

Abstract
TLR 4 stimulation of innate immune cells induces a MyD88-independent signaling pathway that leads to the production of IFN-β. In this study, we demonstrate glycogen synthase kinase 3-β (GSK3-β) plays a fundamental role in this process. Suppression of GSK3-β activity by either pharmacological inhibition, small interfering RNA-mediated gene silencing, or ectopic expression of a kinase-dead GSK3-β mutant enhanced IFN-β production by TLR4-stimulated macrophages. Conversely, ectopic expression of a constitutively active GSK3-β mutant severely attenuated IFN-β production. GSK3-β was found to negatively control the cellular levels of the transcription factor c-Jun and its nuclear association with ATF-2. Small interfering RNA-mediated knockdown of c-Jun levels abrogated the ability of GSK3-β inhibition to augment IFN-β, demonstrating that the ability of GSK3 to control IFN-β production was due to its ability to regulate c-Jun levels. The ability of GSK3 inhibition to control IFN-β production was confirmed in vivo as mice treated with a GSK3 inhibitor exhibited enhanced systemic levels of IFN-β upon LPS challenge. These findings identify a novel regulatory pathway controlling IFN-β production by TLR4-stimulated innate immune cells.
TLRs are type I transmembrane receptors involved in the recognition of highly conserved microbial components (1). Activation of TLRs on innate immune cells can result in the recruitment of different downstream signaling adaptors that impart selectivity on the repertoire of cytokines produced (1). In this regard, the TLR4-signaling pathway can activate distinct innate immune responses via the recruitment of the adaptor molecules TIRAP-MyD88 or TRAM-TRIF (2–5). The production of pro- and anti-inflammatory cytokines by TLR4-stimulated innate immune cells has been shown to be dependent upon signaling events initiated by TIRAP-MyD88 (2, 3, 6). In contrast, the recruitment of the adaptor molecules TRAM and TRIF mediate a signaling cascade involving the activation of the two noncanonical IκB kinases, TBK-1 and IKK-ε, as well as the phospho-specific post translational modifications of the transcription factors NF-κB, ATF-2/c-Jun, and IRF-3 that culminates in the production of type I IFNs, including IFN-β (4, 5, 7–9). Although the molecular mechanisms regulating NF-κB and IRF-3 activity, as well as their involvement in controlling IFN-β production by TLR4-stimulated innate cells have been well described, the upstream signaling events that regulate the levels and activity of the transcriptional complex, ATF-2/c-Jun, and the role this complex plays in controlling IFN-β production by TLR4-stimulated cells is poorly understood.
Stimulation of TLR4 can activate the PI3K pathway, which restrains the MyD88-dependent production of proinflammatory cytokines (10–12). In the presence of MyD88, the ability of the PI3K pathway to negatively regulate the production of proinflammatory cytokines, while augmenting the levels of the anti-inflammatory cytokine, IL-10, is due to its ability to inactivate the constitutively active serine/threonine kinase, glycogen synthase kinase 3-β (GSK3-β)3 in TLR4-stimulated cells (11, 13–15). The serine 9 mediated inactivation of GSK3-β results in the alteration of the transcriptional complex involving the coactivator of transcription CBP, and the transcription factors CREB and NF-κB (14). Although GSK3-β has been shown to regulate MyD88-dependent cytokine responses, whether GSK3-β plays a functional role in the regulation of the prototypical MyD88-independent cytokine, IFN-β, is currently unknown.
In the present study, we show that GSK3-β activity plays a fundamental role in regulating IFN-β production. Specifically, we show that the phosphorylation of GSK3-β (S9) in LPS-stimulated macrophages occurs in the absence of MyD88. Inhibition of GSK3-β activity potently augmented the levels of IFN-β in LPS-stimulated innate immune cells, whereas the ectopic expression of a constitutively active GSK3-β mutant reduced IFN-β production. Inhibition of GSK3-β was found to control the cellular levels of the transcription factor c-Jun and this was demonstrated to be necessary for the ability of GSK3 to control IFN-β production. The functional role of GSK3-β in regulating IFN-β was confirmed in vivo in which the inhibition of GSK3-β potently enhanced the systemic levels of IFN-β in mice administered LPS. Taken together, these findings identify GSK3-β as a critical regulatory kinase controlling IFN-β production.
