

Abstract
Phosphorylation of myosin regulatory light chain (RLC) by myosin light chain kinase (MLCK) and myosin binding protein-C (cMyBP-C) by protein kinase A (PKA) independently accelerate the kinetics of force development in ventricular myocardium. However, while MLCK treatment has been shown to increase the Ca2+ sensitivity of force (pCa50), PKA treatment has been shown to decrease pCa50, presumably due to cardiac troponin I phosphorylation. Further, MLCK treatment increases Ca2+-independent force and maximum Ca2+-activated force, whereas PKA treatment has no effect on either force. To investigate the structural basis underlying the kinase-specific differential effects on steady-state force, we used synchrotron low-angle X-ray diffraction to compare equatorial intensity ratios (I1,1/I1,0) to assess the proximity of myosin cross-bridge mass relative to actin and to compare lattice spacings (d1,0) to assess the inter-thick filament spacing in skinned myocardium following treatment with either MLCK or PKA. As we showed previously, PKA phosphorylation of cMyBP-C increases I1,1/I1,0 and, as hypothesized, treatment with MLCK also increased I1,1/I1,0, which can explain the accelerated rates of force development during activation. Importantly, interfilament spacing was reduced by ∼2 nm (Δ 3.5%) with MLCK treatment, but did not change with PKA treatment. Thus, RLC or cMyBP-C phosphorylation increases the proximity of cross-bridges to actin, but only RLC phosphorylation affects lattice spacing, which suggests that RLC and cMyBP-C modulate the kinetics of force development by similar structural mechanisms; however, the effect of RLC phosphorylation to increase the Ca2+ sensitivity of force is mediated by a distinct mechanism, most probably involving changes in interfilament spacing.
Introduction
In cardiac muscle, myosin ATPase activity is set principally by the myosin isoform expressed (α- or β-myosin heavy chain (MHC)). The rates of cross-bridge attachment and detachment, however, are finely tuned in order to optimize cardiac performance on a beat-to-beat basis. The cardiac isoforms of the myofilament proteins, myosin binding protein-C (cMyBP-C) and troponin I (cTnI), are phosphorylated rapidly by protein kinase A (PKA) over time courses similar to that for the increase in twitch force and kinetics during β1-adrenergic stimulation (England, 1983). In contrast, under physiological conditions the level of regulatory light chain (RLC) phosphorylation is maintained relatively constant by a balance of phosphorylation and phosphatase-induced dephosphorylation, and is regulated by α1-adrenergic tone (Chan et al. 2008). In this regard, RLC phosphorylation may normally contribute to the basal levels of the strength and speed of contraction, but during β1-adrenoreceptor stimulation, phosphorylation of cTnI and cMyBP-C may act to further tune cardiac function to increase the strength and speed of contraction and also to facilitate rapid filling of the ventricles.
RLC and cMyBP-C, which both bind to MHC, are thought to modulate the position of myosin heads relative to actin depending on their phosphorylation state, and thereby regulate the likelihood of myosin cross-bridge interaction with actin. RLC has been proposed to stabilize the α-helical neck region of the myosin head for force transmission (Rayment et al. 1993), whereas cMyBP-C may stabilize thick filament structure through its interactions with the thick filament backbone and/or titin (Moos et al. 1975; Furst et al. 1992; Labeit et al. 1992; Zoghbi et al. 2008). RLC phosphorylation may affect cross-bridge disposition through molecular interactions involving charge repulsion with the thick filament (Metzger et al. 1989; Sweeney et al. 1994), and similarly, the addition of a negatively charged phosphate within the cMyBP-C phosphorylation motif would be expected to disrupt or alter the interaction between the N-terminal C1–C2 region of cMyBP-C and myosin subfragment (S2), and thereby allow cross-bridges to move towards actin (Hofmann et al. 1991; Gautel et al. 1995; Kunst et al. 2000). Changes in myosin head proximity to actin by either phosphorylation would be expected to influence the rate of cross-bridge cycling, as the disposition of cross-bridge mass during the early phase of contraction is predictive of the rate of force development (Matsubara, 1980; Pearson et al. 2007). While there are several targets of PKA, we have recently shown that the accelerated rate of force development in myocardium following PKA-mediated phosphorylation (Stelzer et al. 2006a) is probably due to a movement of myosin heads away from the thick filament backbone and towards myosin binding sites on actin induced by phosphorylation of cMyBP-C but not cTnI or titin (Colson et al. 2008) since these structural and contractile effects were not observed in myocardium from a cMyBP-C−/− null mouse model (Harris et al. 2002). The effect of PKA to decrease the Ca2+ sensitivity of force has been primarily attributed to cTnI phosphorylation, which causes a faster dissociation of Ca2+ from troponin C (Robertson et al. 1982; Solaro, 1995; Zhang et al. 1995; Chandra et al. 1997). Electron microscopy studies of isolated thick filaments have shown that RLC phosphorylation also moves cross-bridges away from the thick filament backbone (Sweeney et al. 1994; Levine et al. 1996), thereby increasing the rate of myocardial force development (Stelzer et al. 2006b); however, such movement of cross-bridges due to RLC phosphorylation have yet to be observed in intact or skinned cardiac muscle. In the absence of the thick filament backbone, the unloaded shortening velocity of myosin decreases by 20% upon RLC phosphorylation, which suggests that phosphorylation also leads to an increase in the myosin duty cycle (Greenberg et al. 2009).
Following from this background, we predicted that phosphorylation of either RLC or cMyBP-C results in a similar displacement of cross-bridges away from the thick filament backbone to increase the probability of cross-bridge binding to actin. However, it seems likely that there are also differences in the molecular structure of myosin cross-bridges following phosphorylation which account for the increase in Ca2+ sensitivity of force only when RLC is phosphorylated. To investigate these possibilities, we used synchrotron low-angle X-ray diffraction to assess the disposition of myosin cross-bridges by the ratio of the 1,1 to the 1,0 equatorial reflections (I1,1/I1,0) and the interfilament spacing between neighbouring thick filaments in wild-type (WT) myocardium in response to treatment with myosin light chain kinase (MLCK) or PKA. In addition, the effects of the kinase treatments on the rate of force development (ktr, the rate constant of force redevelopment) and the Ca2+ sensitivity of force were examined using skinned myocardium under identical activating conditions.
Methods
Experimental animals
Mice (70; SV/129-strain; 6 to 12 months old) of either sex were obtained from Taconic Farms (Germantown, NY, USA). All procedures involving animal care and handling was conducted in accordance with institutional guidelines using protocols approved by the Animal Care and Use Committee of the University of Wisconsin School of Medicine and Public Health. We have read the detailed explanation of The Journal of Physiology policy and relevant UK regulations regarding animal experimentation as described in Drummond (2009) and our procedures are in compliance with the policies and regulations described in that article.
Experimental solutions
Experimental solution compositions were calculated using the computer program of Fabiato (1988) and the stability constants listed by Godt & Lindley (1982) corrected to pH 7.0 and 22°C. Unless otherwise stated, all solutions contained (mm): 100 N,N-bis[2-hydroxyethyl]-2aminoethanesulfonic acid (Bes), 15 creatine phosphate, 5 dithiothreitol (DTT), 1 free Mg2+ and 4 MgATP. In addition, (a) pCa 9.0 solution contained 7 EGTA, and 0.02 CaCl2, (b) pCa 4.5 solution contained 7 EGTA, and 7.01 CaCl2, and (c) pre-activating solution contained 0.07 EGTA. Ionic strength of all solutions was adjusted to 180 mm using potassium propionate. A range of solutions containing different [Ca2+]free were prepared by mixing pCa 9.0 and pCa 4.5 solutions. The catalytic subunit of bovine smooth muscle myosin light chain kinase (MLCK) was prepared as described previously (Nagamoto & Yagi, 1984) and stored at −80°C until used in an experiment. The catalytic subunit of bovine protein kinase A (PKA) was purchased from Sigma (no. P2645).
