

Abstract
We report here the isolation of five new compounds, dictazole A–B (1–2) and dictazoline C-E (5–7). Evidence is presented for the direct conversion of the cyclobutyl analogue 1 to its cyclohexyl constitutional isomer 5, via a vinyl cyclobutane rearrangement.
Marine sponges belonging to the family Thorectidae, and genus Smenospongia in particular, are well-known sources of indole alkaloids.1 Consistent with these observations, we recently reported the isolation of two compounds, dictazoline A (3) and B (4),2 from a Panamanian sponge identified as S. cerebriformis (Duchassaing & Michelotti, 1864) (order Dictyoceratida: family Thorectidae). Related alkaloids3 are proposed to be Diels-Alder adducts of aplysinopsin (8),4 but attempts to affect this transformation have been unsuccessful.3b Baran et al. have demonstrated the related alkaloid ageliferin (11), originally proposed to be formed via a Diels-Alder reaction of hymenidin,5,6 can be efficiently synthesized via a vinyl cyclobutane rearrangement of sceptrin (9) (Scheme 1A).7 This elegant synthesis supports an alternative unprecedented biosynthetic proposal for this dimeric compound.7
Scheme 1.

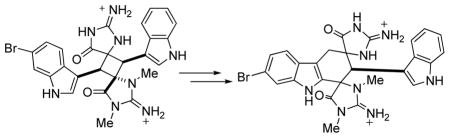
Vinyl Cyclobutane Rearrangements of Sceptrin (9) and Dictazole A (1).
We report here the isolation of dictazole A (1), B (2) and dictazolines C-E (5–7) from the same extract which provided 3 and 4. In addition, we present evidence for the direct conversion of 1 to the constitutional isomer 5, presumably via a vinyl cyclobutane rearrangement (Scheme 1B). In this case, the rearrangement of the cyclobutyl ring system involves an indole rather than the imidazole ring found in 9. These results suggest a more general role for this reaction in the biosynthesis of marine alkaloids and represent only the second example of a vinyl cyclobutane rearrangement featuring an indole ring.8
LC-MS analyses of the dictazoline-containing extract revealed the presence of several additional brominated metabolites. Extensive chromatographic separations eventually yielded 1, which lacked the expected AB spin system for H2-8 observed in the 1H NMR spectra of 3 and 4. Analyses of the DEPT and multiplicity-edited HSQC spectra confirmed this position in 1 was modified, as the compound contained only methine, methyl and quaternary carbons.
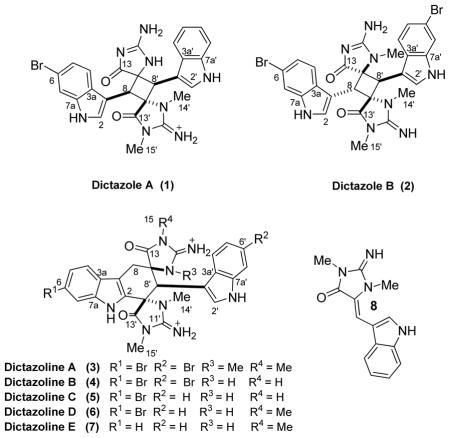
The structure of 1 was defined by analyses of the 2D NMR spectroscopic data (DMSO-d6 and MeOH-d4). Two indole rings substituted at C-3 were easily assembled based on a suite of HMBC and COSY correlations (Table 1). A spiro-2-imino-imidazolidin-4-one ring analogous to those in 4 was deduced based on HMBC correlations from the N-methyls to the adjacent quaternary carbons (H-15′ to C-11′/13′ and H-14′ to C-9′/11′) and carbon chemical shift comparisons with 4 in MeOH-d4. The two nonequivalent methine signals (H-8/H-8′) displayed HMBC correlations to C-9, C-9′, C-13, C-13′, and to either C-8 or C-8′. Together these data indicated 1 contained a cyclobutyl rather than a cyclohexenyl core. Analyses of the ROESY and 1D-DPFGSE NOE spectra established the configurations of C-8, C-8′, and C-9′ based on correlations between H-8′ and H-14′ and between H-8′ and H-8 (See supporting information Figures S13 & S14). The relative configuration of C-9 remains undetermined though as attempts to crystallize our sample were unsuccessful due to decomposition.
