

Abstract
Virus-based nanoparticles (VNPs) from a variety of sources are being developed for biomedical and nanotechnology applications that include tissue targeting and drug delivery. However, the fate of most of those particles in vivo has not been investigated. Cowpea mosaic virus (CPMV), a plant comovirus, has been found to be amenable to the attachment of a variety of molecules to its coat protein, as well as to modification of the coat protein sequence by genetic means. We report here the results of studies of the bio-distribution, toxicology, and pathology of CPMV in mice. Plasma clearance and tissue biodistribution were measured using CPMV particles derivatized with lanthanide metal complexes. CPMV particles were cleared rapidly from plasma, falling to undetectable levels within 20 minutes. By 30 minutes the majority of the injected VNPs were trapped in the liver and to a lesser extent the spleen with undetectable amounts in other tissues. At doses of 1 mg, 10 mg and 100 mg per kg body weight, no toxicity was noted and the mice appeared to be normal. Hematology was essentially normal, although with the highest dose examined, the mice were somewhat leukopenic with relative decreases in both neutrophils and lymphocytes. Histological examination of spleen showed cellular infiltration, which upon flow cytometry analyses revealed elevated B lymphocytes on the first day following virus administration that subsequently subsided. Microscopic evaluation of various other tissues revealed a lack of apparent tissue degeneration or necrosis. Overall, CPMV appears to be a safe and non-toxic platform for in vivo biomedical applications.
1. Introduction
Recently, there has been increasing focus in materials science on producing nanometer-sized particles for a diverse variety of applications. One such effort is to create targeted nanodevices with multifunctional capabilities to enable early diagnosis and offer superior treatment of diseases such as cancer and cardiovascular disease. Strategies being developed to generate “smart” tissue-specific nanoparticles or nanodevices include the use of nanospheres, semiconductor quantum dots, dextrans, liposomes, antibodies, dendrimers, or viral particles [1, 8, 14, 15, 26, 33, 36, 38, 40, 41, 52]. These formulations are often comprised of targeting moieties along with cytotoxic drugs and/or imaging agents. Virus particles are attractive as self-assembled and uniform nanostructures with well-defined geometries that are tailorable at the atomic level by genomic manipulation. Being primarily protein-based, they have a natural biocompatibility, particularly viruses that do not cause human disease such as plant viruses or bacteriophages. Virus capsids are relatively rigid structures, making it feasible to display molecules or epitopes in precise spatial distributions at the nanoscale, a difficult task using inorganic or lipid materials[15, 52]. Viral nanoparticles (VNPs) currently in development include cowpea mosaic virus (CPMV) [9, 29, 51, 58–60], cowpea chlorotic mottle virus (CCMV) [2], bacteriophages (MS2 [3, 8, 23, 43], M13 [28, 31, 32] Qβ [4]) and polyoma virus [1]. A significant challenge in the design of new nanoparticle formulations is monitoring the plasma circulation, biodistribution and toxicity in vivo, which is required to establish the biocompatibility of new nanomaterials prior to extensive development of a formulation strategy.
CPMV is a plant virus that forms a 31 nm-diameter icosahedral particle. The viral genome is bipartite with each particle encapsidating a positive-sense single stranded RNA molecule. Substantial quantities of virus, in the range of 0.8–1.0 mg/gram of infected leaves, can be easily generated and purified by infection of black-eyed pea plants (Vigna unguiculata) [44]. Introduction of sequences in the genome facilitates peptide display on the virus capsid surface [13], the structure of which is known to 2.8Å resolution [10, 30]. CPMV is stable over a wide range of temperature, pH and buffers [59, 60]. A wide variety of compounds have been conjugated to the CPMV surface [9, 59]. For more specific and efficient linkage of molecules to the capsid, the copper(I)-mediated azide-alkyne cycloaddition (CuAAC) reaction was developed for bioconjugation [51, 58]. For these reasons, CPMV-based nanoparticles are attractive candidates for bio-nanotechnology applications.
While there are many VNP platforms in development, the behavior of VNPs in vivo is not well characterized. Previously we showed that CPMV was found in a wide variety of tissues or tissue vasculature for 1–3 days after intravenous administration, but these studies were not quantitative [46]. Here we report the plasma clearance kinetics and biodistribution of CPMV following intravenous administration. CPMV particles were decorated with a gadolinium chelate, or, alternatively, Gd+3 and Tb+3 ions were complexed directly to the capsid by association to the nucleoprotein [45]. Clearance and biodistribution analyses of CPMV particles were performed by measuring the presence of the Gd and Tb metals in plasma and in tissue homogenates using inductively coupled plasma-optical emission spectrometry (ICP-OES). In addition, the toxicity and pathology induced by intravenous injection of CPMV particles in mice were evaluated by necropsy, hematology and tissue histopathology.
2. Materials and Methods
2. 1. Materials
All chemicals and buffers were purchased from either Sigma-Aldrich, (Saint Louis, MO) or Acros/Fisher, (Fairlawn, NJ) or Invitrogen (Carlsbad, CA). Unless otherwise indicated, “buffer” or “HEPES buffer” denotes 0.1 M HEPES, pH 7.0. Cowpea mosaic virus was prepared in Vigna unguiculata seedlings (Burpee, Inc., Warminster, PA). Virus samples were purified by ultracentrifugation using 50.2Ti, SW 28 or TL-100.3 rotors (Beckman, Fullerton, CA). The purity of each CPMV preparation was analyzed using a Sepharose 6 size exclusion column by FPLC (AKTA Explorer; GE Healthcare, Piscataway, NJ). UV absorbance measurements were made with a Beckman DU-80 spectrophotometer. Transmission electron microscopy was performed with a CM 100 electron microscope (Philips, Mahwah, NJ)
Adult Balb/c mice, approximately 8 to 10 weeks of age and with an average body weight of 25 grams were obtained from the TSRI rodent breeding colony. All experiments were performed according to protocols approved by the TSRI Institutional Animal Care and Use Committee (IACUC).
ICP-OES was performed on a Varian VISTA AX CCD simultaneous spectrometer (Palo Alto, CA) equipped with a Teflon nebulizer and sample uptake tubing. Tissue samples were lyophilized (Virtis, Gardiner, NY) and then digested overnight in concentrated nitric acid (70% double-distilled purified, Sigma-Aldrich). Calibration curves for Gd and Tb were established using metal standards purchased from Inorganic Ventures Inc. (Lakewood, NJ); all samples were spiked with 10 ppm yttrium chloride internal standard to normalize differences in nebulization.