Skinned myocardial preparations
Adult mice were injected intraperitoneally with 5000 units heparin (kg body weight−1) and after 5 min were anaesthetized by inhalation of isofluorane. After deep anaesthesia was established, mice were killed by creating a pneumothorax. Hearts were rapidly excised and rinsed of blood in a Petri dish containing Ringer's solution (mm: 120 NaCl, 19 NaHCO3, 1.2 Na2HPO4, 1.2 MgSO4, 5 KCl, 1 CaCl2 and 10 glucose; pH 7.4; 22°C) pre-equilibrated with 95% O2–5% CO2.
To isolate trabeculae (800–1400 μm × 100–250 μm) for use in X-ray experiments, a heart was pinned to the floor of a dissecting dish containing Sylgard, and incisions were made through the posterior wall of the right ventricle to directly expose trabeculae to the perfusion of fresh Ringer's solution containing 30 mm 2,3-butanedione monoxime (BDM) (two solution changes) (Olsson et al. 2004). BDM acts as a chemical phosphatase in myocardium to dephosphorylate RLC to a uniformly low level. After 30 min, unbranched trabeculae that extended from the free wall of the right ventricle to the atrioventricular ring, and whose midsections were free from attachment to walls, were dissected from the right ventricle, and secured to wooden dowels using two loops of 8-0 suture. Trabeculae were then bathed for 12–16 h in ice-cold relaxing solution (mm: 100 KCl, 10 imidazole, 5 MgCl2, 2 EGTA and 4 ATP; pH 7.0) containing 250 μg ml−1 saponin and 1% (v/v) Triton X-100, and washed in fresh ice-cold relaxing solution for 1 h. Skinned preparations were then stored at −20°C for up to 4 days in fresh relaxing solution containing 50% (v/v) glycerol.
To obtain multicellular myocardial preparations (600–900 μm × 100–250 μm) for use in mechanical experiments, hearts were perfused with fresh Ringer's solution containing 30 mm BDM, and myocardial preparations were then prepared by homogenization and skinned as described earlier (Patel et al. 2001).
X-ray diffraction experiments and data analysis
Trabeculae were mounted in a simple X-ray chamber (Farman et al. 2006) and sarcomere length (SL) was set to ∼2.15 μm as described previously (Colson et al. 2008). X-ray measurements were made under relaxed conditions (pCa 9.0) to avoid possible confounding effects due to attachment of cross-bridges to thin filaments in activated muscle. X-ray experiments were performed at 22°C using the small-angle instrument on the BioCAT undulator-based beamline 18-D at the Advanced Photon Source, Argonne National Laboratory (Irving et al. 2000; Colson et al. 2007). The high X-ray flux density and low beam divergence of this instrument are highly advantageous for small-angle X-ray studies of small specimens such as mouse trabeculae (Irving et al. 2000; Colson et al. 2008).
Diffraction patterns were collected on a CCD-based X-ray detector (PCCD 168080, Aviex L.L.C., Napierville, IL, USA) and the spacings of the 1,0 and 1,1 equatorial reflections were converted to d1,0 lattice spacings using Bragg's Law, as described previously (Irving et al. 2000; Colson et al. 2008). d1,0 lattice spacing was then multiplied by 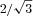 to yield the centre-to-centre distance between thick filaments, i.e. the inter-thick filament spacing (IFS). Intensities of the 1,0 and 1,1 equatorial reflections were determined from one-dimensional projections along the equator and analysed independently by three people and the results averaged (Irving & Millman, 1989). I1,1/I1,0 intensity ratios can be used to estimate shifts of mass (presumably cross-bridges) from the region of the thick filament to region of the thin filament.
to yield the centre-to-centre distance between thick filaments, i.e. the inter-thick filament spacing (IFS). Intensities of the 1,0 and 1,1 equatorial reflections were determined from one-dimensional projections along the equator and analysed independently by three people and the results averaged (Irving & Millman, 1989). I1,1/I1,0 intensity ratios can be used to estimate shifts of mass (presumably cross-bridges) from the region of the thick filament to region of the thin filament.
Mechanical experiments and data analysis
Skinned preparations were transferred from the Petri dish to a stainless-steel experimental chamber (Moss et al. 1983) containing relaxing solution. The ends of each of the preparations were attached to the arms of a motor (model 350; Cambridge Technology, Cambridge, MA, USA) and force transducer (model 403; Cambridge Technology). The chamber assembly was then placed on the stage of an inverted microscope (Olympus) as described earlier (Patel et al. 2001). Bitmap images of the preparations were acquired using an AGP 4X/2X graphics card and associated software (ATI Technologies) and were used to assess mean sarcomere length (SL) during the course of each experiment. Changes in force and motor position were sampled (16-bit resolution, DAP5216a, Microstar Laboratories) at 2.0 kHz using SLControl software developed in this laboratory (http://www.slcontrol.com). Data were saved to computer files for later analysis. At the start of each experiment, the skinned myocardial preparations were stretched to a mean sarcomere length of ∼2.2 μm. The protocol for simultaneous determination of Ca2+-activated force and ktr was a modification of the multistep protocol developed by Brenner & Eisenberg (1986), as described previously (Chase et al. 1994; Patel et al. 2001). Briefly, measurement of ktr involves a mechanical slack–restretch manoeuver to detach bound myosin cross-bridges from actin in steadily Ca2+-activated skinned myocardium. Each skinned preparation was transferred from relaxing to activating solutions of varying free Ca2+ (i.e. pCa 6.2–4.5) and allowed to generate steady-state force. The myocardial preparation was rapidly (<2 ms) slackened by 20% of its original length, resulting in a rapid reduction of force to near zero (i.e. <5% of steady isometric force). This was followed by a brief period of unloaded shortening (i.e. 20 ms) after which the preparation was rapidly restretched to its original length. Force development following the slack–restretch manoeuver and force recovery to the original steady-state value reflect the rate of myosin cross-bridge cycling between weakly bound and strongly bound force generating states (Brenner & Eisenberg, 1986).
Kinase treatments
MLCK
The skinned preparations were incubated (∼22°C) in pre-activating solution first containing 5 mm BDM for 10 min, and then containing 5 mm BDM, 67 μm CaCl2, 12 μm calmodulin and 0.62 μm MLCK for 30 min, and finally in pCa 9.0 solution (30 min, 3 solution changes).
PKA
The skinned preparations were first incubated for 1 h (∼22°C) in a solution of pCa 9.0 with 1 U PKA μl−1, and then in pCa 9.0 solution (30 min, 3 solution changes).
Interfilament lattice spacing and the equatorial intensity ratio or steady-state isometric force and ktr were then measured as described above.
