–6. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinol. 1997;138:4489–4492. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 52.Kielar D, Clark JS, Ciechanowicz A, Kurzawski G, Sulikowski T, Naruszewicz M. Leptin receptor isoforms expressed in human adipose tissue. Metabolism. 1998;47:844–7. doi: 10.1016/s0026-0495(98)90124-x. [DOI] [PubMed] [Google Scholar]
- 53.Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–35. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- 54.Fekete C, Mihaly E, Luo LG, Kelly J, Clausen JT, Mao Q, Rand WM, Moss LG, Kuhar M, Emerson CH, Jackson IM, Lechan RM. Association of cocaine- and amphetamine-regulated transcript-immunoreactive elements with thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and its role in the regulation of the hypothalamic-pituitary-thyroid axis during fasting. J Neurosci. 2000;20:9224–34. doi: 10.1523/JNEUROSCI.20-24-09224.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim MS, Small CJ, Stanley SA, Morgan DG, Seal LJ, Kong WM, Edwards CM, Abusnana S, Sunter D, Ghatei MA, Bloom SR. The central melanocortin system affects the hypothalamo-pituitary thyroid axis and may mediate the effect of leptin. J Clin Invest. 2000;105:1005–1011. doi: 10.1172/JCI8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menyhert J, Wittmann G, Lechan RM, Keller E, Liposits Z, Fekete C. Cocaine- and amphetamine-regulated transcript (CART) is colocalized with the orexigenic neuropeptide Y and agouti-related protein and absent from the anorexigenic alpha-melanocyte-stimulating hormone neurons in the infundibular nucleus of the human hypothalamus. Endocrinology. 2007;148:4276–81. doi: 10.1210/en.2007-0390. [DOI] [PubMed] [Google Scholar]
- 57.Stephens TW, Basinski M, Bristow PK, Bue-Valleskey JM, Burgett SG, Craft L, Hale J, Hoffmann J, Hsiung HM, Kriauciunas A, et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377:530–2. doi: 10.1038/377530a0. [DOI] [PubMed] [Google Scholar]
- 58.Mizuno TM, Makimura H, Silverstein J, Roberts JL, Lopingco T, Mobbs CV. Fasting regulates hypothalamic neuropeptide Y, agouti-related peptide, and proopiomelanocortin in diabetic mice independent of changes in leptin or insulin. Endocrinology. 1999;140:4551–7. doi: 10.1210/endo.140.10.6966. [DOI] [PubMed] [Google Scholar]
- 59.Mizuno TM, Mobbs CV. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology. 1999;140:814–7. doi: 10.1210/endo.140.2.6491. [DOI] [PubMed] [Google Scholar]
- 60.Fekete C, Kelly J, Mihaly E, Sarkar S, Rand WM, Legradi G, Emerson CH, Lechan RM. Neuropeptide Y has a central inhibitory action on the hypothalamic-pituitary-thyroid axis. Endocrinology. 2001;142:2606–13. doi: 10.1210/endo.142.6.8207. [DOI] [PubMed] [Google Scholar]
- 61.Fekete C, Sarkar S, Rand WM, Harney JW, Emerson CH, Bianco AC, Beck-Sickinger A, Lechan RM. Neuropeptide Y1 and Y5 receptors mediate the effects of neuropeptide Y on the hypothalamic-pituitary-thyroid axis. Endocrinology. 2002;143:4513–9. doi: 10.1210/en.2002-220574. [DOI] [PubMed] [Google Scholar]
- 62.Sawchenko PE, Pfeiffer SW. Ultrastructural localization of neuropeptide Y and galanin immunoreactivity in the paraventricular nucleus of the hypothalamus in the rat. Brain Res. 1988;474:231. doi: 10.1016/0006-8993(88)90438-6. [DOI] [PubMed] [Google Scholar]
- 63.Everitt BJ, Hokfelt T, Terenius L, Tatemoto T, Mutt V, Goldstein M. Differential co-existence of neuropeptide Y (NPY)-like immunoreactivity with catecholamuines in the central nervous system of the rat. Neuroscience. 1984;11:443. doi: 10.1016/0306-4522(84)90036-8. [DOI] [PubMed] [Google Scholar]
- 64.Mihaly E, Fekete C, Legradi G, Lechan RM. Hypothalamic dorsomedial nucleus neurons innervate thyrotropin-releasing hormone-synthesizing neurons in the paraventricular nucleus. Brain Res. 2001;891:20–31. doi: 10.1016/s0006-8993(00)03094-8. [DOI] [PubMed] [Google Scholar]
- 65.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci U S A. 1998;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singru PS, Fekete C, Lechan RM. Neuroanatomical evidence for participation of the hypothalamic dorsomedial nucleus (DMN) in regulation of the hypothalamic paraventricular nucleus (PVN) by alpha-melanocyte stimulating hormone. Brain Res. 2005;1064:42–51. doi: 10.1016/j.brainres.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 67.Hoyda TD, Samson WK, Ferguson AV. Adiponectin depolarizes parvocellular paraventricular nucleus neurons controlling neuroendocrine and autonomic function. Endocrinology. 2009;150:832–40. doi: 10.1210/en.2008-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hollenberg AN, Monden T, Flynn TR, Boers ME, Chen O, Wondisford FE. The human thyrotropin releasing hormone gene is regulated by thyroid hormone response elements. Mol Endocrinol. 1995;9:540–550. doi: 10.1210/mend.9.5.7565802. [DOI] [PubMed] [Google Scholar]
- 69.Satoh T, Yamada M, Iwasaki T, Mori M. Negative regulation of the gene for the preprothyrotropin-releasing hormone from the mouse by thyroid hormone requires additional factors in conjunction with thyroid hormone receptors. J Biol Chem. 1996;271:27919–26. doi: 10.1074/jbc.271.44.27919. [DOI] [PubMed] [Google Scholar]
- 70.Diaz-Gallardo MY, Cote-Velez A, Carreon-Rodriguez A, Charli JL, Joseph-Bravo P. Phosphorylated Cyclic-AMP-Response Element-Binding Protein and Thyroid Hormone Receptor Have Independent Response Elements in the Rat Thyrotropin-Releasing Hormone Promoter: An Analysis in Hypothalamic Cells. Neuroendocrinology. 2009 doi: 10.1159/000228833. [DOI] [PubMed] [Google Scholar]
- 71.Guo F, Bakal K, Minokoshi Y, Hollenberg AN. Leptin signaling targets the thyrotropin-releasing hormone gene promoter in vivo. Endocrinology. 2004;145:2221–7. doi: 10.1210/en.2003-1312. [DOI] [PubMed] [Google Scholar]
- 72.Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci USA. 1996;93:6231–6235. doi: 10.1073/pnas.93.13.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, Lai CF, Tartaglia LA. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA. 1996;93:8374–8378. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bjørbæk C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 75.Ihle JN. Cytokine receptor signalling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 76.Vaisse C, Halaas JL, Horvath CM, Darnell JJE, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nature Genetics. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 77.Hollenberg AN, Forrest D. The thyroid and metabolism: the action continues. Cell Metab. 2008;8:10–2. doi: 10.1016/j.cmet.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 78.Lechan RM, Wu P, Jackson IMD, Wolfe H, Cooperman S, Mandel G, Goodman RH. Thyrotropin-releasing hormone precursor: characterization in rat brain. Science. 1986;231:159–161. doi: 10.1126/science.3079917. [DOI] [PubMed] [Google Scholar]