Materials and Methods
Mice and reagents
C57BL/6 mice were purchased from The Jackson Laboratory. B6.MyD88−/− mice were a gift from Shizuo Akira (via Ross Kedl, 3M Corporation) and were backcrossed >6 generations onto the C57BL/6 background. Mice were housed in a specific pathogen-free facility at the University of Louisville School of Medicine and the University of Louisville Institutional Animal Care and Use Committee approved all animal protocols. Ultra pure LPS from Escherichia coli was purchased from Invivogen. All Abs and recombinant cytokines were obtained from Cell Signaling Technology and R&D Systems, respectively. The anti-HA Ab used for immunoblots was purchased from eBioscience. The GSK3-specific inhibitor SB216763 was previously characterized and was shown to be highly specific for GSK3 without discernible effects on a panel of 24 other kinases (16). SB216763 was purchased from Tocris. Small interfering RNAs (siRNAs) were purchased from Dharmacon. The plasmid pcDNA3-GSK3β(S9A) and pcDNA3-GSK3β(K85A) were obtained from Addgene (plasmid numbers 14754 and 14755) and originally created by Dr. James Woodgett’s laboratory (17). The nuclear levels of NF-κB p65 and IRF-3 were measured using the TransAM kit purchased from Active Motif. The amount of nuclear NF-κB p65 or IRF-3 was normalized by the absorbance at 450 nm from 10 μg (NF-κB p65) or 20 μg of nuclear lysate (IRF-3).
Cell preparation
Bone marrow derived macrophages (BM-DM) were prepared by culturing bone marrow from the femurs/tibiae of 6–10-wk-old mice in RPMI 1640 containing 5% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 50 U/ml penicillin, 50 μg/ml streptomycin, 10 ng/ml M-CSF, and 30% L929 conditioned medium. Nonadherent cells were collected after 24 h and cultured for 7 days in Costar ultra low attachment polystyrene culture dishes with a medium change on day 4. BM-DM were >85% CD11b+, as demonstrated by flow cytometry.
In vivo levels of IFN-β
Male C57BL/6 mice (8–12 wk of age; 20–28 g body weight) were injected i.p. with a 5 μg/g of LPS in 100 μl of PBS containing 0.1% DMSO. Mice were analyzed for systemic levels of IFN-β 6 h after being administered LPS in the presence of 0.1% DMSO or 10 μg/g of SB216763. The Institutional Animal Care and Use Committee of the University of Louisville approved all studies.
Transfections and IFN-β production
BM-DM were transfected with nontargeting control (Ctrl) siRNA, siRNA-c-Jun, siRNA-GSK3-β, pcDNA3-GSK3β(S9A), pcDNA3-GSK3β(K85A), or pcDNA3 (empty vector control) using Lipofectamine RNAiMAX (Invitrogen), or Lipofectamine LTX (Invitrogen) following the manufacturer’s protocol. The levels of total c-Jun and GSK3-β protein were assessed by Western blot on day 3. In 96-well plates, 2 × 105 BM-DM were cultured and pretreated for 2 h with 0.01% DMSO (organic solvent control for SB216763) or the GSK3 inhibitor SB216763 (SB216763 at 12 μM) before LPS (1 μg/ml) stimulation. Transfected BM-DM were stimulated with LPS on day 3 post transfection. Cell-free supernatants were assayed for IFN-β levels by ELISA 20 h after the addition of LPS (R&D Systems).