For fluorescence activated cell sorting (FACS) analyses, each mouse spleen was pushed through a 100 μm mesh filter (Becton-Dickinson (BD) Pharmingen, San Diego, CA) and analyzed with antibodies purchased from BD or e-Biosciences (San Diego, CA). Cells were examined using a FACS Calibur instrument (BD) and data were analyzed using Flow Jo software (Treestar, San Carlos, CA). Screening for apoptotic cells in tissue paraffin sections were performed using the Apoptag kit (Millipore, Temecula, CA)
2. 2. Methods
2. 2. 1 CPMV production and purification
Purified CPMV particles were prepared as described previously [46]. Briefly, the infected Vigna unguiculata leaf material homogenized in phosphate buffer (0.1M potassium phosphate, pH 7.0) was extracted with a mixture of chloroform and n-butanol. From the aqueous fraction, the virus was precipitated with 8% polyethylene glycol-8000 and 0.2 M sodium chloride. The precipitate was ultra-centrifuged at 160000 × g for 3 hrs, in a 50.2 Ti rotor through a 40% sucrose cushion and the pellet obtained was purified by ultra-centrifugation over a 10–40% sucrose gradient at 104000 × g for 2 hrs in SW 28 rotor. Following gradient separation, virus bands were collected and ultra-centrifuged (160000 × g for 3 hrs in 50.2 Ti rotor) to obtain the purified virus pellet, which was resuspended in endotoxin-free phosphate buffered saline (PBS). The purity of each CPMV sample was determined by size-exclusion FPLC using a Sepharose 6 column. Virus concentration in mg/mL was determined by absorbance at 260 nm, 0.1 mg/mL giving an optical density of 0.8. Virus samples were also examined by electron microscopy using a Philips CM-100 electron microscope. A 10 μL drop of virus sample was placed on a carbon-coated copper grid for 1 minute, which was then washed three times in sterile filtered water, stained with 10 μL drops of 2% uranyl acetate for 2 minutes, and allowed to dry before being examined under the electron microscope.
2. 2. 2. CPMV Maximum Tolerable Dose Study
Native CPMV particles were resuspended to 25 mg/mL in PBS. At concentrations higher than 30 mg/mL the virus tended to aggregate and precipitate from solution. Each group consisted of 2 to 5 mice. PBS (control) or CPMV in PBS was administered intravenously in 100 μL total volume. Three different doses of CPMV were prepared as 0.25 mg/mL, 2.5 mg/mL, and 25 mg/mL corresponding to approximately 1, 10, and 100 mg/kg body weight per mouse, respectively.
2. 2. 3. Clinical signs monitored, time points and rating system
Each mouse was examined for lack of movement, ataxia, hunched posture, ruffled fur, hypothermia, dehydration, dyspnea, tachypnea, seizure and sustained rapid movement around the cage. The mice were examined at 0–30 mins, each hour from 1 to 8 hours, and again at 24 h, in all cases using a labeling system that prevented the examiner from knowing the identity of each animal. A 0 to 3 scale was used for each of the above parameters was on a 0 to 3 scale: 0 = no indication of the problem, 1 = slight indication of the problem, 2 = serious indication of the problem, 3 = extreme indication of the problem. In a separate experiment, the body weights of mice administered with identical CPMV doses were measured every week for four weeks.
2.2. 6. Plasma pharmacokinetics and bio-distribution of CPMV
CPMV particles were either labeled with Gd+3 or Tb+3 ions by binding to nucleoprotein sites on the interior of the particle or by covalent attachment of a Gd(DOTA) derivative to the external surface of the capsid by the CuAAC reaction [45]. The former was accomplished by mixing a solution of 50 mM GdCl3 or TbCl3 in HEPES buffer containing 30 mM EDTA with a 5 mg/mL solution of CPMV in buffer. A white gelatinous solution was produced that was dialyzed against a solution of 10 mM EDTA in HEPES buffer for 48 hrs with 2 changes of the buffer, against buffer alone for 12 hours, and then finally against 0.1 M phosphate, pH 7.0 for 12 hours. The virus was then subjected to a 10–40% sucrose gradient separation and an ultracentrifugation as described above. The clear pellet was dissolved in endotoxin-free PBS and metal loading was determined by ICP-OES analysis compared to calibrated standards, and by independent measurement of virus concentration. A complete account of this methodology is described elsewhere [45].
Alternatively, a Gd(DOTA) derivative bearing a pendant alkyne group was attached to the virus particles using the copper-mediated azide-alkyne cycloaddition reaction, again as described more fully elsewhere [45], and particles were purified on sucrose gradients as above. The average number of Gd complexes attached to each virion was determined by ICP-OES and measurement of protein concentration.
In order to test whether CPMV-Gd and CPMV-Tb particles were stable in plasma, each was incubated at 1 mg/mL concentration in either 1 mL of freshly isolated mouse plasma or PBS for 4 hrs at room temperature. The samples were then ultracentrifuged on a 40 % sucrose cushion solution using a TL-100.3 rotor for 3 hrs at 160,000 × g. The supernatant and the pellet fractions were analyzed by ICP-OES for the presence of Gd or Tb. The resuspended pellets were also examined by size-exclusion chromatography and electron microscopy (EM). In all cases, intact particles were recovered in high yields with no loss in the amount of entrapped lanthanide metal ion.
To measure plasma circulation time, animals were injected intravenously with 100 μL of lanthanide-labeled CPMV particles (containing various amounts of Gd or Tb as indicated in the results section). Heparinized blood samples were collected by cardiac puncture at time points indicated in Figure 2. Cells were removed by centrifugation at 500 × g for 10 minutes and the resulting plasma isolated. For tissue bio-distribution studies, mouse organs were collected at least 30 minutes after CPMV injection. Each organ was lyophilized and then digested with nitric acid for 18 to 24 hrs. A small amount of insoluble material was occasionally observed following acid digestion and was removed by centrifugation or by filtering through a plug of glass wool in a 1 or 3 mL syringe. Each of the processed tissue samples and plasma samples were then examined by ICP-OES for the presence of Gd and Tb. Acid digestion of tissue [55] and the application of ICP-OES for the determination of Gd concentration in biological samples have been reported previously [18], and those instrument parameters and analytical methods were used here. Calibration plots for Gd or Tb in the concentration range of 0 to 1000 ppb were constructed for each instrument session. The amount of virus in each sample was quantitatively determined from the measured lanthanide concentration and the predetermined loadings of lanthanide for each virus sample. The percentage of virus detected relative to that injected was then easily derived from the known amount of material introduced to each animal. A similar methodology has been reported for the estimation of Gd-containing lipid nanoparticles that are retained in a tumor after in vivo administration [39]. The practical limit of Gd or Tb detection by ICP-OES for the sample volumes used in our experiments is 1–100 ppb, depending on the instrument. The total blood volume for each mouse was assumed to be 7% of the measured body weight [5], which varied somewhat among individual animals. The concentration of labeled virus was calculated using the following formula:
Figure 2.