Table 1.
NMR Spectroscopic Data for 1 in DMSO-d6
| C/H No. | δC | δH, mult (J in Hz) | HMBC | ROESY |
|---|---|---|---|---|
| 2 | 124.6,CH | 7.15, s | H-8, H-15′ | |
| 3 | 106.5,C | H-2, H-4, H-8 | ||
| 3a | 126.4,C | H-2, H-4, H-5, H-7, H-8 | ||
| 4 | 119.5,CH | 7.25, d (8.3) | H-8, H-14′ | |
| 5 | 121.9,CH | 7.07, d (8.3) | ||
| 6 | 114.2,C | H-4, H-5, H-7 | ||
| 7 | 114.3,CH | 7.54, s | ||
| 7a | 136.3,C | H-2, H-4 | ||
| 8 | 43.4,CH | 4.46, s | H-8′ | H-4, H-2, H-14′ |
| 9 | 67.2,C | H-8′, H-8, H-10 | ||
| 10 | 8.16, s | H-2′ | ||
| 11 | 170.9,C | H-10 | ||
| 13 | 188.4,C | H-8, H-8′, H-10 | ||
| 2′ | 123.6,CH | 7.13, s | H-8, H-10 | |
| 3′ | 105.9,C | H-2′, H-4′, H-8′ | ||
| 3a′ | 127.4,C | H-2′, H-4′, H-5′, H-7′, H-8′ | ||
| 4′ | 117.7,CH | 7.31, d (8.0) | H-8, H-14′ | |
| 5′ | 119.1,CH | 6.95, t (8.0) | ||
| 6′ | 121.6,CH | 7.05, t (8.0) | ||
| 7′ | 111.7,CH | 7.32, d (8.0) | ||
| 7a′ | 135.3,C | H-2′, H-4′, H-6′ | ||
| 8′ | 43.6,CH | 4.49, s | H-8′ | H-2′, H-14′ |
| 9′ | 72.7,C | H-8, H-8′, H-14′ | ||
| 11′ | 153.5,C | H-14′, H-15′ | ||
| 13′ | 172.5,C | H-8, H-8′, H-15′ | ||
| 14′ | 25.8,CH3 | 3.21, s | H-4′, H-8′,H-4, H-8 | |
| 15′ | 25.1,CH3 | 2.73, s | H-2 |
The 13C NMR spectrum of 1 was strongly dependent on the NMR solvent. Specifically, in MeOH-d4 the “amide” C-13 and “guanidino” C-11 resonated as expected at 173.8 and 157.0 ppm, respectively, but in DMSO-d6 these signals shifted downfield significantly to 188.4 (C-13) and 170.9 ppm (C-11). A solvent-dependent tautomerization between 2-amino-imidazolone (1) and 2-imino-imidazolidinone (13, Figure 1) explained these observations, as in the former tautomer (1) the lone pair on the “amide” nitrogen resided in a sp2 orbital perpendicular to the π-system. These chemical shift assignments were consistent with spectroscopic data reported for the creatinine derivatives 14 and 15.9
Figure 1.

Solvent-dependent Tautomerization.