RT-PCR, immunoprecipitation, immunoblots, and statistical analysis
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) and real-time PCR was performed using an ABI 7500 system. GAPDH was used as the endogenous control and fold increase was calculated according to ΔΔCT method. At the indicated time points, cells were harvested and analyzed by immunoblot or immunoprecipitation as previously described (12, 14). The Kodak 4000 MM image system was used for obtaining all images and densitometer scans of the blots. The mouse True Blot kit (eBioscience) was used for all immunoprecipitations according to the manufacturer’s protocol. A rabbit isotype control IgG Ab (Cell Signaling Technology) was used for all immunoprecipitations to ensure that the immunoprecipitation of ATF-2 and subsequent immunodetection of c-Jun were not due to nonspecific interference. Data are expressed as mean ± SD of a minimum of three experiments. Statistical significance between groups was evaluated by ANOVA and the Tukey multiple comparison test (Instat Program). Differences were considered significant at p < 0.05.
Results
TLR4-mediated phosphorylation of GSK3β (S9) occurs in the absence of MyD88
LPS stimulation of innate immune cells has been shown to promote GSK3-β inactivation via the phosphorylation of serine 9 (S9) (11, 14). To assess whether MyD88 is required for LPS to induce GSK3-β (S9) phosphorylation, wild-type and MyD88-deficient cells were compared in their abilities to phosphorylate GSK3-β (S9) upon LPS stimulation (Fig. 1A). Both wild-type and MyD88-deficient macrophages demonstrated increased phospho-GSK3β (S9) levels after 30 and 60 min of culture in the presence of LPS, as compared with nonstimulated cells, (Fig. 1A). The phosphorylation of GSK3-β (S9) in both wild-type and MyD88-deficient cells was abrogated by the use of the PI3K inhibitors LY294002 or wortmannin (data not shown). A comparison of the ratios of phospho-GSK3-β (S9) to that of total β-actin was similar between wild-type and MyD88-deficient cells (Fig. 1, B and C). These results demonstrate that the phosphorylation of GSK3-β (S9) in LPS-stimulated macrophages occurs in the absence of MyD88.
FIGURE 1.
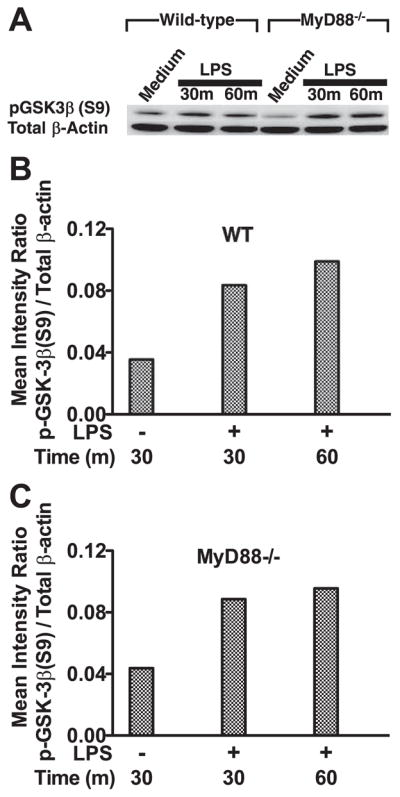
Stimulation of macrophages with LPS induces the phosphorylation of GSK3-β (S9) in both wild-type and MyD88-deficient macrophages. A, Wild-type and MyD88-deficient macrophages were stimulated with 1 μg/ml LPS for 30 or 60 min. To assess phospho-GSK3-β (S9), 15 μg of total cell lysate was resolved on LDS-PAGE, immunoblotted with an anti-phospho GSK3-β (S9) Ab, and developed by ECL. Immunoblots were stripped and reprobed with an Ab to total β-actin to ensure equal protein loading. B and C, Densitometer scans of phospho-GSK3-β (S9) and total β-actin were performed and recorded as the ratio of phospho-GSK3-β (S9):total β-actin. Data are representative of three separate experiments.