- 79.Lechan RM, Wu P, Jackson IMD. Immunolocalization of the thyrotropin-releasing hormone prohormone in the rat central nervous system. Endocrinol. 1986b;119:1210–1216. doi: 10.1210/endo-119-3-1210. [DOI] [PubMed] [Google Scholar]
- 80.Lee SL, Stewart K, Goodman RH. Structure of the gene encoding rat thyrotropin releasing hormone. J Biol Chem. 1988;263:16604–16609. [PubMed] [Google Scholar]
- 81.Masanobu Y, Radovick S, Wondisford FE, Nakayama Y, Weintraub BD, Wilber JF. Cloning and structure of human genomic and hypothalamic cDNA and encoding human preprothyrotropin-releasing hormone. Mol Endocrinol. 1990;4:551–556. doi: 10.1210/mend-4-4-551. [DOI] [PubMed] [Google Scholar]
- 82.Zhou A, Bloomquist BT, Mains RE. The prohormone convertases PC1 and PC2 mediate distinct endoproteolytic cleavages in a strict temporal order during proopiomelanocortin biosynthetic processing. J Biol Chem. 1993;268:1763–1769. [PubMed] [Google Scholar]
- 83.Scamuffa N, Calvo F, Chretien M, Seidah NG, Khatib AM. Proprotein convertases: lessons from knockouts. Faseb J. 2006;20:1954–63. doi: 10.1096/fj.05-5491rev. [DOI] [PubMed] [Google Scholar]
- 84.Romero A, Cakir I, Vaslet CA, Stuart RC, Lansari O, Lucero HA, Nillni EA. Role of a pro-sequence in the secretory pathway of prothyrotropin-releasing hormone. J Biol Chem. 2008;283:31438–48. doi: 10.1074/jbc.M803413200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fricker L. Carboxypeptidase E. Ann Rev Physiol. 1988;50:309–321. doi: 10.1146/annurev.ph.50.030188.001521. [DOI] [PubMed] [Google Scholar]
- 86.Varlamov O, Wu F, Shields D, Fricker LD. Biosynthesis and packaging of carboxypeptidase D into nascent secretory vesicles in pituitary cell lines. J Biol Chem. 1999;274:14040–5. doi: 10.1074/jbc.274.20.14040. [DOI] [PubMed] [Google Scholar]
- 87.Fricker LD. Neuropeptidomics to study peptide processing in animal models of obesity. Endocrinology. 2007;148:4185–90. doi: 10.1210/en.2007-0123. [DOI] [PubMed] [Google Scholar]
- 88.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: the structure of peptidylglycine alpha-hydroxylating monooxygenase. Science. 1997;278:1300–5. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 89.Perez de la Cruz I, Nillni EA. Intracellular sites of prothyrotropin-releasing hormone processing. J Biol Chem. 1996;271:22736–22745. doi: 10.1074/jbc.271.37.22736. [DOI] [PubMed] [Google Scholar]
- 90.Burgess TL, Kelly RB. Constitutive and regulated secretion of proteins. Ann Rev Cell Biol. 1987;3:243–293. doi: 10.1146/annurev.cb.03.110187.001331. [DOI] [PubMed] [Google Scholar]
- 91.Arvan P, Castle D. Sorting and storage during secretory granule biogenesis: looking backward and looking forward. Biochem J. 1998;332:593–610. doi: 10.1042/bj3320593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Glombik MM, Gerdes HH. Signal-mediated sorting of neuropeptides and prohormones: secretory granule biogenesis revisited. Biochimie. 2000;82:315–26. doi: 10.1016/s0300-9084(00)00195-4. [DOI] [PubMed] [Google Scholar]
- 93.Seidah NG, Chretien M. Eukaryotic protein processing: endoproteolysis of precursor proteins. Curr Opin Biotechnol. 1997;8:602–607. doi: 10.1016/s0958-1669(97)80036-5. [DOI] [PubMed] [Google Scholar]
- 94.Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2:31–9. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 95.Gorr SU, Jain RK, Kuehn U, Joyce PB, Cowley DJ. Comparative sorting of neuroendocrine secretory proteins: a search for common ground in a mosaic of sorting models and mechanisms. Mol Cell Endocrinol. 2001;172:1–6. doi: 10.1016/s0303-7207(00)00342-7. [DOI] [PubMed] [Google Scholar]
- 96.Chanat E. Mechanism of sorting of secretory proteins and formation of secretory granules in neuroendocrine cells. C R Seances Soc Biol Fil. 1993;187:697–725. [PubMed] [Google Scholar]
- 97.Glombik MM, Kromer A, Salm T, Huttner WB, Gerdes HH. The disulfide-bonded loop of chromogranin B mediates membrane binding and directs sorting from the trans-Golgi network to secretory granules. EMBO J. 1999;15:1059–1070. doi: 10.1093/emboj/18.4.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cool DR, Fenger M, Snell CR, Loh PY. Identification of the sorting signal motif within the pro-opiomelanocortin for the regulated secretory pathway. J Biol Chem. 1995;270:8723–8729. doi: 10.1074/jbc.270.15.8723. [DOI] [PubMed] [Google Scholar]
- 99.Brakch N, Cohen P, Boileau G. Processing of human prosomatostatin in AtT-20 cells: S-28 and S-14 are generated in different secretory pathways. Biochem Biophys Res Commun. 1994;205:221–9. doi: 10.1006/bbrc.1994.2653. [DOI] [PubMed] [Google Scholar]
- 100.Brechler Y, Chu WN, Baxter JD, Thibault G, Reudelhuber TL. J Biol Chem. 1996;271:20636–20640. doi: 10.1074/jbc.271.34.20636. [DOI] [PubMed] [Google Scholar]
- 101.Rovere C, Luis J, Lissitzky JC, Basak A, Marvaldi J, Chretien M, Seidah NG. The RGD motif and the C-terminal segment of proprotein convertase 1 are critical for its cellular trafficking but not for its intracellular binding to integrin alpha5beta1. J Biol Chem. 1999;274:12461–12467. doi: 10.1074/jbc.274.18.12461. [DOI] [PubMed] [Google Scholar]
- 102.Mulcahy LR, Barker AJ, Nillni EA. Disruption of disulfide bond formation alters the trafficking of prothyrotropin releasing hormone (proTRH)-derived peptides. Regul Pept. 2006;133:123–33. doi: 10.1016/j.regpep.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 103.Mulcahy LR, Vaslet CA, Nillni EA. Prohormone-convertase 1 processing enhances post-Golgi sorting of prothyrotropin-releasing hormone-derived peptides. J Biol Chem. 2005;280:39818–26. doi: 10.1074/jbc.M507193200. [DOI] [PubMed] [Google Scholar]
- 104.Perello M, Stuart R, Nillni EA. Prothyrotropin-releasing Hormone Targets Its Processing Products to Different Vesicles of the Secretory Pathway. J Biol Chem. 2008;283:19936–47. doi: 10.1074/jbc.M800732200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nillni EA, Sevarino KA, Jackson IM. Processing of proTRH to its intermediate products occurs before the packing into secretory granules of transfected AtT20 cells. Endocrinology. 1993;132:1271–7. doi: 10.1210/endo.132.3.8440188. [DOI] [PubMed] [Google Scholar]
- 106.Nillni EA, Sevarino KA, Jackson IM. Identification of the thyrotropin-releasing hormone-prohormone and its posttranslational processing in a transfected AtT20 tumoral cell line. Endocrinology. 1993;132:1260–70. doi: 10.1210/endo.132.3.8440187. [DOI] [PubMed] [Google Scholar]