Several related analogues were also identified in the crude extract. In most cases, simple inspection of the 1H NMR spectra in conjunction with HRMS data enabled the planar structures to be proposed (See supporting information for tabulated NMR data). Briefly, compound 2 was bromo-10-N-methyl 1, with a molecular formula of C27H24Br2N8O2. The additional N-methyl group that was assigned as H-14, based on HMBC correlations, facilitated the assignment of the relative configuration of 2. In 1D-DPFGSE NOE experiments, correlations were observed between H-8 and H-14 and between H-8′ and H-14′ (See supporting information Figures S32 & S33). No correlation was present between H-8 and H-8′ for 2, which contrast sharply with 1, suggesting different relative configurations of the two compounds.10 Additional circumstantial evidence in support of the epimeric nature of 1 and 2 was the notable chemical shift difference observed for these methines in DMSO-d6 (Δδ1–2C-8 −0.9; Δδ1–2C-8′ −2.5; Δδ1–2H-8 −0.51; Δδ1–2H-8′ −0.63). As deduced by the ESI-MS data, compound 5 was a constitutional isomer of 1. In contrast to 1 though, the 1H NMR spectrum of 5 contained diagnostic signals for the H2-8 AB system of the cyclohexenyl ring, which in conjunction with 2D NMR data, established the planar structure. Compound 6 was 12-N-methyl-5, based on the extra methylamide resonance in the 1H NMR spectrum, the HMBC correlations to C-13 and C-11 from the new methyl resonance, and HR-ESI MS data. Finally, 7 was desbromo-6, (See supporting information). The relative configurations at C-8′ and 9′ of 5-7 were established after analyses of their 2D ROESY spectra, while the configuration of C-9 was deduced by comparison with 13C NMR data for 3 and 4.2
Dictazole A inhibited the aspartic protease BACE1 (memapsin 2). This protease is widely believed to have a central role in the pathology of Alzheimer’s disease.11 As such, pharmacological intervention that reduces BACE1 activity should be therapeutically beneficial. Dictazole A inhibited BACE1-mediated cleavage of amyloid precursor protein (APP) in a dose dependent manner with an IC50 value of 50 μg/mL. Interestingly, the 2-imino-imidazolidinone moiety within the dictazoles is common in several BACE1 inhibitors and has led to the suggestion that this privileged subunit is responsible for the observed activity against BACE1.12
Compounds 1 and 2 are unusual. The closest related alkaloids containing cyclobutane rings are sceptrin (9) and orthidine E.13 Baran et al. have proposed a biosynthesis of 11 involving a dicationic diradical vinyl cyclobutane rearrangement (Scheme 1) of 9.14 Evidence for this hypothesis includes computational data15 and the direct microwave conversion of 9 to 11.7 To date, no other potential examples of this biosynthetic rearrangement have been demonstrated.
Given these results, the isomers 1 and 5 are intriguing. The cyclobutyl alkaloid 1 could be a precursor to 5 via a related reaction (Scheme 1). Rearrangement of 1 via the intermediate 12 would result in ring expansion to the cyclohexenyl derivative 5 after double bond isomerization. In this case, the rearrangement would involve an indole rather than a 2-amino-imidazole ring and the electron deficient intermediate 12 would be stabilized by the pendant 2-imino-imidazolidinone moiety as compared to a 2-amino-imidazole. Circumstantial evidence is the relative abundance of the two isolated compounds. As is the case with 9 and 11, the cyclobutyl derivative 1 is isolated in higher yields than the cyclohexenyl analogue 5.
To examine the feasibility of this transformation, two 100 μg aliquots were prepared from the same sample of 1. One sample was dissolved in water, sealed, and heated in a microwave at 200 °C for one minute, similar to the conditions reported for sceptrin.16 The other sample was heated to 150 °C in methanol. While no product was observed in the methanol reaction mixture, remarkably, careful analysis of the aqueous reaction mixture by LC-MS (Figure 2B) revealed a new peak with the same retention time, m/z ratio, and M+2 isotope pattern as 5. HR-ESI mass spectrometry provided pseudomolecular ion peaks at 559.1174 and 561.1144 in approximately a 1:1 ratio that corresponded to the expected molecular formulae (errors of 5 and 7 ppm, respectively). The product was not observed in the starting material (see supporting information Figure S54) or in the methanol control prepared from the same sample of 1.16 It should be noted that an identical solvent dependency was observed for the conversion of 9 to 11. While the conversion proceeded smoothly in water, sceptrin decomposed when heated methanol.17 Due to the limited amount of 1 isolated, the yield of this transformation has not been optimized and the products have not been characterized by NMR. The tentative identification of 5 in the reaction mixture, therefore rests on the standard practice of comparing the retention time and the ionization pattern of an unknown with a standard. It is possible though that another isomer, for example, derived from a single bond scission of the cyclobutane ring, may coelute with 5. As such, final confirmation of this transformation will likely require the synthesis of 1 to provide sufficient material to address these issues.
Figure 2.