Phosphoprotein staining
At the conclusion of each X-ray or mechanical experiment, the myocardial preparations were cut free at the points of t-clip or suture attachment and placed in Rehydration/Sample buffer (Bio-Rad; catalogue no. 163-2106). The samples were then stored at −80°C until subsequent analysis of the phosphorylation state of RLC by two-dimensional gel electrophoresis using pre-cast immobilized pH gradient (IPG) polyacrylamide gel strips (Bio-Rad) as previously described (Gorg et al. 2000). Briefly, the samples were loaded into the iso-electric focusing (IEF) gel strips in the first dimension (pH 4–7) containing 6 mm urea, 4% acrylamide–bisacrylamide (30% acrylamide–bisacrylamide solution, Bio-Rad), 67 mm DTT and 0.2% ampholyte (Bio-Rad; catalogue no. 163-2094), and were electrofocused for at least 50,000 V h. For the second dimension, each IPG strip was equilibrated in SDS buffer, placed onto a 10.5–14% Tris-HCl Criterion gel (Bio-Rad) and electrophoresed until the tracking dye ran off the gel. Gels were incubated in Sypro-Ruby (Molecular Probes; S-12000; total protein stain) protein stain to detect myofibrillar proteins, as described previously (Colson et al. 2008). Per cent RLC phosphorylation (phosphorylated protein in the phosphorylated RLC band as a per cent of total protein in the unphosphorylated and phosphorylated RLC bands) was quantified using the UVP BioImaging System and LaserPix software (Bio-Rad).
Both the phosphoproteins and myofibrillar proteins in untreated and PKA-treated wild-type (WT) myocardium were examined using the method described previously (Colson et al. 2008), with minor modifications. Pro-Q Diamond (Molecular Probes; P-33300; phosphoprotein stain) and Sypro-Ruby were used to compare the PKA-treated phosphorylation levels (PL) of cMyBP-C and cTnI. Pro-Q Diamond intensity as a fraction of Sypro-Ruby intensity was used to compare the PL of cMyBP-C and cTnI in PKA-treated myocardium relative to control, e.g. PL(WT+PKA/WTcontrol) =[Pro-Q intensity(WT+PKA)/Sypro intensity(WT+PKA)]/[Pro-Q intensity(WTcontrol)/Sypro intensity(WTcontrol)] (Tong et al. 2008).
Statistics
Data are expressed as means ±s.e.m. An unpaired t test was used as a test of significance. P values < 0.05 were taken as indicating significant differences.
Results
MLCK treatment
Assessment of RLC phosphorylation
The preparations used in this study were isolated from murine hearts that were first incubated in the presence of BDM in order to dephosphorylate RLC and thereby establish a uniformly low baseline level of RLC phosphorylation prior to X-ray and mechanical measurements. Two-dimensional (SDS–PAGE vs. IEF) gel analysis of skinned myocardium showed that BDM treatment resulted in nearly undetectable RLC phosphorylation and that subsequent treatment with MLCK resulted in significant phosphorylation of RLC (Fig. 1A). Ratiometic analysis of densitometric scans of the two RLC spots in the gels indicated that 40 ± 4% of total RLC was phosphorylated (RLC-P/(RLC + RLC-P)), which is consistent with physiological levels in vivo (Holroyde et al. 1979; Morano, 1999) and with levels due to kinase treatment of skinned myocardium (Sanbe et al. 1999; Stelzer et al. 2006b) reported previously.
Figure 1.

Phosphate incorporation into myofibrillar proteins of skinned murine myocardium following treatment with MLCK or PKA A, SYPRO Ruby-stained two-dimensional SDS–PAGE/IEF gels were used to determine percentage phosphorylation of RLC in skinned myocardium, as shown in these representative gels. The gel in the top panel shows that 2,3-butanedione monoxime (BDM) resulted in little or no detectable phosphorylation of RLC (one spot; RLC), while the gel in the bottom panel shows that subsequent treatment with MLCK resulted in significant phosphorylation of RLC (two spots; RLC and RLC-P). Densitometric scans of the two RLC spots in the gels indicated that ∼40% of total RLC was phosphorylated (RLC-P), consistent with in vivo levels reported previously (Holroyde et al. 1979). When the 2nd dimension was run, a myofibrillar preparation was run in an adjacent lane as a molecular weight standard for RLC. B, SYPRO Ruby- (SR) and Pro-Q Diamond- (PQ) stained gels were used to determine levels of myofibrillar protein phosphorylation with (+) and without (−) PKA treatment, as shown in this representative 10% SDS–PAGE: SR, stained gel for relative abundance of proteins; PQ, stained gel specific for relative abundance of phosphorylated proteins. PKA induced significant incorporation of phosphate in cTnI and cMyBP-C in myocardium. Cardiac troponin T (cTnT), cardiac troponin I (cTnI), myosin essential light chain (ELC) and RLC are also labelled.
Effects of MLCK treatment of murine myocardium on steady-state mechanical properties
In murine myocardium, MLCK treatment significantly increased both the resting force (1.15 ± 0.12 vs. 0.57 ± 0.06 mN mm−2) and maximal Ca2+-activated force (21.82 ± 1.84 vs. 13.28 ± 1.26 mN mm−2) (Table 1). The force–pCa relationships from MLCK-treated myocardium were to the left of those from untreated myocardium (Fig. 2A). Thus, MLCK treatment significantly increased the Ca2+ sensitivity of force, i.e. the pCa50 was significantly higher (pCa 5.92 ± 0.02 vs. 5.81 ± 0.01), and the steepness of the force–pCa relationship was markedly reduced (nH: 3.20 ± 0.11 vs. 4.09 ± 0.07) (Table 1).
Table 1.
Effects of MLCK and PKA treatment on mechanical properties of WT skinned myocardium
| Resting force | Maximum Ca2+- activated force | Hill coefficient (nH) | Ca2+-sensitivity of force (pCa50) | Maximum rate of force redevelopment | |
|---|---|---|---|---|---|
| Treatment | (mN mm−2) | (mN mm−2) | (s−1) | ||
| Control (n= 9) | 0.57 ± 0.06 | 13.29 ± 1.26 | 4.09 ± 0.07 | 5.81 ± 0.01 | 26.07 ± 0.95 |
| + MLCK (n= 11) | 1.15 ± 0.12* | 21.82 ± 1.84* | 3.20 ± 0.11* | 5.92 ± 0.02* | 38.85 ± 1.17* |
| + PKA (n= 9) | 0.57 ± 0.08 | 12.95 ± 1.57 | 4.07 ± 0.06 | 5.69 ± 0.01* | 27.69 ± 1.14 |
Data are means ±s.e.m. Resting force was measured at pCa 9.0. Maximum force and the rate constant of force redevelopment (ktr) were measured at pCa 4.5. pCa50 and nH values were derived by fitting the force–pCa relationships with the Hill equation,as described in Methods. *Significant differences (P < 0.05) between values recorded in skinned myocardial preparations that were untreated (control) or treated with either MLCK or PKA.
Figure 2.