- 107.Dikeakos JD, Reudelhuber TL. Sending proteins to dense core secretory granules: still a lot to sort out. J Cell Biol. 2007;177:191–6. doi: 10.1083/jcb.200701024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seidah N, Marcinkiewicz M, Benjannet S, Gaspar L, Beaubien G, Mattei M, Lazure C, Mbikay M, Chretien M. Cloning and primary sequence of a mouse candidate prohormone convertase PC1 homologous to PC2, furin, and Kex2: distinct chromosomal localization and messenger RNA distribution in brain and pituitary compared to PC2. Mol Endocrinol. 1991;5:111–122. doi: 10.1210/mend-5-1-111. [DOI] [PubMed] [Google Scholar]
- 109.Seidah NG, Gaspar L, Mion P, Marcinkiewicz M, Mbikay M, Chretien M. cDNA sequence of two distinct pituitary proteins homologous to Kex2 and furin gene products: Tissue-specific mRNAs encoding candidates for pro-hormone processing proteinases. DNA. 1990;9:415–424. doi: 10.1089/dna.1990.9.415. [DOI] [PubMed] [Google Scholar]
- 110.Seidah NG, Chretien M, Day R. The family of substilisin/kexin like pro-protein and prohormone convertase: divergent of shared functions. Biochimie. 1994;76:197–209. doi: 10.1016/0300-9084(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 111.Winsky-Sommerer R, Benjannet S, Rovere C, Barbero P, Seidah NG, Epelbaum J, Dournaud P. Regional and cellular localization of the neuroendocrine prohormone convertases PC1 and PC2 in the rat central nervous system. J Comp Neurol. 2000;424:439–60. [PubMed] [Google Scholar]
- 112.Schafer MH, Day R, Cullinan WE, Chretien M, Seidah N, Watson S. Gene Expression of Prohormone and Proprotein Convertases in the Rat CNS: A Comparative in situ Hybridization Analysis. J Neurosci. 1993;13:1258–1279. doi: 10.1523/JNEUROSCI.13-03-01258.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhu X, Zhou A, Dey A, Norrbom C, Carroll R, Zhang C, Laurent V, Lindberg I, Ugleholdt R, Holst JJ, Steiner DF. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A. 2002;99:10293–8. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Furuta M, Yano H, Zhou A, Rouille Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci U S A. 1997;94:6646–51. doi: 10.1073/pnas.94.13.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laurent V, Kimble A, Peng B, Zhu P, Pintar JE, Steiner DF, Lindber I. Mortality in 7B2 null mice can be rescued by adrenalectomy: involvement of dopamine in ACTH hypersecretion. Proc Natl Acad Sci U S A. 2002;99:3087–3092. doi: 10.1073/pnas.261715099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Westphal CH, Muller L, Zhou A, Zhu X, Bonner-Weir S, Schambelan M, Steiner DF, Lindberg I, Leder P. The neuroendocrine protein 7B2 is required for peptide hormone processing in vivo and provides a novel mechanism for pituitary Cushing's disease. Cell. 1999;96:689–700. doi: 10.1016/s0092-8674(00)80579-6. [DOI] [PubMed] [Google Scholar]
- 117.Zhu X, Orci L, Carroll R, Norrbom C, Ravazzola M, Steiner DF. Severe block in processing of proinsulin to insulin accompanied by elevation of des-64,65 proinsulin intermediates in islets of mice lacking prohormone convertase 1/3. Proc Natl Acad Sci U S A. 2002;99:10299–304. doi: 10.1073/pnas.162352799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Webb GC, Dey A, Wang J, Stein J, Milewski M, Steiner DF. Altered proglucagon processing in an alpha-cell line derived from prohormone convertase 2 null mouse islets. J Biol Chem. 2004;279:31068–75. doi: 10.1074/jbc.M404110200. [DOI] [PubMed] [Google Scholar]
- 119.Friedman TC, Loh YP, Cawley NX, Birch NP, Huang SS, Jackson IM, Nillni EA. Processing of prothyrotropin-releasing hormone (Pro-TRH) by bovine intermediate lobe secretory vesicle membrane PC1 and PC2 enzymes. Endocrinology. 1995;136:4462–72. doi: 10.1210/endo.136.10.7664666. [DOI] [PubMed] [Google Scholar]
- 120.Nillni EA. Neuroregulation of ProTRH biosynthesis and processing. Endocrine. 1999;10:185–99. doi: 10.1007/BF02738618. [DOI] [PubMed] [Google Scholar]
- 121.Nillni EA, Aird F, Seidah NG, Todd RB, Koenig JI. PreproTRH(178-199) and two novel peptides (pFQ7 and pSE14) derived from its processing, which are produced in the paraventricular nucleus of the rat hypothalamus, are regulated during suckling. Endocrinology. 2001;142:896–906. doi: 10.1210/endo.142.2.7954. [DOI] [PubMed] [Google Scholar]
- 122.Lloyd DJ, Bohan S, Gekakis N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum Mol Genet. 2006;15:1884–93. doi: 10.1093/hmg/ddl111. [DOI] [PubMed] [Google Scholar]
- 123.Jackson R, Creemers JWM, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O'Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human convertase 1 gene. Nature Genet. 1997;16:303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 124.Challis BG, Pritchard LE, Creemers JW, Delplanque J, Keogh JM, Luan J, Wareham NJ, Yeo GS, Bhattacharyya S, Froguel P, White A, Farooqi IS, O'Rahilly S. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum Mol Genet. 2002;11:1997–2004. doi: 10.1093/hmg/11.17.1997. [DOI] [PubMed] [Google Scholar]
- 125.Naggert JK, Fricker LD, Varlamov D, Nishina PM, Rouillie Y, Steiner DF, Carroll RJ, Paigen BJ, Leiter EH. Hyperinsulinemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nature Genetics. 1995;10:135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- 126.Luo L, Luo JZ, Jackson IM. Thyrotropin-releasing hormone (TRH) reverses hyperglycemia in rat. Biochem Biophys Res Commun. 2008;374:69–73. doi: 10.1016/j.bbrc.2008.06.111. [DOI] [PubMed] [Google Scholar]
- 127.Bacova Z, Najvirtova M, Krizanova O, Hudecova S, Zorad S, Strbak V, Benicky J. Effect of neonatal streptozotocin and thyrotropin-releasing hormone treatments on insulin secretion in adult rats. Gen Physiol Biophys. 2005;24:181–97. [PubMed] [Google Scholar]
- 128.O'Rahilly S, Farooqi IS, Yeo GS, Challis BG. Minireview: human obesity-lessons from monogenic disorders. Endocrinology. 2003;144:3757–64. doi: 10.1210/en.2003-0373. [DOI] [PubMed] [Google Scholar]
- 129.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O'Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Voss-Andreae A, Murphy JG, Ellacott KL, Stuart RC, Nillni EA, Cone RD, Fan W. Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology. 2007;148:1550–60. doi: 10.1210/en.2006-1389. [DOI] [PubMed] [Google Scholar]
- 131.Bianco AC, Sheng XY, Silva JE. Triiodothyronine amplifies norepinephrine stimulation of uncoupling protein gene transcription by a mechanism not requiring protein synthesis. J Biol Chem. 1988;263:18168–75. [PubMed] [Google Scholar]