GSK3β negatively regulates TLR4-induced IFN-β production
Because LPS induced the phosphorylation of GSK3-β (S9) in the absence of MyD88 (Fig. 1), we next investigated whether GSK3 played a role in the production of the MyD88-independent cytokine, IFN-β. Pharmacological inactivation of GSK3 using the GSK3 inhibitor SB216763 (16) resulted in the loss of the GSK3-specific substrate phospho-glycogen synthase (Ser640/641), demonstrating the ability of SB216763 to inactivate endogenous GSK3 activity in macrophages (Fig. 2A). GSK3-inactivated macrophages stimulated with LPS produced significantly (p < 0.05) more IFN-β protein and mRNA, as compared with cells stimulated with LPS alone (Fig. 2, B and C). Moreover, LPS stimulation of macrophages treated with GSK3-β-specific siRNA (>80% total GSK3-β protein knockdown, (Fig. 2D) exhibited more than a 3-fold increase (p < 0.05) in secreted IFN-β levels, as compared with cells treated with control siRNA and stimulated with LPS (Fig. 2E). TLR4-stimulation of macrophages expressing a kinase dead mutant of GSK3-β (K85A) inhibited endogenous GSK3 activity (Fig. 2, F and G) in macrophages and produced significantly (p < 0.05) elevated levels of IFN-β, as compared with empty vector transfected cells stimulated with LPS (Fig. 2H). In contrast, the ectopic expression of a constitutively active GSK3-β (S9A) mutant severely attenuated the production of IFN-β by LPS stimulated macrophages, as compared with empty vector transfected cells stimulated with LPS (Fig. 2H). Taken together, these results demonstrate that GSK3-β activity plays a fundamental role in controlling the production of IFN-β by TLR4-stimulated macrophages.
FIGURE 2.

GSK3-β negatively controls IFN-β production by LPS-stimulated macrophages. A, Macrophages treated with the GSK3 inhibitor SB216763 exhibited a loss in the phosphorylation levels of the GSK3-specific substrate glycogen synthase (Ser640/641). Inhibition of GSK3 augmented the mRNA (B) and protein levels (C) of IFN-β produced by TLR4-stimulated macrophages. siRNA-mediated knockdown of GSK3-β protein levels (D) increased the production of IFN-β (E) by LPS-stimulated (1 μg/ml) macrophages. F, HA expression levels in nontransfected macrophages or macrophages transfected with a kinase dead (K85A) or constitutively active (S9A) plasmid encoding GSK3-β. Levels of HA were detected by Western blot 48 h after transfection. G, Expression of the kinase dead (K85A) GSK3 mutant in macrophages inhibited the endogenous phosphorylation of the GSK3-specific substrate β-catenin (Ser33/37/Thr41). H, As compared with empty vector control macrophages stimulated with LPS (1 μg/ml), the constitutively active GSK3-β (S9A) and kinase dead GSK3-β (K85A) negatively and positively, respectively, regulated IFN-β production by macrophages stimulated with LPS (1 μg/ml). *, Statistically significant differences at p < 0.05 between the indicated groups. Results represent the mean ± SD of three separate experiments.
GSK3 controls the nuclear levels of c-Jun/ATF-2 complexes by regulating total c-Jun levels
We next examined whether serine/threonine phosphorylation by constitutively active GSK3 could be negatively affecting a downstream signaling molecule involved in the control of IFN-β production. Previous studies have shown that c-Jun is a component of the IFN-β enhanceosome (18) and that GSK3 can phosphorylate the transcription factor c-Jun on threonine 239 that, in turn, promotes c-Jun degradation (19, 20). We therefore tested whether GSK3 inhibition could affect the levels of phospho-c-Jun (Thr239) and total c-Jun in LPS-stimulated macrophages (Fig. 3, A and B). Inactivation of GSK3 abrogated the ability of LPS stimulation to augment the levels of phospho-c-Jun (Thr239) in macrophages (Fig. 3A). Furthermore, the total levels of c-Jun were discernibly increased in GSK3-inactivated macrophages after 60 and especially 120 min of culture in the presence of LPS, as compared with nonstimulated or LPS-stimulated cells (Fig. 3B).