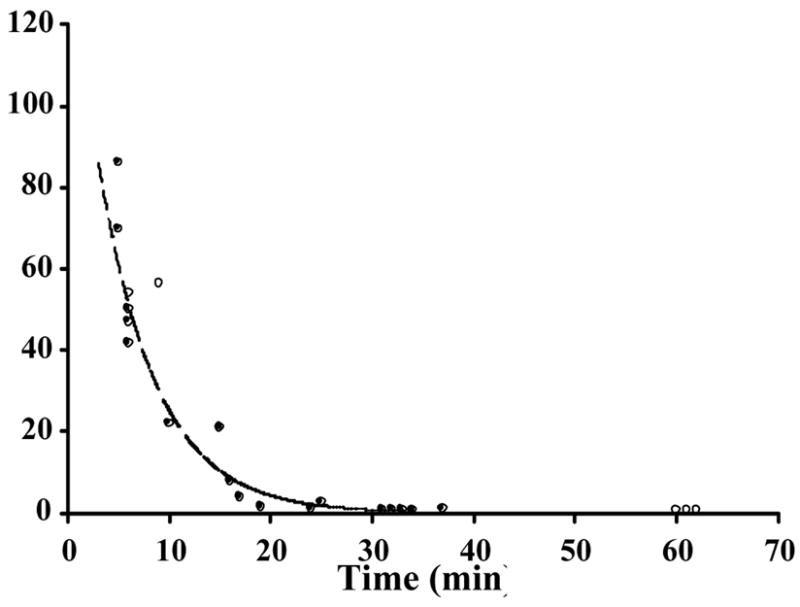

Pharmacokinetics of CPMV in plasma samples. CPMV-particles decorated with either (a) 143 (●) or 190 (○) Gd(DOTA) molecules on the exterior surface of each particle, with solid and dashed lines showing the trend line respectively or (b) a mixture of particles containing 80±20 Gd+3 (◇) or Tb+3 (■) ions bound to encapsulated RNA, were injected intravenously into mice, with dashed and solid lines showing the trend line respectively. Blood samples obtained from sacrificed mice at various times were analyzed for the metal concentration to quantitate the amount of bio-available virus particles. Percentage of injected sample is shown on the y-axis and the times of sample collection are shown on the x-axis. Each time point represents a single mouse.
For plasma samples, the percent of virus detected was obtained by:
For tissue samples, the percent virus detected was obtained by:
2.2. 4. Hematology and Pathology
Hematology of blood samples collected by cardiocentesis was performed on each mouse (2 per each group) at each time point mentioned above. Carcasses were immediately placed on wet ice and transported to the pathology laboratory where comprehensive post-mortem examinations were performed and representative tissues fixed in 10% buffered formalin for histology examination. All major organ systems and tissues were examined, including brain, spinal cord, sciatic nerve, skeletal muscle, femur, turbinate, trachea, lung, heart, aorta, pulmonary artery, thymus, spleen, lymph node, bone marrow, kidney, thyroid, adrenal, ovary, female reproductive tract, mammary gland, skin, eye, lacrimal gland, salivary gland, liver, pancreas, stomach, upper jejunum, lower jejunum/ileum, cecum and colon. These tissues were subsequently trimmed for histology and embedded in paraffin. Tissue sections were cut 3 μm thick, mounted on glass slides, stained with hematoxylin and eosin (H&E) and examined using light microscopy. Screening for apoptotic cells in paraffin sections from liver, spleen, lung, kidneys and heart was also performed using the Apoptag kit according to the manufacturer’s instructions.
2. 2. 5. Flow cytometry
Mice were injected intravenously with 10 mg/kg of CPMV and euthanized (5 mice per day) on day 1, day 2, day 3 or day 5 post-injection. Single-cell suspensions of whole spleens were prepared by passing the tissue through 100 μm mesh filters into RPMI-1640 cell culture medium. The resulting cell suspension was centrifuged at 800 × g and the pellet was resuspended with 0.8% ammonium chloride for 10 minutes to induce red blood cell lysis. The cells were pelleted again and resuspended in FACS buffer (PBS without calcium chloride and magnesium chloride and with 1% fetal calf serum, 25 mM HEPES, 1 mM EDTA, pH 7.0). One million cells of each preparation were then incubated with purified anti-mouse CD16/CD32 (Fc gamma III/II receptor) to block Fc receptors, followed by incubation with phycoerythrin (PE)-conjugated anti-mouse CD4, and Alexa Fluor 488-conjugated anti-mouse CD8 antibodies to detect CD4 and CD8 T-lymphocytes respectively. To estimate natural killer (NK) cells and B-lymphocytes, cells were treated with PE labeled- antibodies to NK 1.1 and B220 respectively, in separate tubes. All antibody incubations were performed in FACS buffer. After staining, cells washed in FACS buffer followed by fixation in 2% paraformaldehyde in FACS buffer were subjected to flow cytometry analysis in the FACS Calibur instrument. Data was subjected to Students t-test (Microsoft Excel) for determination of statistical significance.
3. Results
To measure the plasma clearance kinetics and biodistribution of CPMV in vivo, a method to sensitively detect the particles in plasma and tissue samples was developed. CPMV was labeled with the lanthanide metals gadolinium (Gd) or terbium (Tb) using two different strategies. In the first method, a Gd(DOTA)-alkyne molecule (Figure 1A) was conjugated to a CPMV-azide derivative using the copper-mediated azide-alkyne cycloaddition (CuAAC) reaction as previously described [45]. Following virus purification, the number of Gd ions per particle was determined by ICP-OES. In two independent experiments with different concentrations of reagents, CPMV particles containing 143 or 190 Gd-DOTA complexes per particle were generated and are referred to as CPMV-DOTA-Gd. (The subsequent measurements of particle biodistribution took into account the differing Gd loadings per particle.). In a complementary approach, we took advantage of a natural affinity of lanthanide metals for nucleic acids [6, 57], and found that Gd and Tb ions interact stably with the viral genomic RNA (and probably also the associated protein) inside the particles (Figure 1C) [45]. Wild-type CPMV was soaked with either GdCl3 or TbCl3 in the presence of EDTA, the excess metal was dialyzed away, and the number of bound Gd+3 or Tb+3 ions per virus particle was determined using ICP-OES following virus purification. For each preparation, an average of 80±20 Gd+3 or Tb+3 per particle was observed; these samples were designated CPMV-Gd or CPMV-Tb.
Figure 1.

a. Structure of Gd(DOTA)-alkyne, prepared as described elsewhere [45]. b. Schematic representation of a CPMV particle with Gd(DOTA) (solid circles) presented on the exterior surface and c. Schematic representation of a CPMV particle with Gd+3 or Tb+3 ions (solid circles) chelated to the nucleoprotein and genome-capsid interface.