- 132.Fekete C, Sarkar S, Rand WM, Harney JW, Emerson CH, Bianco AC, Lechan RM. Agouti-related protein (AGRP) has a central inhibitory action on the hypothalamic-pituitary-thyroid (HPT) axis; comparisons between the effect of AGRP and neuropeptide Y on energy homeostasis and the HPT axis. Endocrinology. 2002;143:3846–53. doi: 10.1210/en.2002-220338. [DOI] [PubMed] [Google Scholar]
- 133.Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E, Zambre Y, Pipeleers D, Friedman TC. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest. 2000;105:955–65. doi: 10.1172/JCI7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li QL, Jansen E, Friedman TC. Regulation of prohormone convertase 1 (PC1) by gp130-related cytokines. Mol Cell Endocrinol. 1999;158:143–52. doi: 10.1016/s0303-7207(99)00168-9. [DOI] [PubMed] [Google Scholar]
- 135.Sanchez VC, Goldstein J, Stuart RC, Hovanesian V, Huo L, Munzberg H, Friedman TC, Bjorbaek C, Nillni EA. Regulation of hypothalamic prohormone convertases 1 and 2 and effects on processing of prothyrotropin-releasing hormone. J Clin Invest. 2004;114:357–69. doi: 10.1172/JCI21620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Perello M, Stuart RC, Varslet CA, Nillni EA. Cold Exposure Increases the Biosynthesis and Proteolytic Processing of Prothyrotropin Releasing Hormone in the Hypothalamic Paraventricular Nucleus via Beta-adrenoreceptors. Endocrinology. 2007 doi: 10.1210/en.2007-0522. [DOI] [PubMed] [Google Scholar]
- 137.Nillni EA, Lee A, Legradi G, Lechan RM. Effect of precipitated morphine withdrawal on post-translational processing of prothyrotropin releasing hormone (proTRH) in the ventrolateral column of the midbrain periaqueductal gray. J Neurochem. 2002;80:874–84. doi: 10.1046/j.0022-3042.2002.00763.x. [DOI] [PubMed] [Google Scholar]
- 138.Legradi G, Rand WM, Hitz S, Nillni EA, Jackson IM, Lechan RM. Opiate withdrawal increases ProTRH gene expression in the ventrolateral column of the midbrain periaqueductal gray. Brain Res. 1996;729:10–19. doi: 10.1016/s0006-8993(96)00210-7. [DOI] [PubMed] [Google Scholar]
- 139.Li QL, Jansen E, Brent GA, Friedman TC. Regulation of prohormone convertase 1 (PC1) by thyroid hormone. Am J Physiol Endocrinol Metab. 2001;280:E160–70. doi: 10.1152/ajpendo.2001.280.1.E160. [DOI] [PubMed] [Google Scholar]
- 140.Li QL, Jansen E, Brent GA, Naqvi S, Wilber JF, Friedman TC. Interactions between the prohormone convertase 2 promoter and the thyroid hormone receptor. Endocrinology. 2000;141:3256–66. doi: 10.1210/endo.141.9.7674. [DOI] [PubMed] [Google Scholar]
- 141.Shen X, Li QL, Brent GA, Friedman TC. Thyroid hormone regulation of prohormone convertase 1 (PC1): regional expression in rat brain and in vitro characterization of negative thyroid hormone response elements. J Mol Endocrinol. 2004;33:21–33. doi: 10.1677/jme.0.0330021. [DOI] [PubMed] [Google Scholar]
- 142.Shen X, Li QL, Brent GA, Friedman TC. Regulation of regional expression in rat brain PC2 by thyroid hormone/characterization of novel negative thyroid hormone response elements in the PC2 promoter. Am J Physiol Endocrinol Metab. 2005;288:E236–45. doi: 10.1152/ajpendo.00144.2004. [DOI] [PubMed] [Google Scholar]
- 143.Perello M, Friedman T, Paez-Espinosa V, Shen X, Stuart RC, Nillni EA. Thyroid hormones selectively regulate the posttranslational processing of prothyrotropin-releasing hormone in the paraventricular nucleus of the hypothalamus. Endocrinology. 2006;147:2705–16. doi: 10.1210/en.2005-1609. [DOI] [PubMed] [Google Scholar]
- 144.Nillni EA. Regulation of prohormone convertases in hypothalamic neurons: implications for prothyrotropin-releasing hormone and proopiomelanocortin. Endocrinology. 2007;148:4191–200. doi: 10.1210/en.2007-0173. [DOI] [PubMed] [Google Scholar]
- 145.Jansen E, Ayoubi TA, Meulemans SM, Van de Ven WJ. Neuroendocrine-specific expression of the human prohormone convertase 1 gene. Hormonal regulation of transcription through distinct cAMP response elements. J Biol Chem. 1995;270:15391–7. doi: 10.1074/jbc.270.25.15391. [DOI] [PubMed] [Google Scholar]
- 146.Krulich L. Neurotransmitter control of thyrotropin secretion. Neuroendocrinol. 1982;35:139–147. doi: 10.1159/000123369. [DOI] [PubMed] [Google Scholar]
- 147.Terry LC. Regulation of thyrotropin secretion by central epinephrine system. Neuroendocrinol. 1986;42:102. doi: 10.1159/000124258. [DOI] [PubMed] [Google Scholar]
- 148.Grimm Y, Reichlin S. Thyrotropin-releasing hormone (TRH): neurotransmitter regulation of secretion by mouse hypothalamic tissue in vitro. Endocrinology. 1973;93:626–631. doi: 10.1210/endo-93-3-626. [DOI] [PubMed] [Google Scholar]
- 149.Tapia-Arancibia L, Arancibia S, Astier H. Evidence for alpha1-adrenergic stimulatory control of in vitro release of immunoreactive thyrotropin-releasing hormone from rat median eminence: in vivo corroboration. Endocrinol. 1985;116:2314. doi: 10.1210/endo-116-6-2314. [DOI] [PubMed] [Google Scholar]
- 150.Bjorklund A, Lindvall O. Catecholaminergic brain stem regulatory systems. American Physiological Society; Bethesda, MD: 1986. [Google Scholar]
- 151.Schettini G, Quattrone A, DiRenzo G, Lombardi G. Effect of 6-hydroxydopamine treatment on TSH secretion in basal and cold-stimulated conditions in the rat. Eur J Pharmacol. 1979;56:153–157. doi: 10.1016/0014-2999(79)90445-x. [DOI] [PubMed] [Google Scholar]
- 152.Arancibia S, Tapia-Arancibia L, Astier H, Assenmacher I. Physiological evidence for alpha1-adrenergic facilitatory control of the cold-induced TRH release in the rat, obtained by push-pull cannulation of the median eminence. Neurosci Lett. 1989;100:169. doi: 10.1016/0304-3940(89)90679-4. [DOI] [PubMed] [Google Scholar]
- 153.Zoeller RT, Kabeer N, Albers HE. Cold exposure elevates cellular levels of messenger ribonucleic acid encoding thyrotropin-releasing hormone in paraventricular nucleus despite elevated levels of thyroid hormones. Endocrinology. 1990;127:2955–62. doi: 10.1210/endo-127-6-2955. [DOI] [PubMed] [Google Scholar]
- 154.Arancibia S, Tapia-Arancibia L, Assenmacher I, Astier H. Direct evidence of short-term cold-induced TRH release by the median eminence in unanesthietized rat. Neuroendocrinology. 1983;37:225–228. doi: 10.1159/000123547. [DOI] [PubMed] [Google Scholar]
- 155.Rondeel JMM, deGreef WJ, Hop WCJ, Rowland DL, Visser TJ. Effect of cold exposure on the hypothalamic release of thyrotropin-releasing hormone and catecholamines. Neuroendocrinol. 1991;54:477–481. doi: 10.1159/000125940. [DOI] [PubMed] [Google Scholar]