FIGURE 3.

GSK3 negatively affects the levels of nuclear c-Jun/ATF-2 heterodimer complexes by controlling total c-Jun levels. Inhibition of GSK3 attenuates the levels of phospho-c-Jun (Thr239) (A) and increases total c-Jun levels (B) in LPS-stimulated macrophages. GSK3 inhibition increases the association of total (C) and phospho-c-Jun (Ser63/73) (D) to ATF-2 in LPS-stimulated macrophages. In contrast to the effect of GSK3 inhibition of c-Jun, GSK3 inhibition did not discernibly affect the nuclear levels of the transcription factors NF-κB (E) or IRF-3 (F). A and B, Cell lysates were prepared at the given time points, and 15 μg of total protein was analyzed by immunoblot using Abs to c-Jun (Thr239) or total c-Jun, stripped, and reprobed with an Ab to total β-actin to ensure equal protein loading. C and D, Nuclear lysates were prepared at the given time points, a rabbit isotype control (IC) IgG or total ATF-2 was immunoprecipitated, and associated total (C) and phospho-c-Jun (Ser63/73) (D) were determined by immunoblot. C and D, Total ATF-2 was monitored by immunoblot between groups to ensure equivalent pull-down of ATF-2. C and D, No immunoreactive bands were detected at 43 or 48 kDa (c-Jun) or 65–75 kDa (ATF-2) when a rabbit isotype control (IC) Ab was used for immunoprecipitation. E and F, The transcription factor binding levels of NF-κB p65 (E) using 10 μg of nuclear lysate or IRF-3 (F) using 20 μg of nuclear lysate were obtained from macrophages stimulated with LPS for 6h. A–D, Data are representative of three separate experiments. E and F, Data represent the mean ± SD of three separate experiments.
Because c-Jun has been shown to form a transcriptional complex with ATF-2 (18), we next examined whether GSK3-inhibition influenced the nuclear levels of c-Jun/ATF-2 complexes. Pull-down of nuclear ATF-2 and subsequent probing for associated c-Jun by Western blot demonstrated that GSK3 inhibition increased the total levels of c-Jun associated with ATF-2, as compared with the levels observed in cells stimulated with only LPS (Fig. 3C). Due to the transcriptional activity of c-Jun being regulated by phosphorylation on serines 63 and 73 (21), we also assessed the phosphorylation levels of these residues (Fig. 3D). Immunoprecipitation of nuclear ATF-2 and immunoblotting for associated levels of phospho-c-Jun (Ser63/Ser73) demonstrated that the levels of c-Jun (Ser63) and c-Jun (Ser73) were both highly elevated, as compared with macrophages stimulated with LPS alone (Fig. 3D). No discernible differences were observed in the levels of ATF-2 between groups (Fig. 3, C and D). In contrast to the increased nuclear levels of c-Jun observed in GSK3-inhibited macrophages stimulated with LPS, no significant changes in the nuclear levels of NF-κB p65 or IRF-3 were observed (Fig. 3, E and F). Therefore, active GSK3 negatively regulates the total cellular levels of c-Jun as well as the nuclear levels of c-Jun associated with ATF-2 in LPS-stimulated macrophages.