Gd(DOTA) has an extremely high association constant and the complexes were covalently attached to the particles, so the Gd ions do not dissociate from CPMV-DOTA-Gd under biological conditions. Since the preparation of CPMV-Gd and CPMV-Tb included extensive dialysis to wash away excess metal, we suspected that the lanthanide ions remaining inside the CPMV particle were similarly unable to leach away during the mouse experiments. To further substantiate this point, samples of CPMV-Gd or CPMV-Tb were incubated in fresh mouse plasma or in PBS. After 4 hours, the virus was isolated by ultracentrifugation and the pellet and supernatant prepared for ICP-OES. In both the cases, the lanthanide loadings remained constant with no detectable signal in the supernatant, showing that metal dissociation is negligible for the CPMV-Gd or CPMV-Tb particles during this time frame. The morphologies of the labeled and wild type particles were identical by electron microscopy and size exclusion chromatography [45].
The plasma clearance kinetics of CPMV-DOTA-Gd, CPMV-Gd, and CPMV-Tb were determined in mouse by intravenous injection of 100 μL of each formulation, followed by ICP-OES determination of Gd or Tb present in plasma collected over a time course of approximately 1 hour. Figures 2a and 2b show the amount of labeled virus remaining in plasma for CPMV-DOTA-Gd and CPMV-Gd/Tb, respectively. The observed clearance profiles were identical for CPMV-DOTA-Gd with two different loadings of 143 or 190 per particle, the former tested in two independent sets of mice and the latter tested in one set, each set comprising 5–10 animals (Fig 2a). The clearance kinetics were likewise very similar to that shown by the injection of a mixture of CPMV-Gd and CPMV-Tb, each with 80±20 metal ions on the particle interior (Fig. 2b). The simultaneous detection of Gd and Tb by ICP-OES in each serum sample provides an easy way to improve the precision of the quantitative measurements, since two sets of data are obtained for the same amount of effort. A third experiment, involving injections of CPMV-Gd and CPMV-Tb separately into two groups of 5–10 mice, gave virtually identical results. Based on these multiple experiments, the half-life of CPMV particles in plasma was determined to range from 4 minutes (CPMV-DOTA-Gd) to 7 minutes (CPMV-Gd or Tb), with >95% of the particles removed within 30 minutes. The close correspondence of the clearance curves for CPMV-DOTA-Gd, CPMV-Gd, and CPMV-Tb showed that the nature and location of the metal labels – particularly the placement of Gd(DOTA) complexes on the exposed outer surface of the virus capsid – did not affect the plasma clearance kinetics.
The biodistribution of CPMV within various tissues following intravenous dosing was determined by the quantitation of lanthanide ions in the liver, spleen, kidney and lungs. Since the CPMV particles were removed from plasma after 25–30 minutes, a 30-minute time point was used for tissue biodistribution analyses. More than 90% of the virus, whether externally (CPMV-DOTA-Gd) or internally (CPMV-Gd/Tb) labeled, was localized in the liver (Figure 3). A small but measurable amount of CPMV was also found in the spleen, accounting for less than 3% of the total injected dose (Figure 3). There were no detectable levels of CPMV in other tissue samples tested. In a separate experiment, the heart and the head of the femur bone were analyzed, and no lanthanide ions were found (data not shown).
Figure 3.
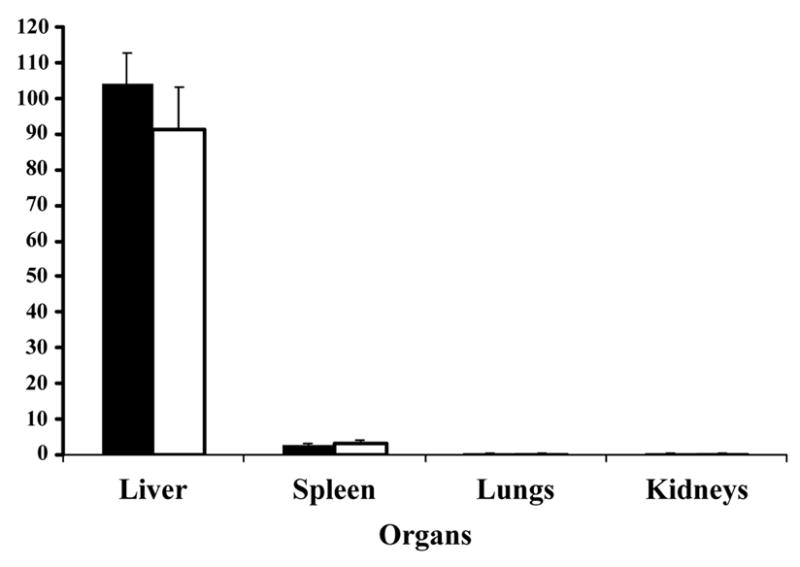
Bio-distribution of CPMV in vivo in mice. CPMV-particles decorated with Gd(DOTA) on the exterior (solid bars) or Gd+3/Tb+3 ions on the interior (white bars) of the virus capsid were injected intravenously into mice. Various organs as indicated were collected after at least 30 minutes and analyzed for metal content to quantitate the amount of virus.
To determine whether administration of CPMV was associated with toxicity in vivo, mice were inoculated intravenously with 1, 10, or 100 mg/kg bwt of unlabeled CPMV or with vehicle (PBS) alone. Careful monitoring at different time points through 24 hours (as indicated in the methods section) showed no clinical signs different from those of saline-injected mice at each of the doses investigated. Grossly, the animals appeared essentially normal, though all were somewhat pale, possibly associated with the terminal cardiocentesis. Hematology was essentially normal in all animals, although the mice receiving 10 and 100 mg/kg CPMV were somewhat leukopenic (white blood cell counts ≤1,200/μL) with relative decreases in both neutrophils and lymphocytes.
Histological examination of tissues comparing PBS and CPMV-inoculated animals included brain, spinal cord, sciatic nerve, skeletal muscle, femur, turbinate, trachea, lung, heart, aorta, pulmonary artery, thymus, spleen, lymph node, bone marrow, kidney, thyroid, adrenal, ovary, female reproductive tract, mammary gland, skin, eye, lacrimal gland, salivary gland, liver, pancreas, stomach, upper jejunum, lower jejunum/ileum, cecum and colon. No pathological changes were noted. Microscopic findings were similar between CPMV-inoculated and control animals, with no signs of overt toxicity (tissue degeneration or necrosis) in any of the examined tissues. Specific detection of apoptotic cells in liver, spleen, lung, kidneys and heart was also not observed (data not shown). In particular, the liver appeared to be normal with no evidence of pathology (Figure 4).