Effects of MLCK and PKA treatment on Ca2+ sensitivity of force and apparent cooperativity in murine skinned myocardium Force–pCa relationships were measured in skinned myocardium that was untreated (control; filled circles) or treated with either MLCK (filled upward triangles) or PKA (filled downward triangles). Fitting the mean data with the Hill equation yielded: pCa50= 5.81 and nH= 4.09 for control (continuous line), pCa50= 5.92 and nH= 3.20 for MLCK-treated (dashed line) and pCa50= 5.69 and nH= 3.96 for PKA-treated (dotted line) myocardium. B, ktr–pCa relationships in murine skinned myocardium. A ktr–pCa relationship was obtained by initially activating the skinned myocardium in solution of pCa 4.5, and then in a series of submaximally activating solutions between pCa 6.2 and 5.4. To assess any decline in the maximal rate of force development, the preparation was activated with solution of pCa 4.5 at the end of each experimental protocol, which was less than 10%. The reference value of maximal ktr for each activation was obtained by interpolation between the initial and final measurements of maximal ktr. The apparent rate constants of force redevelopment (ktr) were estimated by linear transformation of the half-time of force redevelopment, i.e. ktr= 0.693/t1/2, as described previously (Regnier 1998; Fitzsimons 2001b). C, ktr–relative force relationships in murine skinned myocardium. Data are shown with force expressed relative to the maximum force for each condition. ktr–normalized force relationships were measured in control (filled circles), MLCK-treated (filled upward triangles) and PKA-treated (filled downward triangles) preparations. Relative force was calculated by normalizing force values at each pCa to the maximum force value in solution of pCa 4.5 for each of the unpaired treatment groups.
Control and MLCK-treated myocardium exhibited activation-dependent (Fig. 2B–C) changes in the rate of force redevelopment (ktr). MLCK treatment caused the myocardium to redevelop both sub-maximal (Fig. 2B–C) and maximum forces (38.85 ± 1.17 vs. 26.07 ± 0.95 s−1; Fig. 2B–C; Table 1) at significantly faster rates. This suggests that MLCK treatment speeds cross-bridge cycling kinetics at all levels of activation in myocardium and may accelerate the cooperative recruitment of cross-bridges to force-generating states.
Effects of MLCK treatment of WT myocardium on the distribution of cross-bridge mass between the thick and thin filaments and on interfilament spacing
Intensity ratios, I1,1/I1,0, determined from the 1,0 and 1,1 equatorial reflections in diffraction patterns from WT mouse trabeculae (Fig. 3) were found to be significantly greater (P < 0.03) in trabeculae treated with MLCK (0.33 ± 0.03, n= 11) than in untreated trabeculae (0.23 ± 0.02, n= 11) (Table 2). These results suggest that in resting myocardium MLCK causes a net transfer of cross-bridge mass from the region of the thick filament towards the thin filaments, such that the average cross-bridge position is further away from the surface of the thick filament and closer to the thin filament. It should be noted that, in principle, the equatorial intensities can also be used as indicators of changes in myosin head shape (Lymn, 1978).
Figure 3.

Effects of MLCK and PKA treatments on intensities and spacings of 1,0 and 1,1 equatorial peaks in WT skinned myocardium A, X-ray diffraction patterns from skinned myocardium that was untreated (control; top panel) or treated with either MLCK (middle panel) or PKA (bottom panel). The ratio of intensities of the 1,0 and 1,1 equatorial reflections can be used to estimate shifts of cross-bridge mass from the region of the thick filament to the region of the thin filament. B, representative intensity traces along the equator of X-ray patterns of skinned myocardium that was untreated (control; top panel) or treated with either MLCK (middle panel) or PKA (bottom panel). The pixel intensity along the y-axis is labelled from 0 to 1000 arbitrary units and the peak profiles were not otherwise modified.
Table 2.
Summary data from peak intensity analyses of X-ray equatorial patterns from murine skinned myocardium
| I1,1/I1,0 | d1,0 | IFS | |
|---|---|---|---|
| Treatment | (nm) | (nm) | |
| Control (n= 11) | 0.23 ± 0.02 | 46.0 ± 0.3 | 53.1 ± 0.3 |
| + MLCK (n= 11) | 0.33 ± 0.03* | 44.4 ± 0.3* | 51.3 ± 0.3* |
| + PKA (n= 17) | 0.33 ± 0.03* | 46.1 ± 0.1 | 53.2 ± 0.1 |
Data are means ±s.e.m.I1,1/I1,0 is the ratio of the intensities of the 1,0 and 1,1 X-ray equatorial reflections. The distance between the 1,0 and 1,1 reflections were converted to d1,0 lattice spacings using Bragg's law. Inter-thick filament spacing (IFS) was calculated by multiplying the lattice spacing, d1,0, by 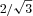 (Irving & Millman, 1989). *Significant differences (P < 0.05) between WT skinned myocardial preparations that were untreated (control) or treated with either MLCK or PKA. I1,1/I1,0 and d1,0 data from PKA-treated myocardium were previously published elsewhere (Colson et al. 2008); however, the diffraction patterns were collected during the same experimental sessions as control and MLCK-treated myocardium.
(Irving & Millman, 1989). *Significant differences (P < 0.05) between WT skinned myocardial preparations that were untreated (control) or treated with either MLCK or PKA. I1,1/I1,0 and d1,0 data from PKA-treated myocardium were previously published elsewhere (Colson et al. 2008); however, the diffraction patterns were collected during the same experimental sessions as control and MLCK-treated myocardium.
The separation of the 1,0 and 1,1 equatorial reflections in diffraction patterns was converted to d1,0 lattice spacings to determine the effects of MLCK treatment on the interfilament spacing (IFS). Compared to control, MLCK treatment significantly decreased the d1,0 lattice spacing (46.0 ± 0.3 nm, n= 11, vs. 44.4 ± 0.3 nm, n= 11; P < 0.002) (Table 2) in resting myocardium, suggesting that the average distance between neighbouring thick filaments is reduced by RLC phosphorylation.
PKA treatment
Assessment of cMyBP-C and cTnI phosphorylation
Gel analysis of protein phosphorylation using SYPRO-Ruby and Pro-Q Diamond staining showed that PKA treatment of myocardium phosphorylated cMyBP-C, which increased from a baseline arbitrarily set to PL(control/control) = 1 to PL(control/control)) = 1.7 ± 0.2 with PKA treatment, and cTnI, which increased from a baseline arbitrarily set to PL(control/control) = 1 to PL(control/control) = 2.8 ± 0.2 with PKA treatment (Fig. 1B), similar to results reported previously (Colson et al. 2008).
Effects of PKA treatment of murine myocardium on steady-state mechanical properties
Although PKA treatment had no significant effect on resting force, maximum force or the steepness of the force–pCa relationship in murine myocardium (Table 1), PKA treatment decreased the Ca2+ sensitivity of force (pCa50= pCa 5.69 ± 0.01 vs. 5.81 ± 0.01; Fig. 2A). PKA-treated myocardium also exhibited activation-dependent (Fig. 3B) changes in the rate of force redevelopment (ktr). PKA treatment increased rates of force development at sub-maximal (Fig. 2C) but not maximum forces (27.69 ± 1.14 vs. 26.07 ± 0.95 s−1; Fig. 2C; Table 1). This suggests that PKA treatment accelerates cross-bridge cycling kinetics at sub-maximal but not maximal activation in WT myocardium.