GSK3 inhibits TLR-4 induced IFN-β production by regulating c-Jun levels
To determine whether the increased c-Jun levels observed in GSK3 inhibited cells played a functional role in the ability of GSK3 to modulate IFN-β levels by LPS-stimulated macrophages, c-Jun levels were knocked down by transfecting macrophages with c-Jun-specific siRNA (Fig. 4A). Transfection with c-Jun-specific siRNA reduced c-Jun levels by >90%, as compared with non-transfected cells or cells transfected with control siRNA (Fig. 4A). The knockdown in c-Jun levels abrogated the ability of GSK3-inhibition to significantly elevate the levels of IFN-β produced by LPS-stimulated macrophages, as compared with control siRNA transfected cells stimulated with LPS (Fig. 4B). In contrast, GSK3-inhibited macrophages transfected with control siRNA and stimulated with LPS exhibited a significant (p < 0.05) increase in IFN-β levels, as compared with cells treated with control siRNA and stimulated with LPS (Fig. 4B). Thus, the ability of GSK3 to regulate the production of IFN-β by LPS-stimulated macrophages is dependent upon GSK3’s ability to increase the cellular levels of c-Jun.
FIGURE 4.

GSK3 controls IFN-β production by regulating c-Jun levels in LPS-stimulated macrophages. A and B, The siRNA-mediated knockdown in c-Jun protein levels abrogates the ability of GSK3 inhibition to augment IFN-β production by LPS-stimulated macrophages (1 μg/ml). A, Total c-Jun levels were determined 72 h post transfection by immunoblot. B, Cell-free supernatants were harvested 20 h after LPS stimulation (1 μg/ml) and analyzed for IFN-β levels by ELISA. *, Statistically significant differences at p < 0.05 between the indicated groups. Results represent the mean ± SD of three separate experiments.
GSK3 regulates the in vivo production of IFN-β
We next wanted to determine whether inhibiting GSK3 in vivo could modulate the induction of IFN-β in mice given a sublethal dose of LPS. For this, mice were administered the GSK3 inhibitor SB216763 or DMSO 2 h before receiving LPS. The systemic levels of IFN-β were monitored in mice 6 h after being given LPS (Fig. 5). Mice administered the GSK3 inhibitor SB216763 and challenged with LPS exhibited a significant increase in the levels of IFN-β, as compared with control mice that received DMSO and LPS (Fig. 5). No detectable levels of IFN-β were observed in mice given DMSO or SB216763 alone (Fig. 5). These findings demonstrate targeting GSK3 in vivo potently increases the levels of IFN-β upon LPS challenge.
FIGURE 5.
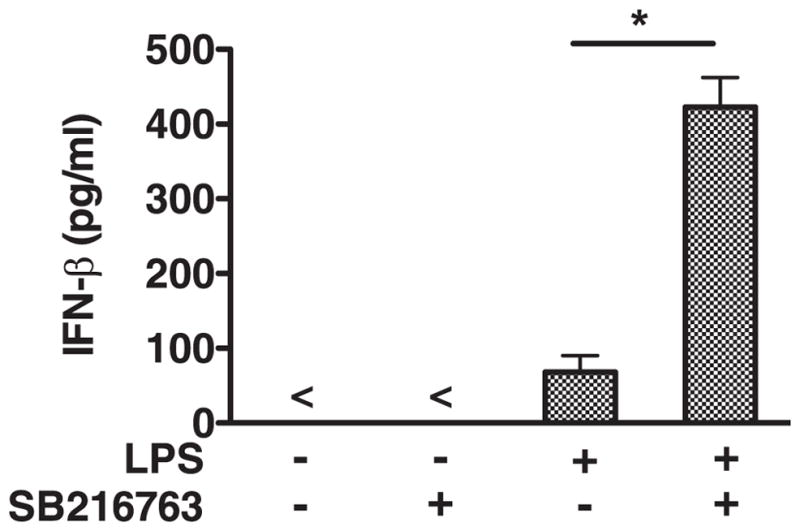
Inhibition of GSK3 augments the systemic levels of IFN-β by mice given LPS. Intraperitoneal administration of the GSK3 inhibitor SB216763 (10 μg/g) significantly increased the levels of IFN-β by mice given 5 μg/g of LPS. Results represent the mean ± SD of five mice/group. In vivo levels of IFN-β in plasma were measured by ELISA 6 h after mice were given LPS. Sham-immunized mice were given PBS containing 0.1% DMSO. *, Statistically significant differences at p < 0.05 compared with control mice given LPS containing 0.1% DMSO. All groups of mice were given PBS containing 0.1% DMSO with or without LPS or SB216763.