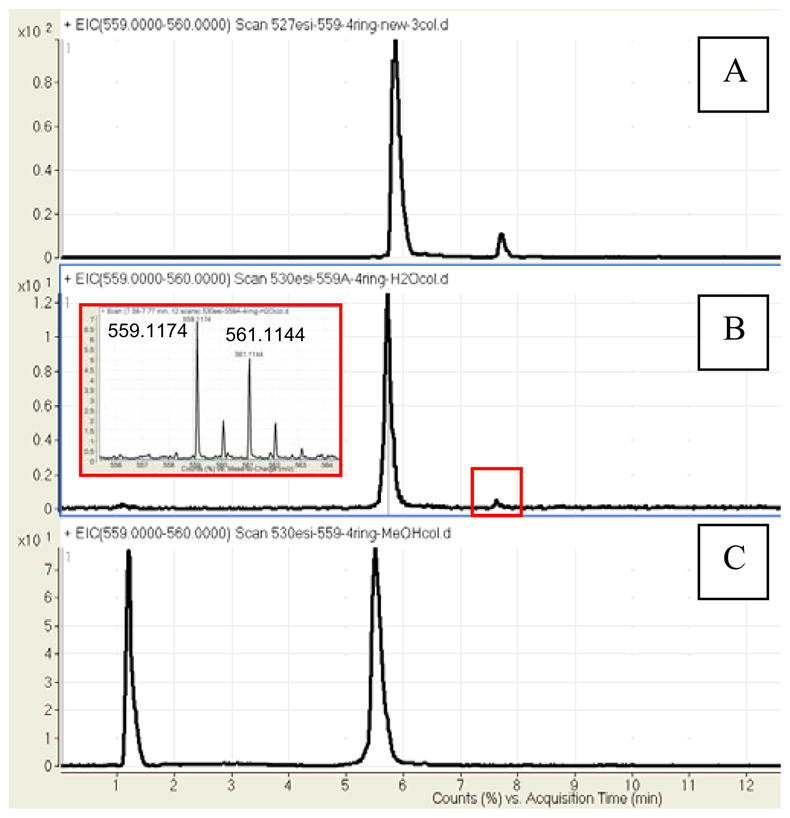
LC-MS Extracted Ion Chromatograms (m/z 559–560) (A) Standards 1 (major) and 5 (minor) (B) Crude microwave reaction in H2O of pure 1 after one minute at 200 °C (C) Crude microwave reaction in MeOH of pure 1 after one minute at 150 °C; the peak at one minute is a result of deliberate overloading of the HPLC column to ensure 5 is not present in that reaction mixture.
These results deserve comment. First, the reaction mixture containing the product 5 was comprised mostly of starting material and fragmentation products. Specifically, pseudomolecular ions consistent with (E)- and (Z)-isomers of 16–18 were present.18 Baran et al. have noted the interconversion of 9 to 11 is strongly dependent on the counterion, with the highest yields obtained with formate or acetate salts.15 It is possible the low yield of our reaction is attributable to a similar counterion dependency with the formate salt being less than ideal for this substrate.
These results suggest the possible involvement of a vinyl cyclobutane rearrangement in the biosynthesis of 3–7, as opposed to the Diels-Alder reaction suggested by Mancini et al. for the cycloaplysinopsins.19 Interestingly, during the isolation of this latter class of compounds, a constitutional isomer of cycloaplysinopsin A was identified by LC-MS that was attributed to a diastereomeric Diels-Alder adduct. Our results raise the possibility that this uncharacterized metabolite may instead be a cyclobutyl isomer.
Based on NMR experiments with chiral shift reagents in CDCl3, the same group proposed that cycloaplysinopsin was a scalemic mixture. We attempted to duplicate these experiments with 1. Unfortunately, 1 is not soluble in CDCl3 and attempts to titrate this compound with Eu(fod)3 in CD3CN have been unsuccessful. This failure is due to the hygroscopic nature of the solvent required and the trace amounts of 1 remaining (200 μg).20
To the best of our knowledge, the conversion of 1 to 5 is only the second example of a vinyl cyclobutane rearrangement involving an indole ring and the first for a natural product. Our data suggests this rearrangement may play a larger role in the biosynthesis of alkaloids from marine invertebrates than previously appreciated, and suggests a possible route towards the synthesis of this family of compounds.
