

SUMMARY
We report the crystal structures of the phosporylated pyruvate dehydrogenase (E1p) component of the human pyruvate dehydrogenase complex (PDC). The complete phosphorylation at Ser264-α (site 1) of a variant E1p protein was achieved using robust pyruvate dehydrogenase kinase 4 free of the PDC core. We show that unlike its unmodified counterpart, the presence of a phosphoryl group at Ser264-α prevents the cofactor thiamine diphosphate-induced ordering of the two loops carrying the three phosphorylation sites. The disordering of these phosphorylation loops is caused by a previously unrecognized steric clash between the phosphoryl group at site 1 and a nearby Ser266-α, which nullifies a hydrogen-bonding network essential for maintaining the loop conformations. The disordered phosphorylation loops impede the binding of lipoyl domains of the PDC core to E1p negating the reductive acetylation step. This results in the disruption of the substrate channeling in the PDC, leading to the inactivation of this catalytic machine.
INTRODUCTION
The human pyruvate dehydrogenase complex (PDC) catalyzes the oxidative decarboxylation of pyruvate to produce acetyl-CoA and NADH (Reaction 1), and is the gate-keeper enzyme that strategically links glycolysis to the Krebs cycle and lipogenic pathways (Patel and Roche, 1990; Reed, 2001).
| (1) |
PDC is a 9.5 × 106-dalton multi-enzyme complex organized around a dodecahedral core formed by 60 combined dihydrolipoyl transacetylase (E2p) and E3-binding protein (E3BP) subunits. To this E2p/E3BP core, multiple copies of the thiamin diphosphate (ThDP)-dependent pyruvate dehydrogenase (E1p) component, the FAD-containing dihydrolipoamide dehydrogenase (E3) component, one to two copies each of pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP) are tethered via non-covalent interactions (Reed, 2001; Roche and Hiromasa, 2007). Reaction steps catalyzed by the three catalytic components (E1p, E2p and E3) of PDC, which are linked through substrate channeling, are as follows:
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
The E1p component catalyzes the ThDP-mediated decarboxylation of pyruvate (Reaction 2) and the reductive acetylation of a lipoyl group ([lipS2]) covalently linked to the lipoyl-bearing domains (LBDs) of E2p and E3BP (Reaction 3). The acetylated LBDs ([CH3-CO-S-lipSH]) serve as “swinging arms” (Perham, 2000) to transfer the acetyl group from E1p to the E2p active site, where it is converted to acetyl-CoA (Reaction 4). Finally, the E3 component re-oxidizes the dihydrolipoyl moiety on LBDs ([lip(SH)2]) with NAD+ as the ultimate electron acceptor (Reactions 5 and 6).
Phosphorylation/inactivation of the PDC by pyruvate dehydrogenase kinase (PDK), discovered in late 1960’s, was among the first examples of enzyme regulation by reversible phosphorylation (Linn et al., 1969). Since then, phosphorylation of the PDC has been shown to play a pivotal role in regulating carbohydrate and lipid metabolism (Harris et al., 2002; Sugden and Holness, 2003). Starvation and diabetes increase phosphorylation that inactivates PDC, leading to impaired glucose oxidation (Holness et al., 2000; Kwon et al., 2004; Wu et al., 1999). Prevention of PDC phosphorylation by the specific PDK inhibitor dichloroacetate increases levels of reactive oxygen species in mitochondria, which promotes the expression of a mitochondria-K+ channel axis, leading to cellular apoptosis and the inhibition of tumor growth (Bonnet et al., 2007; Cairns et al., 2007). Therefore, the regulation of the PDC flux by reversible phosphorylation is potentially a novel drug target for obesity, type 2 diabetes and cancer (Pan and Mak, 2007; Sugden, 2008; Sugden and Holness, 2003).
To date, four PDK isoforms (1 to 4) in mammalian mitochondria have been identified (Popov et al., 1997). PDK isoforms are recruited to the PDC by preferentially binding to the inner LBD (L2) of the E2p subunit with concomitant increased kinase activity (Roche and Hiromasa, 2007). Co-localization of PDKs with the E1p substrate bound to the subunit-binding domain (SBD) of E2p results in enhanced PDK activity (Roche et al., 2003). In addition, conformational changes of PDK3 induced by L2 binding foster stimulation of PDK3 activity through facilitated ADP/ATP exchange, which removes product inhibition exerted by ADP (Bao et al., 2004; Kato et al., 2005). PDK4 has the lowest affinity for L2 and is only minimally stimulated by the E2p/E3BP core (Roche and Hiromasa, 2007). However, PDK4 was recently shown to possess the highest kinase activity among the four PDK isoforms, regardless of the presence of L2 or the E2p/E3BP core (Wynn et al., 2008).
Phosphorylation of the heterotetrameric (α2β2) E1p component by PDK isoforms occurs at three serine residues of the α subunit (Ser264-α: site 1, Ser271-α: site 2 and Ser203-α: site 3) (Figure 1) (Sale and Randle, 1981a; Teague et al., 1979; Yeaman et al., 1978). Crystal structures of human E1p have shown that sites 1 and 2 are located in the conserved phosphorylation loop A (Ph-loop A, residues 259-α – 282-α) (Ciszak et al., 2003), which forms one wall of the E1p active-site channel and helps anchor the cofactor ThDP to the active site. Site 3 is located in a short loop segment (residues 198-α – 205-α), termed phosphorylation loop B (Ph-loop B) adjacent to Ph-loop A. Ph-loop B also provides coordination to a Mg2+ ion chelated by the diphosphate group of ThDP. Phosphorylation of any of the three sites alone inactivates E1p, with site 1 being most rapidly phosphorylated and site 3 the slowest (Korotchkina and Patel, 1995; Sale and Randle, 1981a). Each PDK isoform exhibits different site specificity. Using E1p mutants with single functional phosphorylation sites, it was shown that all four isoforms phosphorylate sites 1 and 2, but only PDK1 phosphorylates site 3 (Kolobova et al., 2001; Korotchkina and Patel, 2001a).