- 156.Légrádi G, Emerson CH, Ahima RS, Flier JS, Lechan RM. Leptin Prevents Fasting-Induced Suppression of Prothyrotropin-Releasing Hormone Messenger RNA in Neurons of the Hypothalamic Paraventricular Nucleus. Endocrinol. 1997;138:2569–2576. doi: 10.1210/endo.138.6.5209. [DOI] [PubMed] [Google Scholar]
- 157.Uribe RM, Redondo JL, Charli JL, Joseph-Bravo P. Suckling and cold stress rapidly and transiently increase TRH mRNA in the paraventricular nucleus. Neuroendocrinology. 1993;58:140–5. doi: 10.1159/000126523. [DOI] [PubMed] [Google Scholar]
- 158.Jansen E, Ayoubi TA, Meulemans SM, Van De Ven WJ. Regulation of human prohormone convertase 2 promoter activity by the transcription factor EGR-1. Biochem J. 1997;328(Pt 1):69–74. doi: 10.1042/bj3280069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Perello M, Stuart RC, Nillni EA. Differential effects of fasting and leptin on proopiomelanocortin peptides in the arcuate nucleus and in the nucleus of the solitary tract. Am J Physiol Endocrinol Metab. 2007;292:E1348–57. doi: 10.1152/ajpendo.00466.2006. [DOI] [PubMed] [Google Scholar]
- 160.Dong W, Seidel B, Marcinkiewicz M, Chretien M, Seidah NG, Day R. Cellular location of the prohormone convertases in the hypothalamic paraventricular and supraoptic nuclei: selective regulation of PC1 in corticotropin releasing hormone parvocellular neurons mediated by glucocorticoids. J Neurosci. 1997;17:563–575. doi: 10.1523/JNEUROSCI.17-02-00563.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Espinosa VP, Ferrini M, Shen X, Lutfy K, Nillni EA, Friedman TC. Cellular colocalization and coregulation between hypothalamic pro-TRH and prohormone convertases in hypothyroidism. Am J Physiol Endocrinol Metab. 2007;292:E175–86. doi: 10.1152/ajpendo.00288.2006. [DOI] [PubMed] [Google Scholar]
- 162.Segerson TP, Kauer J, Wolfe HC, Mobtaker H, Wu P, Jackson IM, Lechan RM. Thyroid hormone regulates TRH biosynthesis in the paraventricular nucleus of the rat hypothalamus. Science. 1987;238:78–80. doi: 10.1126/science.3116669. [DOI] [PubMed] [Google Scholar]
- 163.Lindsley JE, Rutter J. Nutrient sensing and metabolic decisions. Comp Biochem Physiol B Biochem Mol Biol. 2004;139:543–59. doi: 10.1016/j.cbpc.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 164.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–30. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 165.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 166.Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 167.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 168.Flier JS, Maratos-Flier E. Obesity and the hypothalmus: Novel peptides for new pathways. Cell. 1998;92:437–440. doi: 10.1016/s0092-8674(00)80937-x. [DOI] [PubMed] [Google Scholar]
- 169.Flier JS. Clinical review 94: What's in a name? In search of leptin's physiologic role. J Clin Endocrinol Metab. 1998;83:1407–1413. doi: 10.1210/jcem.83.5.4779. [DOI] [PubMed] [Google Scholar]
- 170.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 171.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 172.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 173.Chen H, Chatlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 174.Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougneres P, Lebouc Y, Froguel P, Guy-Grand B. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 175.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–5. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 176.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O'Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 177.Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–31. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- 178.Huo L, Munzberg H, Nillni EA, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic trh gene expression by leptin. Endocrinology. 2004;145:2516–23. doi: 10.1210/en.2003-1242. [DOI] [PubMed] [Google Scholar]
- 179.Fekete C, Legradi G, Mihaly E, Huang QH, Tatro JB, Rand WM, Emerson CH, Lechan RM. alpha-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J Neurosci. 2000;20:1550–8. doi: 10.1523/JNEUROSCI.20-04-01550.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bjørbæk C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and in leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 181.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Woolf EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 182.Legradi G, Emerson CH, Ahima RS, Rand WM, Flier JS, Lechan RM. Arcuate nucleus ablation prevents fasting-induced suppression of ProTRH mRNA in the hypothalamic paraventricular nucleus. Neuroendocrinology. 1998;68:89–97. doi: 10.1159/000054354. [DOI] [PubMed] [Google Scholar]
- 183.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–41. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 184.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 185.Fekete C, Marks DL, Sarkar S, Emerson CH, Rand WM, Cone RD, Lechan RM. Effect of Agouti-Related Protein (Agrp) in Regulation of the Hypothalamic-Pituitary-Thyroid (Hpt) Axis in the Mc4-R Ko Mouse. Endocrinology. 2004 doi: 10.1210/en.2004-0476. [DOI] [PubMed] [Google Scholar]
- 186.Legradi G, Lechan RM. The arcuate nucleus is the major source for neuropeptide Y-innervation of thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 1998;139:3262–70. doi: 10.1210/endo.139.7.6113. [DOI] [PubMed] [Google Scholar]
- 187.Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang N, Larsen PJ, Hastrup S. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature. 1998;393:72–76. doi: 10.1038/29993. [DOI] [PubMed] [Google Scholar]
- 188.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 189.Thornton JE, Cheung CC, Clifton DK, Steiner RA. Regulation of hypothalamic proopiomelanocortin mRNA by leptin in ob/ob mice. Endocrinol. 1997;138:5063–5066. doi: 10.1210/endo.138.11.5651. [DOI] [PubMed] [Google Scholar]
- 190.Tang-Christensen M, Holst JJ, Hartmann B, Vrang N. The arcuate nucleus is pivotal in mediating the anorectic effects of centrally administered leptin. Neuroreport. 1999;10:1183–7. doi: 10.1097/00001756-199904260-00005. [DOI] [PubMed] [Google Scholar]
- 191.Dawson R, Pelleymounter MA, Millard WJ, Liu S, Eppler B. Attenuation of leptin-mediated effects by monosodium glutamate-induced arcuate nucleus damage. Am J Physiol. 1997;273:E202–6. doi: 10.1152/ajpendo.1997.273.1.E202. [DOI] [PubMed] [Google Scholar]
- 192.Perello M, Gaillard RC, Chisari A, Spinedi E. Adrenal enucleation in MSG-damaged hyperleptinemic male rats transiently restores adrenal sensitivity to leptin. Neuroendocrinology. 2003;78:176–84. doi: 10.1159/000072799. [DOI] [PubMed] [Google Scholar]