Discussion
The TLR4-signaling pathway in innate immune cells has been shown to mediate the induction of two distinct molecular pathways based on the usage of MyD88. The ability of TLR4 to recruit TIRAP-MyD88 and TRAM-TRIF to its cytosolic domain results in the production of proinflammatory cytokines and type I IFNs, respectively (4–9). Past studies by our group and others have demonstrated that the levels of MyD88-dependent cytokines, including both pro- and anti-inflammatory cytokines, were intimately controlled by GSK3-β. Specifically, the inactivation of GSK3-β has been shown to negatively influence the levels of proinflammatory cytokines while concurrently augmenting the levels of the anti-inflammatory cytokine IL-10 in response to TLR4-stimulation (13–15). The present study extends these previous findings by demonstrating that the inactivation of GSK3-β (Ser9) occurs in the absence of MyD88 and GSK3-β activity was a critical component of the regulatory mechanism that controlled the levels of IFN-β by TLR4-stimulated cells both in vitro and in vivo. Thus, while the TLR4 signaling complex can mediate the production of both pro- and anti-inflammatory cytokines and IFN-β via the recruitment of distinct adaptor molecules including TIRAP-MyD88 and TRAM-TRIF (4, 5, 7–9), respectively, the capacity of GSK3-β to regulate both MyD88-dependent (13–15, 21) and MyD88-independent cytokine responses highlights the central importance that GSK3-β plays in the regulation of the host innate inflammatory response.
The identification and characterization of the downstream cell-signaling events regulating the IFN-β response have been shown to involve the activation of the kinases TBK-1 and IKK-ε that are involved in the phosphorylation of the transcription factor IRF-3 and its subsequent dimerization and translocation into the nucleus (22–24). The critical importance of TBK-1 and IKK-ε in regulating IFN-β via IRF-3 are highlighted by the findings that cells deficient in IRF-3 are unable to produce IFN-β (25). In addition to IRF-3, studies analyzing the infβ promoter have revealed the presence of additional regulatory molecules that can bind one of the four positive regulatory domains within the ifnβ promoter. In this regard, ATF-2 and c-Jun have been shown to bind the ifnβ promoter via a heterodimeric complex within the positive regulatory domain IV of the ifnβ promoter (18, 26, 27). Although the findings of the present study did not observe any discernible effects of GSK3 inhibition on the activity of IRF-3 (H. Wang and M. Martin, unpublished observations), the ability of GSK3 to regulate the cellular levels of c-Jun were found to be of critical importance for the ability of GSK3 to modulate IFN-β production by LPS-stimulated innate immune cells. Moreover, siRNA-mediated knockdown of c-Jun levels in macrophages reduced the levels of IFN-β produced by cells stimulated with LPS. These findings demonstrate a fundamental role for c-Jun in the regulation of IFN-β and highlight the underlying molecular mechanism by which GSK3 regulates IFN-β by LPS stimulated innate immune cells.