Figure 4.
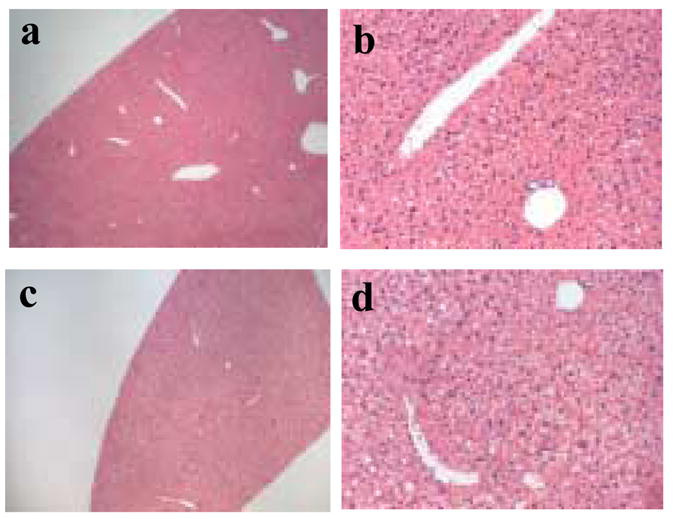
Histological examination of liver tissue. Hematoxylin and eosin stained sections of liver from a saline injected mouse (panel a at 4x and panel b at 20x magnification) and a mouse injected with CPMV particles (panel c at 4x and panel d at 20x magnification).
The only organ where any CPMV-related changes were apparent was the spleen. Here there was apparent hyperplasia in the periarteriolar lymphoid sheath (PALS) of all CPMV-inoculated animals, characterized by increased cell numbers including increased lymphoblast numbers plus the presence of cellular karyorrhexis in these areas (Figure 5). Interestingly, this was not a dose-dependent effect. Therefore we analyzed spleens from mice that were injected with an intermediate dose of 10 mg/kg bwt for the subsequent experiments. FACS analyses of total splenocytes comparing CPMV-inoculated and control animals revealed that the increases in cell numbers observed histologically were primarily associated with an increase in lymphocytes (Table 1). The levels of CD4+ cells and CD8+ cells were essentially normal (Table 1). However, there appeared to be a minor drop in both populations by day 3 that was statistically not significant. While a change in NK cell response was not apparent, a significant increase in B-cell numbers (p = 0.002 by Student’s two tailed t-test) was evident on day 1 following CPMV administration. Recovery from this response was evident by day 5, presumably reflective of virus clearance from the body.
Figure 5.
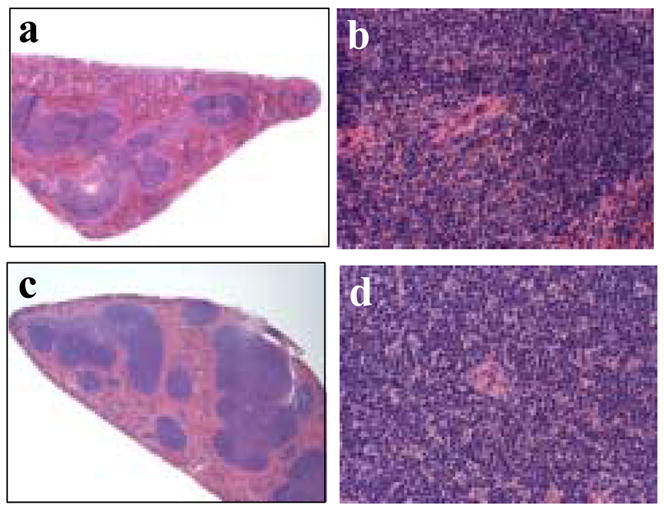
Histological examination of spleen tissue. Hematoxylin and eosin stained sections of a spleen from a saline injected mouse (panel a at 4x and panel b at 40x magnification) compared to sections of spleen from a mouse injected with CPMV particles (panel c at 4x and panel d at 40x magnification).
Table 1.
Spleen cell analyses following CPMV administration measured by flow cytometry. Spleen cell suspensions from mice (n=5 each day) injected either with PBS or CPMV particles (10 mg/kg bwt) were analyzed by FACS analyses using cell specific antibodies as indicated in the methods. The p-value was determined by Students t-test.
| Cell type | PBS | CPMV |
|||
|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 5 | ||
| CD4+ | 43.76±1.55 | 44.46±2.23 | 42.63±3.24 | 38.05±2.49 | 39.68±2.37 |
| CD8+ | 18.65±1.12 | 17.96±1.34 | 19.25±2.22 | 15.88±1.18 | 18.54±1.08 |
| NK | 1.10±0.79 | 2.43±1.54 | ndb | 1.08±0.24 | 0.95±0.20 |
| B | 56.40±9.64 | 97.52±3.34a | nd | 47.32±3.34 | 56.58±1.38 |
statistically significant (p= 0.0002)
not done
DISCUSSION
A variety of nanoparticles including VNPs are being developed for biomedical applications [1, 8, 9, 34, 52], however little information is available about the biological behavior of these particles in vivo. Such studies are important for the design and development of a successful bio-nanoparticle. In the current study we analyzed the behavior of CPMV-based VNPs following intravenous inoculation in mice. Using doses of 1, 10, and 100 mg/kg bwt, no visibly concerned clinical signs were observed. CPMV particles were rapidly cleared from blood circulation within 30 minutes with an average half-life of 4–7 minutes in plasma. The CPMV particles primarily localized to the liver, but with no associated toxicity observed in liver or other tissues. Hemagglutination was not observed following CPMV administration, however, a mild leukopenia was observed that could be in response to the presence of virus in circulation. A small amount of CPMV localized to spleen where it caused a transient increase in the numbers of B-cells. Whether this increase was caused by infiltration or cell proliferation is not known.
The clearance kinetics of viruses have been reported in several instances, including our prior work with CPMV [46] and, most notably, studies of adenovirus (Ad) and bacteriophages [20, 53, 56]. In general, particulate materials including viruses are removed from the bloodstream relatively quickly by the reticulo-endothelial system (RES) of the liver and spleen [17, 42]. The retention time of lanthanide-CPMV particles in the liver is currently unknown, but is an active area of research in our labs. Recent results have shown that particles were still present in the liver in significant quantities even 48 hours post-injection (unpublished results). Ad particles administered intravenously in mice were found in the plasma for 10–15 minutes with a half-life of not more than 5 minutes [20]. particles are human pathogens, which bind to receptors such as coxsackie adenovirus receptor (CAR) that are expressed on a variety of cell types including liver cells. Ad particles appear to be sequestered from circulation in part by RES in mice, presumably by interaction with the scavenger receptors [17, 42] or platelets [54]. Bacteriophages are widely used in biopanning experiments in vivo for identification of tissue-specific biomarkers [27, 35]. When T7 phage is administered intravenously amounts of circulating particles decline to negligible levels within one hour [53]. In contrast, the lambda phage particles survived longer in circulation, and a single amino acid change from lysine to glutamic acid in the coat protein was reported to extend the circulation time by 3 to 4 orders of magnitude [56], suggesting that surface interactions between the phage, immune system and vascular endothelial cells have a significant effect on plasma clearance.