- 193.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–51. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 194.Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, SR B. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinol. 1998;139:4428–4431. doi: 10.1210/endo.139.10.6332. [DOI] [PubMed] [Google Scholar]
- 195.Nijenhuis WA, Oosterom J, Adan RA. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol. 2001;15:164–71. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 196.Gehlert DR. Role of hypothalamic neuropeptide Y in feeding and obesity. Neuropeptides. 1999;33:329–38. doi: 10.1054/npep.1999.0057. [DOI] [PubMed] [Google Scholar]
- 197.Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. Journal of Neuroscience. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Diano S, Naftolin F, Goglia F, Horvath TL. Segregation of the intra- and extrahypothalamic neuropeptide Y and catecholaminergic inputs on paraventricular neurons, including those producing thyrotropin-releasing hormone. Regul Pept. 1998;75-76:117–26. doi: 10.1016/s0167-0115(98)00060-3. [DOI] [PubMed] [Google Scholar]
- 199.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–4. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 200.Wolak ML, DeJoseph MR, Cator AD, Mokashi AS, Brownfield MS, Urban JH. Comparative distribution of neuropeptide Y Y1 and Y5 receptors in the rat brain by using immunohistochemistry. J Comp Neurol. 2003;464:285–311. doi: 10.1002/cne.10823. [DOI] [PubMed] [Google Scholar]
- 201.Broberger C, Visser TJ, Kuhar MJ, Hokfelt T. Neuropeptide Y innervation and neuropeptide-Y-Y1-receptor-expressing neurons in the paraventricular hypothalamic nucleus of the mouse. Neuroendocrinology. 1999;70:295–305. doi: 10.1159/000054490. [DOI] [PubMed] [Google Scholar]
- 202.Toni R, Jackson IM, Lechan RM. Neuropeptide-Y-immunoreactive innervation of thyrotropin-releasing hormone-synthesizing neurons in the rat hypothalamic paraventricular nucleus. Endocrinology. 1990;126:2444–53. doi: 10.1210/endo-126-5-2444. [DOI] [PubMed] [Google Scholar]
- 203.Lecklin A, Lundell I, Salmela S, Mannisto PT, Beck-Sickinger AG, Larhammar D. Agonists for neuropeptide Y receptors Y1 and Y5 stimulate different phases of feeding in guinea pigs. Br J Pharmacol. 2003;139:1433–40. doi: 10.1038/sj.bjp.0705389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Marsh DJ, Hollopeter G, Kafer KE, Palmiter RD. Role of the Y5 neuropeptide Y receptor in feeding and obesity. Nat Med. 1998;4:718–21. doi: 10.1038/nm0698-718. [DOI] [PubMed] [Google Scholar]
- 205.Ogden CL, Carroll MD, McDowell MA, Flegal KM. Obesity among adults in the United States--no statistically significant chance since 2003-2004. NCHS Data Brief. 2007:1–8. [PubMed] [Google Scholar]
- 206.Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132:2087–102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 207.Levin BE. Central regulation of energy homeostasis intelligent design: how to build the perfect survivor. Obesity (Silver Spring) 2006;14 5:192S–196S. doi: 10.1038/oby.2006.307. [DOI] [PubMed] [Google Scholar]
- 208.Blake N, Eckland D, Foster O, Lightman S. Inhibition of hypothalamic thyrotropin-releasing hormone messenger ribonucleic acid during food deprivation. Endocrinol. 1991;129:2714–2718. doi: 10.1210/endo-129-5-2714. [DOI] [PubMed] [Google Scholar]
- 209.Rondeel JM, de Greef WJ, Klootwijk W, Visser TJ. Effects of hypothyroidism on hypothalamic release of thyrotropin-releasing hormone in rats. Endocrinology. 1992;130:651–6. doi: 10.1210/endo.130.2.1733713. [DOI] [PubMed] [Google Scholar]
- 210.van Haasteren GA, Linkels E, Klootwijk W, van Toor H, Rondeel JM, Themmen AP, de Jong FH, Valentijn K, Vaudry H, Bauer K, et al. Starvation-induced changes in the hypothalamic content of prothyrotrophin-releasing hormone (proTRH) mRNA and the hypothalamic release of proTRH-derived peptides: role of the adrenal gland. J Endocrinol. 1995;145:143–53. doi: 10.1677/joe.0.1450143. [DOI] [PubMed] [Google Scholar]
- 211.van Haasteren GA, van Toor H, Klootwijk W, Handler B, Linkels E, van der Schoot P, van Ophemert J, de Jong FH, Visser TJ, de Greef WJ. Studies on the role of TRH and corticosterone in the regulation of prolactin and thyrotrophin secretion during lactation. J Endocrinol. 1996;148:325–36. doi: 10.1677/joe.0.1480325. [DOI] [PubMed] [Google Scholar]
- 212.Flier JS, Harris M, Hollenberg AN. Leptin, nutrition, and the thyroid: the why, the wherefore, and the wiring. J Clin Invest. 2000;105:859–61. doi: 10.1172/JCI9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Goglia F, Silvestri E, Lanni A. Thyroid hormones and mitochondria. Biosci Rep. 2002;22:17–32. doi: 10.1023/a:1016056905347. [DOI] [PubMed] [Google Scholar]
- 214.Bianco AC, Maia AL, da Silva WS, Christoffolete MA. Adaptive activation of thyroid hormone and energy expenditure. Biosci Rep. 2005;25:191–208. doi: 10.1007/s10540-005-2885-6. [DOI] [PubMed] [Google Scholar]
- 215.Bianco AC, Silva JE. Intracellular conversion of thyroxine to triiodothyronine is required for the optimal thermogenic function of brown adipose tissue. J Clin Invest. 1987;79:295–300. doi: 10.1172/JCI112798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.de Jesus LA, Carvalho SD, Ribeiro MO, Schneider M, Kim SW, Harney JW, Larsen PR, Bianco AC. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest. 2001;108:1379–85. doi: 10.1172/JCI13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sims EA, Danforth E., Jr Expenditure and storage of energy in man. J Clin Invest. 1987;79:1019–25. doi: 10.1172/JCI112913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Coppola A, Meli R, Diano S. Inverse Shift In Circulating Corticosterone And Leptin Levels Elevates Hypothalamic Deiodinase Type 2 In Fasted Rats. Endocrinology. 2005 doi: 10.1210/en.2004-1361. [DOI] [PubMed] [Google Scholar]
- 219.Diano S, Naftolin F, Goglia F, Csernus V, Horvath TL. Monosynaptic pathway between the arcuate nucleus expressing glial type II iodothyronine 5′-deiodinase mRNA and the median eminence-projective TRH cells of the rat paraventricular nucleus. J Neuroendocrinol. 1998;10:731–42. doi: 10.1046/j.1365-2826.1998.00204.x. [DOI] [PubMed] [Google Scholar]
- 220.Diano S, Naftolin F, Goglia F, Horvath TL. Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology. 1998;139:2879–84. doi: 10.1210/endo.139.6.6062. [DOI] [PubMed] [Google Scholar]
- 221.Coppola A, Hughes J, Esposito E, Schiavo L, Meli R, Diano S. Suppression of hypothalamic deiodinase type II activity blunts TRH mRNA decline during fasting. FEBS Lett. 2005;579:4654–8. doi: 10.1016/j.febslet.2005.07.035. [DOI] [PubMed] [Google Scholar]