Effects of PKA treatment of murine myocardium on the distribution of cross-bridge mass between the thick and thin filaments and on interfilament spacing
I1,1/I1,0 ratios were found to be significantly greater (P < 0.03) in trabeculae treated with PKA (0.33 ± 0.03, n= 17) than in untreated trabeculae (0.23 ± 0.02, n= 11) (Table 2), consistent with previous results (Colson et al. 2008). These results suggest that in resting myocardium, PKA treatment causes a net transfer of cross-bridge mass from the region of the thick filament towards the thin filaments, such that the average cross-bridge position is further away from the surface of the thick filament and closer to the thin filament. PKA treatment had no effect (46.1 ± 0.1 nm, n= 17 vs. 46.0 ± 0.3 nm, n= 11) (Table 2) on d1,0 lattice spacings, as we reported previously (Colson et al. 2008), suggesting that in resting myocardium PKA treatment (which phosphorylates cMyBP-C as well as cTnI) has no net effect on lattice spacing. The intensity ratios and lattice spacings from PKA-treated myocardium were reported elsewhere previously (Colson et al. 2008). Diffraction patterns from control myocardium were collected during the experimental sessions for both MLCK- and PKA-treatments, and the I1,1/I1,0 and d1,0 lattice spacings from both control data sets were averaged, and results from the subsequent sessions were not different (I1,1/I1,0: 0.23 ± 0.03; d1,0: 46 ± 1 nm) (Colson et al. 2008).
Discussion
Short-term changes in myocardial work capacity can be accomplished by phosphorylation of several key myofilament proteins, including RLC, cMyBP-C and cTnI. Changes in the phosphorylation levels can affect the disposition of myosin cross-bridges and the distance between neighbouring myofilaments, which manifests as altered mechanical performance of the myocardium. We designed the present study to examine the structural effects and corresponding mechanical responses of MLCK and PKA phosphorylations using virtually identical conditions (i.e. SL, temperature, solutions) and treatments (e.g. isolation, skinning, kinases) in both X-ray diffraction and mechanical experiments. We collected data from control, MLCK-treated and PKA-treated preparations during the same experimental sessions in order to facilitate unambiguous comparisons and interpretations of our findings. Importantly, since both RLC and cMyBP-C phosphorylation is thought to modulate cross-bridge disposition, we took care to reduce basal RLC phosphorylation levels to near zero and thereby establish a uniformly low level of RLC phosphorylation.
Effects of MLCK and PKA treatment of WT myocardium on myofilament structure
Our studies provide the first direct evidence that phosphorylated RLC and cMyBP-C have similar effects on the structural and mechanical properties of murine skinned myocardium. The primary result of this study is that following either RLC or MyBP-C phosphorylation, myosin cross-bridges assume positions further from the surface of the thick filament backbone and closer to the thin filament. This conclusion follows from the observation that the I1,1/I1,0 ratio was significantly greater in myocardium treated with MLCK or PKA (0.33 ± 0.03, either treatment) than in untreated myocardium (0.23 ± 0.02) (Table 2), and treatment with either MLCK or PKA accelerated the rate of force redevelopment at a given level of submaximal force (Fig. 2C). This finding suggests that both RLC and cMyBP-C regulate the proximity and interaction of myosin with actin, and their actions are modulated by altering the phosphorylation status of RLC and cMyBP-C. A closer proximity of myosin to actin following phosphorylation of RLC or cMyBP-C may be expected to increase the probability of cross-bridges binding and thereby reduce the time taken for cooperative recruitment of cross-bridges, which could explain the accelerated kinetics of cross-bridge cycling (ktr, Fig. 2B–C; kdf as reported by Stelzer et al. 2006b,c;, where kdf is the rate constant of delayed force development in response to stretch (for reference, see Steiger 1977; Lombardi et al. 1995; Davis & Rodgers 1995)).
The distance between neighbouring thick and thin filaments is widely assumed to be a major determinant of the Ca2+ sensitivity of force, and in situ studies of murine beating hearts suggest that interfilament spacing at end-diastole plays a direct role in modulating the number of cross-bridges that can form attachments at the beginning of the next contraction (Pearson et al. 2007). Treatment with MLCK or PKA increases I1,1/I1,0 to strikingly similar degrees, but only MLCK treatment reduces the lateral spacing of filaments within the lattice. Our novel finding that RLC phosphorylation reduces IFS while phosphorylation of cTnI and cMyBP-C has no net effect on IFS (Table 2) correlates with the observation that RLC phosphorylation significantly increases Ca2+ sensitivity of force in myocardium but PKA phosphorylation decreases the Ca2+ sensitivity of force, primarily because phosphorylated cTnI has reduced Ca2+-binding affinity (Table 1; Fig. 2A; Kranias & Solaro, 1983; Stelzer et al. 2006b,c;). Our observations of structural effects due to RLC or cMyBP-C phosphorylation in myocardium suggest that the movement of thick filaments towards the thin filaments and the movement of cross-bridge mass from the thick filament backbone towards the thin filaments could position cross-bridges in unique orientations that have distinctly different effects on the probabilities of attachment and detachment. Thus, while either type of movement would bring myosin heads closer to actin, different attachment angles or orientations of cross-bridges resulting from phosphorylation or changes in IFS may explain these differential effects on force development. We note that there is an expansion of the myofilament lattice upon skinning and so the extent of the effects on lattice spacing in intact myocardium would probably be less pronounced but qualitatively similar to the effects of RLC phosphorylation on lattice spacing in skinned myocardium presented here. In support of this notion, the extent of the changes in Ca2+ sensitivity of force was to a lesser degree with RLC phosphorylation in intact myocardium (Stull et al. 2002) as compared to skinned myocardium (Stelzer et al. 2006b) and to a lesser degree with PKA phosphorylation at longer sarcomere lengths in intact myocardium (Komukai & Kurihara, 1997).
Effects of MLCK and PKA treatment of WT myocardium on mechanical properties
Our mechanical measurements were made at levels of RLC phosphorylation ranging from <10% in control myocardium to ∼40% in myocardium treated with MLCK, which is near the levels reported in vivo (Holroyde et al. 1979; Metzger et al. 1989). Recently, Scruggs et al. (2009) assessed effects due to similar levels of RLC phosphorylation using transgenic mice (∼15% due to expression of non-phosphorylatable RLC in transgenic mice and ∼50% in basally phosphorylated control mice). Similar increases in maximum force were observed with increased RLC phosphorylation as compared to our measurements following MLCK treatment of myocardium, but in contrast to the leftward shift in the force–pCa relationship following RLC phosphorylation observed here and in numerous studies in both cardiac and skeletal muscle (Fig. 2A; Persechini et al. 1985; Sweeney & Stull, 1986, 1990; Olsson et al. 2004; Stelzer et al. 2006b) these investigators observed no change in the Ca2+ sensitivity of force. We would expect the force–pCa relationship in the non-phosphorylatable RLC transgenic myocardium to be shifted to the right of the control myocardium, but these authors also observed that baseline phosphorylations of cMyBP-C and cTnI were reduced, which could counteract any tendency toward a rightward shift in the force–pCa relationship due to reduced RLC phosphorylation.
In the present study, RLC phosphorylation increased both resting force at pCa 9.0 (by ∼100%) and maximal force at pCa 4.5 (by ∼60%), while decreasing the overall steepness of the force–pCa relationship, which indicates that there is an increase in cross-bridge binding both in the absence and presence of Ca2+. The increase in resting force in the absence of Ca2+ is probably due to the thin filament-activating effects of strong binding cross-bridges, which are greater in cardiac than skeletal muscle (Fitzsimons et al. 2001a). The rate constant of force development, ktr, also increased with RLC phosphorylation at both maximal (by ∼50%) and submaximal Ca2+-activated forces (Fig. 2B–C). Furthermore, since RLC phosphorylation increases the Ca2+ sensitivity of force, i.e. increases the number of cross-bridges bound at a given level of Ca2+ activation, at each level of activation there are fewer cross-bridges available for recruitment, which could explain why RLC phosphorylation speeds contraction to an even greater degree than cMyBP-C phosphorylation. The increases in maximal force and maximal ktr due to RLC phosphorylation indicate that additional cross-bridges are bound (Fitzsimons et al. 2001b), which could accelerate cooperative recruitment of cross-bridges to force-generating states. The bi-directional spread of cooperative activation elicited by RLC phosphorylation is greater than cMyBP-C phosphorylation, which might be expected since RLC is bound to every cross-bridge, whereas cMyBP-C only interacts with heads in every third crown in the C-zone (Zoghbi et al. 2008). It is likely that the reduction in lattice spacing only with RLC phosphorylation, but not with PKA-mediated phosphorylation of cMyBP-C and cTnI, contributes to the increases in maximal force and maximal ktr, which are similarly observed only with RLC phosphorylation but not with PKA phosphorylation.