The relatively rapid clearance of CPMV from plasma is similar to Ad but unlike a phage. Phages and CPMV have evolved to bind and replicate in bacteria and plants respectively, therefore were anticipated to have different circulation half lives in mammalian systems. However, unlike phages, CPMV particles were found to bind to a 54 kDa protein expressed on a variety of mammalian cells including endothelial cells [25]. The rapid removal of CPMV from circulation may be related to this uptake of CPMV in endothelial cells [29]. Furthermore, our laboratory previously reported that CPMV was found in a variety of tissues 1–3 days after administration [46]. In that study, the virus particle was detected by the very sensitive method of RT-PCR. The persistence of a small amount of CPMV over periods of days is likely due to their adhesion to endothelial or other cells at a level that cannot be detected by ICP-OES. It must be noted that unlike Ad there is no evidence that either CPMV or phages replicate in mammalian cells that show virus uptake.
The potential for toxic side effects that may be associated with VNPs related to human pathogens has caused concern for the use of such virus-based platforms. For example, while Ad administration causes gene expression to occur preferentially in the liver [50], a substantial liver injury was seen with administration of approximately 2 × 1011 particles/kg body weight [20] in both non-human primates and humans. Even replication-defective Ad or Ad virus components induced toxic responses in the host. Acute induction of proinflammatory cytokines such as IL-6, IL-1β and TNF-α and elevated serum transaminases are common findings following Ad therapy [7, 11, 12, 16, 22, 37, 48]. These responses are presumably due to interactions of Ad or its components with macrophages and liver Kupffer cells [49]. Viruses such as plant pathogens and bacteriophages are less likely to interact with human cells in a way that causes toxic effects. For example, phage particles that are widely used for biopanning experiments in vivo in mice can be administered intravenously in doses of approximately 4 × 1012 particles/kg bwt with no overt toxicity. Similarly, here we have shown that CPMV based VNP administration of approximately 1016 particles/kg bwt (100 mg/kg bwt) showed no apparent toxicity. Although we did not detect apoptotic cells in the tissue samples we analyzed, we are conducting experiments to identify any such effects at later time points following administration of CPMV.
Immune responses, particularly involving B-lymphocytes, presumably play an important role in the eventual elimination of virus from the body. We have previously shown that unmodified CPMV capsids induce antibody responses [47], and here we report that an increase in B-cell number is observed in spleen following intravenous administration of CPMV. A similar increased B-cell infiltration was also reported in mice following Qβ phage particle intravenous administration [19]. Other studies demonstrate that B-cells participate in elimination of T7 phage particles [53]. Although phage particles, like CPMV, induce anti-capsid antibody responses, it has been shown that M13 phage can effectively target tissues even in the presence of circulating antibodies [21]. It will be interesting to examine whether the clearance of CPMV will be altered in the presence of serum antibodies or complement as seen with repeat injections of PEGylated liposomes [24].
The rapid clearance and liver-selective trafficking of CPMV particles suggests that specific targeting to other tissues or cells would require modification with PEG or other immune masking agents. We previously showed that a strong immune response is mounted in mice against CPMV particles, and that a modest polyethylene glycol (PEG) coating inhibits the induction of the anti-CPMV response [47] as well as reduce virus uptake in endothelial cells, liver and spleen [29]. Interestingly, preliminary studies indicate that coating of CPMV particles with 2000 MW PEG doubles the plasma circulation time (Destito et al., unpublished observation), likely by inhibiting the interaction with endothelial cells. Similarly, adenovirus circulation time was extended by several minutes following coating of the particles with a polymer that presumably repels interactions with cells [20].
As there are many VNPs in development, a detailed study of how each behaves in vivo, and the kind of immune responses they generate, provides valuable information on their potential uses and limitations. CPMV based particles, which can be produced economically in large quantities and can be modified by genetic and chemical means, comprise a nanoparticle scaffold with some promise for in vivo applications. With concerns about toxicity allayed by the studies described here, attention is now turned in our laboratories to the goal of selective targeting by the conjugation of a variety of effector molecules to the capsid exterior.
Acknowledgments
These studies were funded by National Institutes of Health grant R01 CA112075, and by the Skaggs Institute for Chemical Biology. We acknowledge P. Nieto-Gonzalez, A. Reyes and K. Klingensmith, from the Division of Animal Resources at The Scripps Research Institute, for assistance with mouse experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Refrences
- 1.Abbing A, Blaschke UK, Grein S, Kretschmar M, Stark CM, Thies MJ, Walter J, Weigand M, Woith DC, Hess J, Reiser CO. Efficient intracellular delivery of a protein and a low molecular weight substance via recombinant polyomavirus-like particles. J Biol Chem. 2004;279:27410–27421. doi: 10.1074/jbc.M313612200. [DOI] [PubMed] [Google Scholar]
- 2.Allen M, Bulte JW, Liepold L, Basu G, Zywicke HA, Frank JA, Young M, Douglas T. Paramagnetic viral nanoparticles as potential high-relaxivity magnetic resonance contrast agents. Magnetic Resonance Medicine. 2005;54:807–812. doi: 10.1002/mrm.20614. [DOI] [PubMed] [Google Scholar]
- 3.Anderson EA, Issacman S, Peabody DS, Wang EY, Canary JW, Kirshenbaum K. Viral nanoparticles donning a paramagnetic coat: conjugation of MRI contrast agents to the MS2 capsid. Nano Letters. 2006;6:1160–1164. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- 4.Ashcroft AE, Lago H, Macedo JM, Horn WT, Stonehouse NJ, Stockley PG. Engineering thermal stability in RNA phage capsids via disulphide bonds. Journal of nanoscience and nanotechnology. 2005;5:2034–2041. doi: 10.1166/jnn.2005.507. [DOI] [PubMed] [Google Scholar]