- 222.Coppola A, Liu ZW, Andrews ZB, Paradis E, Roy MC, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao XB, Diano S. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5:21–33. doi: 10.1016/j.cmet.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Cettour-Rose P, Burger AG, Meier CA, Visser TJ, Rohner-Jeanrenaud F. Central stimulatory effect of leptin on T3 production is mediated by brown adipose tissue type II deiodinase. Am J Physiol Endocrinol Metab. 2002;283:E980–7. doi: 10.1152/ajpendo.00196.2002. [DOI] [PubMed] [Google Scholar]
- 224.Kohrle J. Local activation and inactivation of thyroid hormones: the deiodinase family. Mol Cell Endocrinol. 1999;151:103–19. doi: 10.1016/s0303-7207(99)00040-4. [DOI] [PubMed] [Google Scholar]
- 225.Sanchez E, Vargas MA, Singru PS, Pascual I, Romero F, Fekete C, Charli JL, Lechan RM. Tanycyte pyroglutamyl peptidase II contributes to regulation of the hypothalamic-pituitary-thyroid axis through glial-axonal associations in the median eminence. Endocrinology. 2009;150:2283–91. doi: 10.1210/en.2008-1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.O'Brian JT, Bybee DE, Burman KD, Osburne RC, Ksiazek MR, Wartofsky L, Georges LP. Thyroid hormone homeostasis in states of relative caloric deprivation. Metabolism. 1980;29:721–7. doi: 10.1016/0026-0495(80)90193-6. [DOI] [PubMed] [Google Scholar]
- 227.Spencer CA, Lum SM, Wilber JF, Kaptein EM, Nicoloff JT. Dynamics of serum thyrotropin and thyroid hormone changes in fasting. J Clin Endocrinol Metab. 1983;56:883–8. doi: 10.1210/jcem-56-5-883. [DOI] [PubMed] [Google Scholar]
- 228.Romijn JA, Adriaanse R, Brabant G, Prank K, Endert E, Wiersinga WM. Pulsatile secretion of thyrotropin during fasting: a decrease of thyrotropin pulse amplitude. J Clin Endocrinol Metab. 1990;70:1631–6. doi: 10.1210/jcem-70-6-1631. [DOI] [PubMed] [Google Scholar]
- 229.Feldt-Rasmussen U. Thyroid and leptin. Thyroid. 2007;17:413–9. doi: 10.1089/thy.2007.0032. [DOI] [PubMed] [Google Scholar]
- 230.Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J Clin Invest. 2003;111:1409–21. doi: 10.1172/JCI17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chan JL, Matarese G, Shetty GK, Raciti P, Kelesidis I, Aufiero D, De Rosa V, Perna F, Fontana S, Mantzoros CS. Differential regulation of metabolic, neuroendocrine, and immune function by leptin in humans. Proc Natl Acad Sci U S A. 2006;103:8481–6. doi: 10.1073/pnas.0505429103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Schurgin S, Canavan B, Koutkia P, Depaoli AM, Grinspoon S. Endocrine and metabolic effects of physiologic r-metHuLeptin administration during acute caloric deprivation in normal-weight women. J Clin Endocrinol Metab. 2004;89:5402–9. doi: 10.1210/jc.2004-1102. [DOI] [PubMed] [Google Scholar]
- 233.Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J Clin Endocrinol Metab. 2002;87:2391–4. doi: 10.1210/jcem.87.5.8628. [DOI] [PubMed] [Google Scholar]
- 234.Lechan RM, Fekete C. Feedback regulation of thyrotropin-releasing hormone (TRH): mechanisms for the non-thyroidal illness syndrome. J Endocrinol Invest. 2004;27:105–19. [PubMed] [Google Scholar]
- 235.Fliers E, Alkemade A, Wiersinga WM, Swaab DF. Hypothalamic thyroid hormone feedback in health and disease. Prog Brain Res. 2006;153:189–207. doi: 10.1016/S0079-6123(06)53011-0. [DOI] [PubMed] [Google Scholar]
- 236.Ramos EJ, Suzuki S, Marks D, Inui A, Asakawa A, Meguid MM. Cancer anorexia-cachexia syndrome: cytokines and neuropeptides. Curr Opin Clin Nutr Metab Care. 2004;7:427–34. doi: 10.1097/01.mco.0000134363.53782.cb. [DOI] [PubMed] [Google Scholar]
- 237.Rondeel JM, Heide R, de Greef WJ, van Toor H, van Haasteren GA, Klootwijk W, Visser TJ. Effect of starvation and subsequent refeeding on thyroid function and release of hypothalamic thyrotropin-releasing hormone. Neuroendocrinology. 1992;56:348–53. doi: 10.1159/000126248. [DOI] [PubMed] [Google Scholar]
- 238.Farooqi IS, Yeo GS, O'Rahilly S. Binge eating as a phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003;349:606–9. author reply 606-9. [PubMed] [Google Scholar]
- 239.Farooqi IS, O'Rahilly S. Recent advances in the genetics of severe childhood obesity. Arch Dis Child. 2000;83:31–4. doi: 10.1136/adc.83.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, O'Rahilly S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271–9. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Persani L. Hypothalamic thyrotropin-releasing hormone and thyrotropin biological activity. Thyroid. 1998;8:941–6. doi: 10.1089/thy.1998.8.941. [DOI] [PubMed] [Google Scholar]
- 242.Ahima RS. Central actions of adipocyte hormones. Trends Endocrinol Metab. 2005;16:307–13. doi: 10.1016/j.tem.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 243.Clement K. Genetics of human obesity. Proc Nutr Soc. 2005;64:133–42. doi: 10.1079/pns2005416. [DOI] [PubMed] [Google Scholar]
- 244.Flier JS, Foster DW. Eating disorders: obesity, anorexia nervosa, and bulimia nervosa. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams Textbook of Endocrinology. 9th. W.B. Saunders Co.; Philadelphia: 1997. pp. 1061–1098. [Google Scholar]
- 245.Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–50. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- 246.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–43. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 247.Korbonits M. Leptin and the thyroid--a puzzle with missing pieces. Clin Endocrinol (Oxf) 1998;49:569–72. doi: 10.1046/j.1365-2265.1998.00594.x. [DOI] [PubMed] [Google Scholar]
- 248.Krotkiewski M. Thyroid hormones in the pathogenesis and treatment of obesity. Eur J Pharmacol. 2002;440:85–98. doi: 10.1016/s0014-2999(02)01420-6. [DOI] [PubMed] [Google Scholar]
- 249.Mantzoros CS. The role of leptin and hypothalamic neuropeptides in energy homeostasis: update on leptin in obesity. Growth Horm IGF Res. 2001;11 A:S85–9. doi: 10.1016/s1096-6374(01)80014-9. [DOI] [PubMed] [Google Scholar]
- 250.Mantzoros CS, Ozata M, Negrao AB, Suchard MA, Ziotopoulou M, Caglayan S, Elashoff RM, Cogswell RJ, Negro P, Liberty V, Wong ML, Veldhuis J, Ozdemir IC, Gold PW, Flier JS, Licinio J. Synchronicity of frequently sampled thyrotropin (TSH) and leptin concentrations in healthy adults and leptin-deficient subjects: evidence for possible partial TSH regulation by leptin in humans. J Clin Endocrinol Metab. 2001;86:3284–91. doi: 10.1210/jcem.86.7.7644. [DOI] [PubMed] [Google Scholar]