The ability of GSK3 activity to differentially regulate the levels of MyD88-dependent pro- and anti-inflammatory cytokines while concurrently controlling the production of the MyD88-independent cytokine, IFN-β, demonstrated the existence of a cross-talk network between these two pathways mediated by GSK3. Interestingly, the downstream molecular mechanism by which GSK3-β regulated MyD88-dependent and MyD88-indpendent cytokine responses are unique. Studies by our laboratory were the first to show active GSK3-β negatively regulated the levels of the anti-inflammatory cytokine IL-10 while simultaneously promoting the production of proinflammatory cytokines by TLR-stimulated innate immune cells (14). Analysis of the mechanism by which GSK3-β influenced the transcriptional control of MyD88-dependent cytokines revealed GSK3 repressed the nuclear association of the transcription factor CREB (Ser133) with the coactivator of transcription CBP. Upon GSK3 inactivation, IL-10 production increased while proinflammatory cytokine production was severely suppressed due to the displacement of NF-κB p65 from CBP by CREB (14). Although the inhibition of GSK3 did exhibit similar effects on the nuclear levels of CREB as has been previously reported (14), knockdown in the levels of CREB within the context of the current study did not affect the ability of GSK3 inhibition to modulate IFN-β production by LPS-stimulated innate immune cells (H. Wang and M. Martin, unpublished observations). However, GSK3 was found to also control c-Jun levels that resulted in increased nuclear levels of ATF-2/c-Jun complexes. siRNA-mediated gene silencing of c-Jun demonstrated that the ability of GSK3 to regulate IFN-β production by TLR4-stimulated macrophages was dependent upon increased c-Jun levels. Thus, the ability of GSK3-β to regulate both MyD88-dependent and MyD88-independent cytokine responses occur via different molecular mechanisms.
In conclusion, we have identified that the constitutively active kinase, GSK3, plays a fundamental role in controlling the production of IFN-β. Our results showed that the production of IFN-β by LPS-stimulated macrophages was regulated by the activity of GSK3-β and its ability to affect the cellular and subsequent nuclear levels of the transcription factor c-Jun associated with ATF-2. Overall, the current findings identify GSK3 as a fundamental kinase involved in the regulation of the MyD88-independent cytokine, IFN-β, and provide a rationale to modulate the levels of IFN-β in vivo.
Acknowledgments
We thank Panagiota Stathopoulou for technical assistance.
Footnotes
This research was supported by Grant R01DE017680 from the National Institute of Dental Research.
Abbreviations used in this paper: GSK3-β, glycogen synthase kinase 3-β; BM-DM, bone marrow derived macrophage; siRNA, small interfering RNA.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 2.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 3.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 7.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 10.Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 11.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 12.Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- 13.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodionova E, Conzelmann M, Maraskovsky E, Hess M, Kirsch M, Giese T, Ho AD, Zoller M, Dreger P, Luft T. GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood. 2007;109:1584–1592. doi: 10.1182/blood-2006-06-028951. [DOI] [PubMed] [Google Scholar]
- 16.Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD, Reith AD. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J Neurochem. 2001;77:94–102. doi: 10.1046/j.1471-4159.2001.t01-1-00251.x. [DOI] [PubMed] [Google Scholar]
- 17.Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 β in intact cells via serine 9 phosphorylation. Biochem J. 1994;303:701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-β enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003;22:3876–3886. doi: 10.1093/emboj/cdg388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 22.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 23.Takahasi K, Fujita T, Inagaki F. Three dimensional structure of IRF-3 and its implication in signal transduction. Tanpakushitsu Kakusan Koso. 2004;49:1280–1287. [PubMed] [Google Scholar]
- 24.Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol. 2003;10:922–927. doi: 10.1038/nsb1001. [DOI] [PubMed] [Google Scholar]
- 25.Stockinger S, Reutterer B, Schaljo B, Schellack C, Brunner S, Materna T, Yamamoto M, Akira S, Taniguchi T, Murray PJ, et al. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol. 2004;173:7416–7425. doi: 10.4049/jimmunol.173.12.7416. [DOI] [PubMed] [Google Scholar]
- 26.Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG. Structure and function of the interferon-β enhanceosome. Cold Spring Harbor Symp Quant Biol. 1998;63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- 27.Panne D, Maniatis T, Harrison SC. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-β enhancer. EMBO J. 2004;23:4384–4393. doi: 10.1038/sj.emboj.7600453. [DOI] [PMC free article] [PubMed] [Google Scholar]