Roles of RLC and cMyBP-C phosphorylation in the contractile properties of myocardium
In the context of a simple two-state model of cross-bridge cycling (Huxley, 1957) in which ktr is the sum of the forward (fapp) and reverse (gapp) rate constants of cross-bridge attachment (Brenner & Eisenberg, 1986), the movement of myosin heads away from the thick filament backbone and towards the thin filament following RLC and cMyBP-C phosphorylation would be expected to increase the likelihood of cross-bridge binding, thereby increasing fapp. Conversely, the reduction in lattice spacing observed only with RLC phosphorylation could contribute to a decreased gapp. This combination would allow for concomitant increases in the rate of force development (due to increased fapp) and the Ca2+ sensitivity of force (due to decreased gapp) by RLC phosphorylation. Recent studies using stretch activation of murine myocardium by Stelzer et al. (2006a,b); are consistent with this idea, in that kdf increases with MLCK and PKA treatments, which suggests that fapp is increased, whereas krel is decreased with MLCK treatment and increased with PKA treatment, which also suggests that gapp is reduced with MLCK treatment and increased with PKA treatment. It is also possible that phosphorylation of RLC and cMyBP-C differentially affects cross-bridge stiffness, which could also impact on attachment and detachment rates. Since cMyBP-C and cTnI are co-phosphorylated by PKA, their individual effects on myocardial structure and mechanical responses are not readily dissociated. Following PKA treatment, the rightward shift in the force–pCa relationship has been attributed primarily to cTnI phosphorylation; however, it is unresolved whether PKA phosphorylation of cMyBP-C in the absence of phosphorylated cTnI increases or decreases Ca2+ sensitivity. To resolve the differential effects of cMyBP-C and cTnI in PKA phosphorylation, future studies will be directed to examine changes in the Ca2+ sensitivity of force and the kinetics of cross-bridge cycling when only cMyBP-C is phosphorylated (and RLC phosphorylation is reduced to a uniform baseline level) using transgenic myocardium lacking phosphorylatable PKA sites in cTnI (Pi, 2002 & Pi, 2003). Recent studies by Stelzer et al. (2007) are consistent with the idea that phosphorylated cMyBP-C accelerates the rates of cross-bridge cycling, which may reduce the time cross-bridges spend in force-generating states, and thereby could contribute to reduce the Ca2+ sensitivity of force.
Increased myocardial work capacity increases stroke volume during ejection and thereby increases cardiac output. The amount of work produced during each heart beat is directly influenced by the rates of force development and relaxation, which are finely tuned by the phosphorylation status of myofibrillar proteins, including RLC, cMyBP-C and cTnI. In this context, phosphorylation of RLC or cMyBP-C would be expected to increase the rate of rise of ventricular pressure during isovolumic contraction, which could increase the fraction of time in systole spent in ejection of blood. Further, during β-adrenergic stimulation, phosphorylation of cMyBP-C and cTnI would speed the relaxation of ventricular pressure and thereby preserve filling time at higher heart rates. In contrast, RLC phosphorylation would be expected to slow relaxation and thereby contribute to stronger, longer duration contractions. Efficient myocardial pump function is dependent on the precise coordination and timing of regional mechanical activation (Markhasin et al. 2003). Thus, an organized regional heterogeneity of RLC phosphorylation levels in the heart could contribute to synchronizing myocardium at the multi-cellular level, whereas the effects of cMyBP-C and cTnI in myocardium during β-adrenergic stimulation would tend to override the contributions of phosphorylated RLC to basal contractility.
Our conclusion that RLC and cMyBP-C appear to be important regulators of contraction reinforces similar conclusions from earlier studies in which mutations in either RLC or cMyBP-C were linked to inherited hypertrophic cardiomyopathies (Watkins et al. 1995; Poetter et al. 1996; Dhandapany et al. 2009). Our findings are also consistent with the idea that altered phosphorylation of RLC and/or cMyBP-C can contribute to compensatory mechanisms and ultimate contractile dysfunction in heritable or acquired myocardial disease in human (van der Velden et al. 2003; El-Armouche et al. 2007) and in mouse models (Sadayappan et al. 2005; Tong et al. 2008; Scruggs et al. 2009). The effects of phosphorylation on contractile properties would be more profound during pathological conditions (van der Velden et al. 2006; Hamdani et al. 2008; Kooij et al. 2009).
In summary, we show that the disposition of myosin cross-bridges and the lateral spacing between myofilaments plays an important role in modulating the strength and speed of myocardial contraction due to MLCK-mediated phosphorylation of RLC and PKA-mediated phosphorylation of cMyBP-C and cTnI. In skinned myocardium, RLC phosphorylation increases the proximity of cross-bridges to the thin filament and also reduces the interfilament spacing, which contributes to an acceleration of the kinetics of cross-bridge cycling, an increase in the amplitude of force at all levels of activation and an increase in the Ca2+ sensitivity of force by increasing the probability of interaction and/or the transition to force-generating states. PKA phosphorylation of cMyBP-C also increases the proximity of cross-bridges to actin but when both cMyBP-C and cTnI are phosphorylated there is no net effect on interfilament spacing. Thus, in modulation of cardiac performance, phosphorylated RLC defines the basal tuning of contractile parameters (Olsson et al. 2004) and phosphorylated cTnI and cMyBP-C define contractile parameters during the β-adrenergic response (Stelzer et al. 2007).
Acknowledgments
This work was supported by an American Heart Association predoctoral fellowship (B.A.C.) and by NIH HL-R37-82900 (R.L.M.). We thank Satchal K. Erramilli and Divya Srinivasa for assistance in analysis of diffraction patterns. Use of the APS was supported by the US DOE, Basic Energy Sciences, Office of Science, under contract No. W-31-109-ENG-38. BioCAT is an NIH-supported Research Center RR-08630. The content is the sole responsibility of the authors and does not necessarily reflect the official views of the NCRR or the NIH.