- 5.Bannerman R. Hematology. III. New York: Academic press; 1983. [Google Scholar]
- 6.Barela TD, Burchett S, Kizer DE. Terbium binding to ribosomes and ribosomal RNA. Biochemistry. 1975;14:4887–4892. doi: 10.1021/bi00693a016. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Gary H, McKinney RL, Rosengart T, Lesser ML, Crystal RG. Systemic interleukin-6 responses following administration of adenovirus gene transfer vectors to humans by different routes. Mol Ther. 2002;6:287–297. doi: 10.1006/mthe.2002.0658. [DOI] [PubMed] [Google Scholar]
- 8.Brown WL, Mastico RA, Wu M, Heal KG, Adams CJ, Murray JB, Simpson JC, Lord JM, Taylor-Robinson AW, Stockley PG. RNA bacteriophage capsid-mediated drug delivery and epitope presentation. Intervirology. 2002;45:371–380. doi: 10.1159/000067930. [DOI] [PubMed] [Google Scholar]
- 9.Chatterji A, Ochoa W, Shamieh L, Salakian SP, Wong SM, Clingon G, Ghosh P, Lint T, Johnson J. Chemical conjugation of heterologous proteins on the surface of cowpea mosaic virus. Bioconjug Chem. 2004;15:807–813. doi: 10.1021/bc0402888. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Stauffacher C, Johnson JE. Capsid Structure and RNA Packaging in Comovirus. Sem in Virol. 1990;1:453–466. [Google Scholar]
- 11.Christ M, Louis B, Stoeckel F, Dieterle A, Grave L, Dreyer D, Kintz J, Ali Hadji D, Lusky M, Mehtali M. Modulation of the inflammatory properties and hepatotoxicity of recombinant adenovirus vectors by the viral E4 gene products. Hum Gene Ther. 2000;11:415–427. doi: 10.1089/10430340050015888. [DOI] [PubMed] [Google Scholar]
- 12.Cotter MJ, Muruve DA. The induction of inflammation by adenovirus vectors used for gene therapy. Front Biosci. 2005;10:1098–1105. doi: 10.2741/1603. [DOI] [PubMed] [Google Scholar]
- 13.Dessens JT, Lomonossoff GP. Cauliflower mosaic virus 35S promoter-controlled DNA copies of cowpea mosaic virus RNAs are infectious on plants. J Gen Virol. 1993;74:889–892. doi: 10.1099/0022-1317-74-5-889. [DOI] [PubMed] [Google Scholar]
- 14.Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, CG Cerveny, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778–784. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- 15.Douglas T, Young M. Viruses: making friends with old foes. Science. 2006;312:873–875. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- 16.Engler H, Machemer T, Philopena J, Wen Sf, Quijano E, Ramachandra M, Tsai V, Ralston R. Acute hepatoxocity of oncolytic adenoviruses in mouse models is associated with expression of wild-type E1a and induction of TNF-alpha. Virology. 2004;328:52–61. doi: 10.1016/j.virol.2004.06.043. [DOI] [PubMed] [Google Scholar]
- 17.Fisher K. Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther. 2006;8:301–313. [PubMed] [Google Scholar]
- 18.Frame EMS, Uzgiris EE. Gadolinium determination in tissue samples by inductively coupled mass spectrometry and inductively coupled plasma atomic emission spectrometry in evaluation of the action of magnetic resonance imaging contrast agents. Analyst. 1998;123:675–679. doi: 10.1039/a708905h. [DOI] [PubMed] [Google Scholar]
- 19.Gatto D, Ruedl C, Odermatt B, Bachmann MF. Rapid response of marginal zone B cells to viral particles. J Immunol. 2004;173:4308–4316. doi: 10.4049/jimmunol.173.7.4308. [DOI] [PubMed] [Google Scholar]
- 20.Green NK, Herbert CW, Hale SJ, Hale AB, Mautner V, Harkins R, Hermiston T, Ulbrich K, Fisher KD, Seymour LW. Extended plasma circulation time and decreased toxicity of polymer-coated adenovirus. Gene Ther. 2004;11:1256–1263. doi: 10.1038/sj.gt.3302295. [DOI] [PubMed] [Google Scholar]
- 21.Hajitou A, Trepel M, Lilley CE, Soghomonyan S, Alauddin MM, Marini FC, 3rd, Restel BH, Ozawa MG, Moya CA, Rangel R, Sun Y, Zaoui K, Schmidt M, von Kalle C, Weitzman MD, Gelovani JG, Pasqualini R, Arap W. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell. 2006;125:385–398. doi: 10.1016/j.cell.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 22.Higginbotham JN, Seth P, Blaese RM, Ramsey WJ. The release of inflammatory cytokines from human peripheral blood mononuclear cells in vitro following exposure to adenovirus variants and capsid. Hum Gene Ther. 2002;13:129–141. doi: 10.1089/10430340152712683. [DOI] [PubMed] [Google Scholar]
- 23.Hooker JM, Kovacs EW, Francis MB. Interior surface modification of bacteriophage MS2. J Am Chem Soc. 2004;126:3718–3719. doi: 10.1021/ja031790q. [DOI] [PubMed] [Google Scholar]
- 24.Ishida T, Ichihara M, Wang X, Kiwada H. Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J Control Release. 2006;115:243–250. doi: 10.1016/j.jconrel.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 25.Koudelka KJ, Rae CS, Gonzalez MJ, Manchester M. Interaction between a 54kD mammalian cell surface protein and Cowpea Mosaic Virus (CPMV) J Virol. 2006;81:1632–1640. doi: 10.1128/JVI.00960-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JJ. Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res. 2005;65:5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- 27.Landon LA, Deutscher SL. Combinatorial discovery of tumor targeting peptides using phage display. J Cell Biochem. 2003;90:509–517. doi: 10.1002/jcb.10634. [DOI] [PubMed] [Google Scholar]
- 28.Lee SW, Mao C, Flynn CE, Belcher AM. Ordering of quantum dots using genetically engineered viruses. Science. 2002;296:892–895. doi: 10.1126/science.1068054. [DOI] [PubMed] [Google Scholar]
- 29.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Viral nanoparticles as tools for intravital vascular imaging. Nat Med. 2006;12:354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin T, Chen Z, Usha R, Stauffacher C, Dai J, Schmidt T, Johnson JE. The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology. 1999;265:20–34. doi: 10.1006/viro.1999.0038. [DOI] [PubMed] [Google Scholar]
- 31.Mao C, Flynn CE, Hayhurst A, Sweeney RY, Qi J, Georgiou G, Iverson B, Belcher AM. Viral assembly of oriented quantum dot nanowires. Proc Natl Acad Sci USA. 2003;100:6946–6951. doi: 10.1073/pnas.0832310100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao C, Solis DJ, Reiss BD, Kottmann ST, Sweeney RY, Hayhurst A, Georgiou G, Iverson B, Belcher AM. Virus-based toolkit for the directed synthesis of magnetic and semiconducting nanowires. Science. 2004;303:213–217. doi: 10.1126/science.1092740. [DOI] [PubMed] [Google Scholar]