Experimental Section
Extraction and Isolation of BMNH 2000.12.11.6
The freeze-dried sponge (114 g) was exhaustively extracted with 1:1 i-PrOH:CH2Cl2 (3 × 3 L) to afford 14.85 g of lipophilic extract. Partitioning using a modified Kupchan procedure yielded hexane (6.07 g), DCM (1.88 g), n-BuOH (2.94 g) and H2O (5.78 g) fractions. The residue from the n-BuOH phase was separated on a Sephadex LH-20 column eluting with MeOH and the resulting fractions were pooled based on TLC analyses into seven fractions. These fractions were subsequently separated by a combination of Si flash chromatography and RP-HPLC to yield 1, 2, and 5–7.
Dictazole A
(1, 4.5 mg, 3.0 × 10−2 % yield): colorless powder; [α]D22 +8.5 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 223 (2.5) 284 (2.4) nm; IR (CaF2) νmax 3337, 1643, 1592, 1352 cm−1; See Table S1 (DMSO-d6) and Table S2 (MeOH-d4) for tabulated spectral data; HRESI-TOFMS m/z 561.1206 [M + H]+ [Calcd for C26H2481BrN8O2+, 561.1185, +3.7 ppm].
Dictazole B
(2, 0.8 mg, 5.0 × 10−3 % yield): colorless powder; [α]D22 −42.5 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 228 (2.5) 288 (1.9) nm; IR (CaF2) νmax 3392, 1653, 1591, 1352 cm−1; See Table S3 for tabulated spectral data; HRESI-TOFMS m/z [M + H]+ 651.0490 [Calcd for C27H2579Br2N8O2+, 651.0467, +3.5 ppm].
Dictazoline C
(5, 1.5 mg, 1.0 × 10−2 % yield): colorless powder; [α]D22 −19.2 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 225 (2.6) 289 (1.9) nm; IR (CaF2) νmax 3542, 1646 cm−1; See Table S4 for tabulated spectral data; HRESI-TOFMS m/z 559.1221 [M + H]+ [Calcd for C26H2479BrN8O2+, 559.1206, +2.8 ppm].
Dictazoline D
(6, 2.5 mg, 1.7 × 10−2 % yield): colorless powder; [α]D22 −1.1 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 283 (9.14) nm; IR (CaF2) νmax 3422, 2930, 1656, 1586 cm−1; See Table S5 for tabulated spectral data; HRESI-TOFMS m/z 573.1352 [M + H]+ [Calcd for C27H2679BrN8O2+, 573.1362, −1.7 ppm].
Dictazoline E
(7, 0.5 mg, 3.4 × 10−3 % yield): colorless powder; [α]D22 −22.5 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 220 (4.6) 283 (3.8) nm; IR (CaF2) νmax 3542, 1646 cm−1; See Table S5 for tabulated spectral data; HRESI-TOFMS m/z 495.2279 [M + H]+ [Calcd for C27H27N8O2+, 495.2257, +4.4 ppm].
Supplementary Material
Acknowledgments
This work was funded by grants from the Victoria S. and Bradley L. Geist Foundation (20070461), the NSF (OCE04-32479), and the NIEHS (P50 ES012740). Upgrades of the NMR instrumentation were provided by the CRIF program of the NSF (CH E9974921) and the Elsa U. Pardee Foundation. The purchase of the LC-MS TOF was funded by the DOD (W911NF-04-1-0344). We thank W. Yoshida (UH) for the NMR data, A. Johnson and G. Jiao (PanThera Biopharma) for assistance with the microwave reactions, and C. Hughes (UCSD) for the CD spectra.