- 251.Danforth E, Jr, Horton ES, O'Connell M, Sims EA, Burger AG, Ingbar SH, Braverman L, Vagenakis AG. Dietary-induced alterations in thyroid hormone metabolism during overnutrition. J Clin Invest. 1979;64:1336–47. doi: 10.1172/JCI109590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Scriba PC, Bauer M, Emmert D, Fateh-Moghadam A, Hofmann GG, Horn K, Pickardt CR. Effects of obesity, total fasting and re-alimentation on L-thyroxine (T4), 3,5,3′-L-triiodothyronine (T3), 3,3′,5′-L-triiodothyronine (rT3), thyroxine binding globulin (TBG), cortisol, thyrotrophin, cortisol binding globulin (CBG), transferrin, alpha 2-haptoglobin and complement C'3 in serum. Acta Endocrinol (Copenh) 1979;91:629–43. doi: 10.1530/acta.0.0910629. [DOI] [PubMed] [Google Scholar]
- 253.Strata A, Ugolotti G, Contini C, Magnati G, Pugnoli C, Tirelli F, Zuliani U. Thyroid and obesity: survey of some function tests in a large obese population. Int J Obes. 1978;2:333–40. [PubMed] [Google Scholar]
- 254.Donders SH, Pieters GF, Heevel JG, Ross HA, Smals AG, Kloppenborg PW. Disparity of thyrotropin (TSH) and prolactin responses to TSH-releasing hormone in obesity. J Clin Endocrinol Metab. 1985;61:56–9. doi: 10.1210/jcem-61-1-56. [DOI] [PubMed] [Google Scholar]
- 255.Sari R, Balci MK, Altunbas H, Karayalcin U. The effect of body weight and weight loss on thyroid volume and function in obese women. Clin Endocrinol (Oxf) 2003;59:258–62. doi: 10.1046/j.1365-2265.2003.01836.x. [DOI] [PubMed] [Google Scholar]
- 256.Michalaki MA, Vagenakis AG, Leonardou AS, Argentou MN, Habeos IG, Makri MG, Psyrogiannis AI, Kalfarentzos FE, Kyriazopoulou VE. Thyroid function in humans with morbid obesity. Thyroid. 2006;16:73–8. doi: 10.1089/thy.2006.16.73. [DOI] [PubMed] [Google Scholar]
- 257.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 258.Hamann A, Matthaei S. Regulation of energy balance by leptin. Exp Clin Endocrinol Diabetes. 1996;104:293–300. doi: 10.1055/s-0029-1211457. [DOI] [PubMed] [Google Scholar]
- 259.Wynne K, Stanley S, McGowan B, Bloom S. Appetite control. J Endocrinol. 2005;184:291–318. doi: 10.1677/joe.1.05866. [DOI] [PubMed] [Google Scholar]
- 260.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–95. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 261.Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–9. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 262.Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-Induced Obesity Causes Severe but Reversible Leptin Resistance in Arcuate Melanocortin Neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 263.Chua SC, Hennessey K, Zeitler P, Leibel RL. The little (lit) mutation cosegregates with the growth hormone releasing factor receptor on mouse chromosome 6. Mammalian Genome. 1993;4:555–9. doi: 10.1007/BF00361384. [DOI] [PubMed] [Google Scholar]
- 264.Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ, Hess JF. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996;13:18–9. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 265.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A. 1997;94:8878–83. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Scarpace PJ, Matheny M, Zhang Y, Tumer N, Frase CD, Shek EW, Hong B, Prima V, Zolotukhin S. Central leptin gene delivery evokes persistent leptin signal transduction in young and aged-obese rats but physiological responses become attenuated over time in aged-obese rats. Neuropharmacology. 2002;42:548–61. doi: 10.1016/s0028-3908(02)00003-5. [DOI] [PubMed] [Google Scholar]
- 267.Lanni A, Moreno M, Lombardi A, de Lange P, Silvestri E, Ragni M, Farina P, Baccari GC, Fallahi P, Antonelli A, Goglia F. 3,5-diiodo-L-thyronine powerfully reduces adiposity in rats by increasing the burning of fats. Faseb J. 2005;19:1552–4. doi: 10.1096/fj.05-3977fje. [DOI] [PubMed] [Google Scholar]
- 268.Collins S, Cao W, Daniel KW, Dixon TM, Medvedev AV, Onuma H, Surwit R. Adrenoceptors, uncoupling proteins, and energy expenditure. Exp Biol Med (Maywood) 2001;226:982–90. doi: 10.1177/153537020122601104. [DOI] [PubMed] [Google Scholar]
- 269.Garcia SI, Landa MS, Porto PI, Alvarez AL, Schuman M, Finkielman S, Pirola CJ. Thyrotropin-releasing hormone decreases leptin and mediates the leptin-induced pressor effect. Hypertension. 2002;39:491–5. doi: 10.1161/hy0202.103049. [DOI] [PubMed] [Google Scholar]
- 270.Landa MS, Garcia SI, Schuman ML, Burgueno A, Alvarez AL, Saravia FE, Gemma C, Pirola CJ. Knocking down the diencephalic thyrotropin-releasing hormone precursor gene normalizes obesity-induced hypertension in the rat. Am J Physiol Endocrinol Metab. 2007;292:E1388–94. doi: 10.1152/ajpendo.00234.2006. [DOI] [PubMed] [Google Scholar]
- 271.Klieverik LP, Coomans CP, Endert E, Sauerwein HP, Havekes LM, Voshol PJ, Rensen PC, Romijn JA, Kalsbeek A, Fliers E. Thyroid Hormone Effects on Whole-Body Energy Homeostasis and Tissue-Specific Fatty Acid Uptake in Vivo. Endocrinology. 2009 doi: 10.1210/en.2009-0297. [DOI] [PubMed] [Google Scholar]
- 272.Lechan RM, Qi Y, Jackson IM, Mahdavi V. Identification of thyroid hormone receptor isoforms in thyrotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Endocrinology. 1994;135:92–100. doi: 10.1210/endo.135.1.7516871. [DOI] [PubMed] [Google Scholar]
- 273.Abel ED, Ahima RS, Boers ME, Elmquist JK, Wondisford FE. Critical role for thyroid hormone receptor beta2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest. 2001;107:1017–23. doi: 10.1172/JCI10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Alkemade A, Friesema EC, Unmehopa UA, Fabriek BO, Kuiper GG, Leonard JL, Wiersinga WM, Swaab DF, Visser TJ, Fliers E. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab. 2005;90:4322–34. doi: 10.1210/jc.2004-2567. [DOI] [PubMed] [Google Scholar]
- 275.Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40128–35. doi: 10.1074/jbc.M300909200. [DOI] [PubMed] [Google Scholar]
- 276.Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–7. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- 277.Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–75. doi: 10.1086/380999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, Bauer K. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology. 2005;146:1701–6. doi: 10.1210/en.2004-1179. [DOI] [PubMed] [Google Scholar]
- 279.Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, Heuer H. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117:627–35. doi: 10.1172/JCI28253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Hollenberg AN. The role of the thyrotropin-releasing hormone (TRH) neuron as a metabolic sensor. Thyroid. 2008;18:131–9. doi: 10.1089/thy.2007.0251. [DOI] [PubMed] [Google Scholar]