Glossary
Abbreviations
- BDM
2,3-butanedione monoxime
- C1
cardiac myosin binding protein-C domain 1
- C2
cardiac myosin binding protein-C domain 2
- C1–C2
the two N-terminal domains of cardiac myosin binding protein-C including the cardiac-specific phosphorylation motif
- CCD
charge-coupled device
- cMyBP-C
cardiac myosin binding protein-C
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- d1,0
lattice spacing
- ELC
myosin essential light chain
- fapp
the rate constant of cross-bridge attachment
- I1,1/I1,0
equatorial intensity ratio
- IEF
iso-electric focusing
- gapp
the rate constant of cross-bridge detachment
- IFS
interfilament spacing
- IPG
immobilized pH gradient
- kdf
the rate constant of delayed force development
- ktr
the rate constant of force redevelopment
- MHC
myosin heavy chain
- MLCK
myosin regulatory light chain
- PL
phosphorylation level
- PQ
Pro-Q Diamond
- RLC
regulatory light chain
- RLC-P
phosphorylated regulatory light chain
- S2
myosin subfragment 2
- SL
sarcomere length
- SR
Sypro-Ruby
- Std
standard
- TG
transgenic
Author contributions
Conception and design of experiments: B.A.C., J.R.P., D.P.F., T.C.I. and R.L.M. Collection, data analysis and interpretation of data: B.A.C., M.R.L., T.B., J.R.P., D.P.F., T.C.I. and R.L.M. Drafting the article or revising it critically for important intellectual content: B.A.C., J.R.P., D.P.F., T.C.I. and R.L.M.
References
- Brenner B, Eisenberg E. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc Natl Acad Sci U S A. 1986;83:3542–3546. doi: 10.1073/pnas.83.10.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki H, Sato N, Chien KR, Kasahara H. Identification of cardiac-specific myosin light chain kinase. Circ Res. 2008;102:571–580. doi: 10.1161/CIRCRESAHA.107.161687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra M, Dong WJ, Pan BS, Cheung HC, Solaro RJ. Effects of protein kinase A phosphorylation on signalling between cardiac troponin I and the N-terminal domain of cardiac troponin C. Biochemistry. 1997;36:13305–13311. doi: 10.1021/bi9710129. [DOI] [PubMed] [Google Scholar]
- Chase PB, Martyn DA, Hannon JD. Isometric force redevelopment of skinned muscle fibres from rabbit activated with and without Ca2+ Biophys J. 1994;67:1994–2001. doi: 10.1016/S0006-3495(94)80682-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson BA, Bekyarova T, Fitzsimons DP, Irving TC, Moss RL. Radial displacement of myosin cross-bridges in mouse myocardium due to ablation of myosin binding protein-C. J Mol Biol. 2007;367:36–41. doi: 10.1016/j.jmb.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson BA, Bekyarova T, Locher MR, Fitzsimons DP, Irving TC, Moss RL. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ Res. 2008;103:244–251. doi: 10.1161/CIRCRESAHA.108.178996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JS, Rodgers ME. Indirect coupling of phosphate release to de novo tension generation during muscle contraction. Proc Natl Acad Sci USA. 1995;92:10482–10486. doi: 10.1073/pnas.92.23.10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, Dobrev D, Eschenhagen T, Carrier L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- England PJ. Cardiac function and phosphorylation of contractile proteins. Philos Trans R Soc Lond B Biol Sci. 1983;302:83–90. doi: 10.1098/rstb.1983.0040. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- Farman GP, Walker JS, de Tombe PP, Irving TC. Impact of osmotic compression on sarcomere structure and myofilament calcium sensitivity of isolated rat myocardium. Am J Physiol Heart Circ Physiol. 2006;291:H1847–H1855. doi: 10.1152/ajpheart.01237.2005. [DOI] [PubMed] [Google Scholar]
- Fitzsimons DP, Patel JR, Campbell KS, Moss RL. Cooperative mechanisms in the activation dependence of the rate of force development in rabbit skinned skeletal muscle fibres. J Gen Physiol. 2001a;117:133–148. doi: 10.1085/jgp.117.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons DP, Patel JR, Moss RL. Cross-bridge interaction kinetics in rat myocardium are accelerated by strong binding of myosin to the thin filament. J Physiol. 2001b;530:263–272. doi: 10.1111/j.1469-7793.2001.0263l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst DO, Vinkemeier U, Weber K. Mammalian skeletal muscle C-protein: purification from bovine muscle, binding to titin and the characterization of a full-length human cDNA. J Cell Sci. 1992;102:769–778. doi: 10.1242/jcs.102.4.769. [DOI] [PubMed] [Google Scholar]
- Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibres of the frog. J Gen Physiol. 1982;80:279–297. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorg A, Obermaier C, Boguth G, Harder A, Scheibe B, Wildgruber R, Weiss W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 2000;21:1037–1053. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1037::AID-ELPS1037>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Greenberg MJ, Mealy TR, Watt JD, Jones M, Szczesna-Cordary D, Moore JR. The molecular effects of skeletal muscle myosin regulatory light chain phosphorylation. Am J Physiol Regul Integr Comp Physiol. 2009;297:R265–R274. doi: 10.1152/ajpregu.00171.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani N, Kooij V, van Dijk S, Merkus D, Paulus WJ, Remedios CD, Duncker DJ, Stienen GJ, Van Der Velden J. Sarcomeric dysfunction in heart failure. Cardiovasc Res. 2008;77:649–658. doi: 10.1093/cvr/cvm079. [DOI] [PubMed] [Google Scholar]
- Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- Hofmann PA, Hartzell HC, Moss RL. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibres. J Gen Physiol. 1991;97:1141–1163. doi: 10.1085/jgp.97.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holroyde MJ, Small DA, Howe E, Solaro RJ. Isolation of cardiac myofibrils and myosin light chains with in vivo levels of light chain phosphorylation. Biochim Biophys Acta. 1979;587:628–637. doi: 10.1016/0304-4165(79)90014-x. [DOI] [PubMed] [Google Scholar]
- Huxley AF. Muscle structure and theories of contraction. Prog Biophys Biophys Chem. 1957;7:255–318. [PubMed] [Google Scholar]
- Irving TC, Konhilas J, Perry D, Fischetti R, de Tombe PP. Myofilament lattice spacing as a function of sarcomere length in isolated rat myocardium. Am J Physiol Heart Circ Physiol. 2000;279:H2568–H2573. doi: 10.1152/ajpheart.2000.279.5.H2568. [DOI] [PubMed] [Google Scholar]
- Irving TC, Millman BM. Changes in thick filament structure during compression of the filament lattice in relaxed frog sartorius muscle. J Muscle Res Cell Motil. 1989;10:385–394. doi: 10.1007/BF01758435. [DOI] [PubMed] [Google Scholar]
- Komukai K, Kurihara S. Length dependence of Ca2+-tension relationship in aequorin-injected ferret papillary muscles. Am J Physiol Heart Circ Physiol. 1997;273:H1068–H1074. doi: 10.1152/ajpheart.1997.273.3.H1068. [DOI] [PubMed] [Google Scholar]
- Kooij V, Boontje N, Zaremba R, Jaquet K, Dos Remedios C, Stienen GJ, Van Der Velden J. Protein kinase C α and ɛ phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res Cardiol. 2009;105:289–300. doi: 10.1007/s00395-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranias EG, Solaro RJ. Coordination of cardiac sarcoplasmic reticulum and myofibrillar function by protein phosphorylation. Fed Proc. 1983;42:33–38. [PubMed] [Google Scholar]
- Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res. 2000;86:51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- Labeit S, Gautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. EMBO J. 1992;11:1711–1716. doi: 10.1002/j.1460-2075.1992.tb05222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine RJ, Kensler RW, Yang Z, Stull JT, Sweeney HL. Myosin light chain phosphorylation affects the structure of rabbit skeletal muscle thick filaments. Biophys J. 1996;71:898–907. doi: 10.1016/S0006-3495(96)79293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi V, Piazzesi G, Ferenczi MA, Thirlwell H, Dobbie I, Irving M. Elastic distortion of myosin heads and Repriming of the working stroke in muscle. Nature. 1995;374:553–555. doi: 10.1038/374553a0. [DOI] [PubMed] [Google Scholar]
- Lymn RW. Myosin subfragment-1 attachment to actinExpected effect on equatorial reflections. Biophys J. 1978;21:93–98. doi: 10.1016/S0006-3495(78)85510-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markhasin VS, Solovyova O, Katsnelson LB, Protsenko Y, Kohl P, Noble D. Mechano-electric interactions in heterogeneous myocardium: development of fundamental experimental and theoretical models. Prog Biophys Mol Biol. 2003;82:207–220. doi: 10.1016/s0079-6107(03)00017-8. [DOI] [PubMed] [Google Scholar]
- Matsubara I. X-ray diffraction studies of the heart. Annu Rev Biophys Bioeng. 1980;9:81–105. doi: 10.1146/annurev.bb.09.060180.000501. [DOI] [PubMed] [Google Scholar]
- Metzger JM, Greaser ML, Moss RL. Variations in cross-bridge attachment rate and tension with phosphorylation of myosin in mammalian skinned skeletal muscle fibres. Implications for twitch potentiation in intact muscle. J Gen Physiol. 1989;93:855–883. doi: 10.1085/jgp.93.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos C, Offer G, Starr R, Bennett P. Interaction of C-protein with myosin, myosin rod and light meromyosin. J Mol Biol. 1975;97:1–9. doi: 10.1016/s0022-2836(75)80017-9. [DOI] [PubMed] [Google Scholar]
- Morano I. Tuning the human heart molecular motors by myosin light chains. J Mol Med. 1999;77:544–555. doi: 10.1007/s001099900031. [DOI] [PubMed] [Google Scholar]
- Moss RL, Swinford AE, Greaser ML. Alterations in the Ca2+ sensitivity of tension development by single skeletal muscle fibres at stretched lengths. Biophys J. 1983;43:115–119. doi: 10.1016/S0006-3495(83)84329-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamoto H, Yagi K. Properties of myosin light chain kinase prepared from rabbit skeletal muscle by an improved method. J Biochem. 1984;95:1119–1130. doi: 10.1093/oxfordjournals.jbchem.a134700. [DOI] [PubMed] [Google Scholar]
- Olsson MC, Patel JR, Fitzsimons DP, Walker JW, Moss RL. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am J Physiol Heart Circ Physiol. 2004;287:H2712–H2718. doi: 10.1152/ajpheart.01067.2003. [DOI] [PubMed] [Google Scholar]
- Patel JR, Fitzsimons DP, Buck SH, Muthuchamy M, Wieczorek DF, Moss RL. PKA accelerates rate of force development in murine skinned myocardium expressing α- or β-tropomyosin. Am J Physiol Heart Circ Physiol. 2001;280:H2732–H2739. doi: 10.1152/ajpheart.2001.280.6.H2732. [DOI] [PubMed] [Google Scholar]
- Pearson JT, Shirai M, Tsuchimochi H, Schwenke DO, Ishida T, Kangawa K, Suga H, Yagi N. Effects of sustained length-dependent activation on in situ cross-bridge dynamics in rat hearts. Biophys J. 2007;93:4319–4329. doi: 10.1529/biophysj.107.111740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persechini A, Stull JT, Cooke R. The effect of myosin phosphorylation on the contractile properties of skinned rabbit skeletal muscle fibers. J Biol Chem. 1985;260:15988–15995. [PubMed] [Google Scholar]
- Pi Y, Kemnitz KR, Zhang D, Kranias EG, Walker JW. Phosphorylation of troponin I controls cardiac twitch dynamics: evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ Res. 2002;90:649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]
- Pi Y, Zhang D, Kemnitz KR, Wang H, Walker JW. Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium. J Physiol. 2003;552:845–857. doi: 10.1113/jphysiol.2003.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Regnier M, Martyn DA, Chase PB. Calcium regulation of tension redevelopment Kinetics with 2-deoxy-ATP or low [ATP] in rabbit skeletal muscle. Biophys J. 1998;74:2005–2015. doi: 10.1016/S0006-3495(98)77907-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD, Solaro RJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257:260–263. [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbe A, Fewell JG, Gulick J, Osinska H, Lorenz J, Hall DG, Murray LA, Kimball TR, Witt SA, Robbins J. Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J Biol Chem. 1999;274:21085–21094. doi: 10.1074/jbc.274.30.21085. [DOI] [PubMed] [Google Scholar]
- Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, Geenen DL, Buttrick PM, Solaro RJ. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem. 2009;284:5097–5106. doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ. Troponin C–troponin I interactions and molecular signalling in cardiac myofilaments. Adv Exp Med Biol. 1995;382:109–115. doi: 10.1007/978-1-4615-1893-8_12. [DOI] [PubMed] [Google Scholar]
- Steiger GJ. Tension transients in extracted rabbit heart muscle preparations. J Mol Cell Cardiol. 1977;9:671–685. doi: 10.1016/s0022-2828(77)80362-3. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ Res. 2006a;98:1212–1218. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J Gen Physiol. 2006b;128:261–272. doi: 10.1085/jgp.200609547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006c;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Walker JW, Moss RL. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res. 2007;101:503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- Stull LB, Leppo MK, Marban E, Janssen PM. Physiological determinants of contractile force generation and calcium handling in mouse myocardium. J Mol Cell Cardiol. 2002;34:1367–1376. doi: 10.1006/jmcc.2002.2065. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Stull JT. Phosphorylation of myosin in permeabilized mammalian cardiac and skeletal muscle cells. Am J Physiol Cell Physiol. 1986;250:C657–C660. doi: 10.1152/ajpcell.1986.250.4.C657. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Stull JT. Alteration of cross-bridge kinetics by myosin light chain phosphorylation in rabbit skeletal muscle: implications for regulation of actin-myosin interaction. Proc Natl Acad Sci U S A. 1990;87:414–418. doi: 10.1073/pnas.87.1.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney HL, Yang Z, Zhi G, Stull JT, Trybus KM. Charge replacement near the phosphorylatable serine of the myosin regulatory light chain mimics aspects of phosphorylation. Proc Natl Acad Sci U S A. 1994;91:1490–1494. doi: 10.1073/pnas.91.4.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Velden J, Narolska NA, Lamberts RR, Boontje NM, Borbely A, Zaremba R, Bronzwaer JG, Papp Z, Jaquet K, Paulus WJ, Stienen GJ. Functional effects of protein kinase C-mediated myofilament phosphorylation in human myocardium. Cardiovasc Res. 2006;69:876–887. doi: 10.1016/j.cardiores.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Van Der Velden J, Papp Z, Boontje NM, Zaremba R, de Jong JW, Janssen PM, Hasenfuss G, Stienen GJ. The effect of myosin light chain 2 dephosphorylation on Ca2+-sensitivity of force is enhanced in failing human hearts. Cardiovasc Res. 2003;57:505–514. doi: 10.1016/s0008-6363(02)00662-4. [DOI] [PubMed] [Google Scholar]
- Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhao J, Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca2+ affinity of cardiac troponin C. J Biol Chem. 1995;270:30773–30780. doi: 10.1074/jbc.270.51.30773. [DOI] [PubMed] [Google Scholar]
- Zoghbi ME, Woodhead JL, Moss RL, Craig R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci U S A. 2008;105:2386–2390. doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]