Footnotes
Supporting Information Available. Experimental details, tabulated NMR data for 1–2, copies of the relevant spectroscopic data, and LC-MS traces from the microwave conversion of 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Blunt JW, Copp BR, Hu WP, Munro MHG, Northcote PT, Prinsep MR. Nat Prod Rep. 2008;25:35–94. doi: 10.1039/b701534h. [DOI] [PubMed] [Google Scholar]
- 2.Dai J, Jiménez JI, Kelly M, Barnes S, Lorenzo P, Williams PG. J Nat Prod. 2008;71:1287–1290. doi: 10.1021/np8001018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Iwagawa T, Miyazaki M, Okamura H, Nakatani M, Doe M, Takemura K. Tetrahedron Lett. 2003;44:2533–2535. [Google Scholar]; b) Mancini I, Guella G, Zibrowius H, Pietra F. Tetrahedron. 2003;59:8757–8762. [Google Scholar]
- 4.Kazlauskas R, Murphy PT, Quinn RJ, Wells RJ. Tetrahedron Lett. 1977;1:61–64. [Google Scholar]
- 5.a) Kobayashi J, Tsuda M, Murayama T, Nakamura H, Ohizumi Y, Ishibashi M, Iwamura M, Ohta T, Nozoe S. Tetrahedron. 1990;46:5579–5586. [Google Scholar]; b) Endo T, Tsuda M, Okada T, Mitsuhashi S, Shima H, Kikuchi K, Mikami Y, Fromont J, Kobayashi J. J Nat Prod. 2004;67:1262–1267. doi: 10.1021/np034077n. [DOI] [PubMed] [Google Scholar]
- 6.For an alternative proposals involving non-concerted cyclizations see Keifer PA, Schwartz RE, Koker MES, Hughes RG, Jr, Rittschof D, Rinehart KL. J Org Chem. 1991;56:2965–2975.Mourabit AA, Potier P. Eur J Org Chem. 2001;2:237–243.
- 7.Baran PS, O’Malley DP, Zografos AL. Angew Chem, Int Ed. 2004;43:2674–2677. doi: 10.1002/anie.200453937. [DOI] [PubMed] [Google Scholar]
- 8.Wenkert E, Moeller PDR, Piettre SR, McPhail AT. J Org Chem. 1987;52:3404–3409. [Google Scholar]
- 9.Krawczyk H, Pietras A, Kraska A. Spectrochim Acta, Part A. 2007;66A:9–16. doi: 10.1016/j.saa.2006.02.035. and references therein. [DOI] [PubMed] [Google Scholar]
- 10.C-9 epimers of compounds related to 3 have been previously reported by Mancini (see Reference 3b).
- 11.Hardy J. Curr Alzheimer Res. 2006;3:71–73. doi: 10.2174/156720506775697098. [DOI] [PubMed] [Google Scholar]
- 12.Hills ID, Vacca JP. Curr Opin Drug Disc. 2007;10:383–391. [PubMed] [Google Scholar]
- 13.Pearce AN, Chia EW, Berridge MV, Maas EW, Page MJ, Harper JL, Webb VL, Copp BR. Tetrahedron. 2008;64:5748–5755. [Google Scholar]
- 14.For a review on vinyl cyclobutane rearrangements see: Baldwin JE, Leber PA. Org Biomol Chem. 2008;6:36–47. doi: 10.1039/b711494j.
- 15.Northrop BH, O’Malley DP, Zografos AL, Baran PS, Houk KN. Angew Chem, Int Ed. 2006;45:4126–4130. doi: 10.1002/anie.200600514. [DOI] [PubMed] [Google Scholar]
- 16.The injection amount in Figure 2a and 2c was approximately 10x the amount injected in Figure 2b.
- 17.O’Malley DP, Li K, Maue M, Zografos AL, Baran PS. J Am Chem Soc. 2007;129:4762–4775. doi: 10.1021/ja069035a. [DOI] [PubMed] [Google Scholar]
- 18.16, m/z 334.0200 (calcd for C14H1379BrN3O2+ 334.0191, +2.7 ppm), m/z 336.0160 (calcd for C14H1381BrN3O2+ 336.0171, −3.2 ppm); 17 m/z 256.1086 (calcd for C14H14N3O2+ 256.1086, 0.0 ppm), m/z 278.0902 (calcd for C14H13N3O2Na+ 278.0902, −1.2 ppm); 18 m/z 320.0033 (calcd for C13H1179BrN3O2+ 320.0035, −0.5 ppm), 321.9998 (calcd for C13H1181BrN3O2+ 322.0014, −5.0 ppm).
- 19.The occurrence of these similar metabolites in two dissimilar sources suggests that the true producer may be microbial.
- 20.The method development required for chiral HPLC analysis of 1 has not been undertaken.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.