- 33.Medina OP, Zhu Y, Kairemo K. Targeted liposomal drug delivery in cancer. Curr Pharm Des. 2004;10:2981–2989. doi: 10.2174/1381612043383467. [DOI] [PubMed] [Google Scholar]
- 34.Merril CR, Scholl D, Adhya SL. The prospect for bacteriophage therapy in Western Medicine. Nature Reviews Drug Discovery. 2003;2:489–497. doi: 10.1038/nrd1111. [DOI] [PubMed] [Google Scholar]
- 35.Mori T. Cancer-specific ligands identified from screening of peptide-display libraries. Curr Pharm Des. 2004;10:2335–2343. doi: 10.2174/1381612043383944. [DOI] [PubMed] [Google Scholar]
- 36.Muldoon LL, Neuwelt EA. BR96-DOX immunoconjugate targeting of chemotherapy in brain tumor models. J Neurooncol. 2003;65:49–62. doi: 10.1023/a:1026234130830. [DOI] [PubMed] [Google Scholar]
- 37.Muruve DA. The innate immune response to adenovirus vectors. Hum Gene Ther. 2004;15:1157–1166. doi: 10.1089/hum.2004.15.1157. [DOI] [PubMed] [Google Scholar]
- 38.Okuda T, Kawakami S, Akimoto N, Niidome T, Yamashita F, Hashida M. PEGylated lysine dendrimers for tumor-selective targeting after intravenous injection in tumor-bearing mice. J Control Release. 2006;116:330–336. doi: 10.1016/j.jconrel.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Oyewumi MO, Yokel RA, Jay M, Coakley T, Mumper RJ. Comparison of cell uptake, biodistribution and tumor retention of folate-coated and PEG-coated gadolinium nanoparticles in tumor-bearing mice. J Control Release. 2004;95:613–626. doi: 10.1016/j.jconrel.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Park JW, Kirpotin DB, Hong K, Shalaby R, Shao Y, Nielsen UB, Marks JD, Papahadjopoulos D, Benz CC. Tumor targeting using anti-her2 immunoliposomes. J Control Release. 2001;74:95–113. doi: 10.1016/s0168-3659(01)00315-7. [DOI] [PubMed] [Google Scholar]
- 41.Pattenden LK, Middelberg AP, Niebert M, Lipin DI. Towards the preparative and large-scale precision manufacture of virus-like particles. Trends Biotechnol. 2005;23:523–529. doi: 10.1016/j.tibtech.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 42.Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14:123–128. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- 43.Pickett GG, Peabody DS. Encapsidation of heterologous RNAs by bacteriophage MS2 coat protein. Nucleic Acids Res. 1993;21:4621–4626. doi: 10.1093/nar/21.19.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porta C, Spall VE, Findlay KC, Gergerich RC, Farrance CE, Lomonossoff G. Cowpea mosaic virus-based chimeras. Effects of inserted peptides on the phenotype, host range, and transmissibility of the modified viruses. Virology. 2003;310:50–63. doi: 10.1016/s0042-6822(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 45.Prasuhn DE, Yeh R, Obenaus A, Manchester M, Finn MG. Viral MRI contrast Agents: Coordination of Gd by native virions and attachment of Gd complexes by azide-alkyne cycloaddition. Chemical Communications. 2006;12:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 46.Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh P, Thomas DM, Estrada MN, Powell E, Finn MG, Manchester M. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343:224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 47.Raja KS, Wang Q, Gonzalez MJ, Manchester M, Johnson JE, Finn MG. Hybrid virus-polymer materials. 1. Synthesis and properties of PEG-decorated cowpea mosaic virus. Biomacromolecules. 2003;4:472–476. doi: 10.1021/bm025740+. [DOI] [PubMed] [Google Scholar]
- 48.Reid T, Warren R, Kirn D. Intravascular adenoviral agents in cancer patients: lessons from clinical trials. Cancer Gene Ther. 2002;9:979–986. doi: 10.1038/sj.cgt.7700539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schiedner G, Bloch W, Hertel S, Johnston M, Molojavyi A, Dries V, Varga G, Van Rooijen N, Kochanek S. A hemodynamic response to intravenous adenovirus vector particles is caused by systemic Kupffer cell-mediated activation of endothelial cells. Hum Gene Ther. 2003;14:1631–1641. doi: 10.1089/104303403322542275. [DOI] [PubMed] [Google Scholar]
- 50.Schiedner G, Clemens PR, Volpers C, Kochanek S. High-capacity gutless adenoviral vectors: Technical aspects and applications. New York, USA: Academic Press; 2002. pp. 429–442. [Google Scholar]
- 51.Sen Gupta S, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Accelerated bioorthogonal cojugation: A practical method for ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconj Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 52.Singh P, Gonzalez MJ, Manchester M. Viruses and their uses in nanotechnology. Drug Development Research. 2006;67:23–41. [Google Scholar]
- 53.Srivastava AS, Kaido T, Carrier E. Immunological factors that affect the in vivo fate of T7 phage in the mouse. J Virol Methods. 2004;115:99–104. doi: 10.1016/j.jviromet.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 54.Stone D, Liu Y, Shayakhmetov D, Li ZY, Ni S, Lieber A. Adenovirus-platelet interaction in blood causes virus sequestration to the reticulo-endothelial system of liver. J Virol. 2007 Feb 14; doi: 10.1128/JVI.02819-06. e-pub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamat SR, Moore DE, Allen BJ. Determination of boron in biological tissues by inductively coupled plasma atomic emission spectrometry. Anal Chem. 1987;59:2161–2164. doi: 10.1021/ac00144a033. [DOI] [PubMed] [Google Scholar]
- 56.Vitiello CL, Merril CR, Adhya S. An amino acid substitution in a capsid protein enhances phage survival in mouse circulatory system more than a 1000-fold. Virus Res. 2005;114:101–103. doi: 10.1016/j.virusres.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 57.Walter NG, Yang N, Burke JM. Probing non-selective cation binding in the hairpin ribozyme with Tb(III) J Mol Biol. 2000;298:539–555. doi: 10.1006/jmbi.2000.3691. [DOI] [PubMed] [Google Scholar]
- 58.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 59.Wang Q, Kaltgrad E, Lin T, Johnson J, Finn M. Natural supramolecular building blocks: wild-type cowpea mosaic virus. Chem Biol. 2002a;9:805–811. doi: 10.1016/s1074-5521(02)00165-5. [DOI] [PubMed] [Google Scholar]
- 60.Wang Q, Lin T, Johnson J, Finn M. Natural supramolecular building blocks: cysteine-added mutants of cowpea mosaic virus. Chem Biol. 2002b;9:813–819. doi: 10.1016/s1074-5521(02)00166-7. [DOI] [PubMed] [Google Scholar]