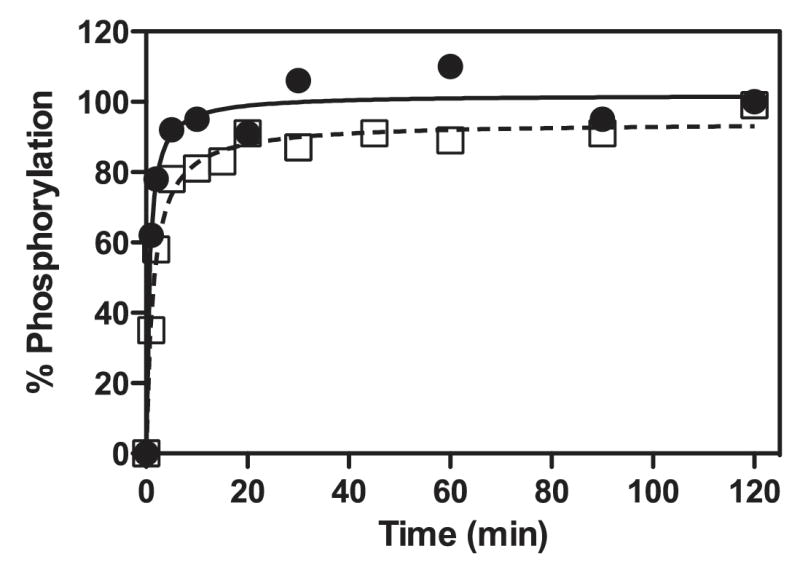
Figure 1. Time Course for Phosphorylation of E1p Variants by PDK4.

E1p variants S1-E1p and S2-E1p contain phosphorylatable site 1 and site 2, respectively. The remaining two phosphorylation sites in each E1p variant were mutated to alanine. S1-E1p (●) and S2-E1p (□) were phosphorylated by PDK4 in the absence of the E2p/E3BP cores as described in Experimental Procedures. The amount of [32P] phosphate incorporated per E1p heterotetramer was converted to % phosphorylation. Each variant E1p heterotetramer has two phosphorylatable serine residues, therefore 100% phosphorylation indicates that both serine residues in the heterotetramer are phosphorylated.
Despite the discovery of PDC regulation by phosphorylation four decades ago and its clinical ramifications for diabetes and cancer, the structural basis for inactivation of the PDC by phosphorylation remains unknown. This presumably has resulted from the difficulty until now to obtain a fully phosphorylated E1p component, due to half-of-the-sites phosphorylation when the reaction is carried out in the presence of the E2p/E3BP core (Kolobova et al., 2001; Korotchkina and Patel, 2001b; Sugden et al., 1978). We have previously shown that the phosphorylation of Ser291-α (site 1) in the related E1b component of the human branched-chain α-ketoacid dehydrogenase complex (BCKDC) blocks the ThDP-induced ordering of the single Ph-loop, which corresponds to Ph-loop A in E1p, carrying both Ser291-α (site 1) and Ser302-α (site 2) (Wynn et al., 2004). This conformational change abolishes the binding of LBD in the E2b component of BCKDC to the E1b component, resulting in the inactivation of BCKDC. However, since the loop in phosphorylated E1b is not visible, the structural basis underlying the disordering of the loop in phosphorylated E1b is not clear. With respect to the human PDC, a recent study showed that the substitution of Ser264-α (site 1) in the E1p component with a bulky, negatively charged glutamate residue impedes active site accessibility, but Ph-loop A in this S264E-α mutant remains fully ordered (Seifert et al., 2007). It was proposed that the inaccessibility of substrates pyruvate and lipoylated LBD, and by extrapolation not a disordered Ph-loop, is responsible for inactivation of the PDC by phosphorylation. However, the bona fide mechanism for PDC inactivation by phosphorylation remains uncertain and controversial, since the structure of phosphorylated E1p has not been determined.
In the present study, we approached the mechanism for inactivation of PDC by deciphering crystal structures of a phophorylated E1p variant, in which sites 2 and 3 are eliminated by alanine substitutions (Korotchkina and Patel, 1995). The complete phosphorylation at Ser264-α (site 1) of this E1p variant was accomplished, for the first time, using isolated PDK4 free of the E2p/E3BP core. The robust basal activity of PDK4 (Wynn et al., 2008) allowed efficient phosphorylation of the E1p variant without geometric constraints imposed by the E2p/E3BP core. Crystal structures of the phosphorylated site 1-only E1p variant reveal the presence of disordered conformations for both Ph-loop A and Ph-loop B, despite the inclusion of the bound cofactor ThDP. Serendipitously, some of the Ph-loops in the phosphorylated E1p mutant become ordered due to crystal packing. This allows a visualization of the phosphoryl group on Ph-loop A, which potentially poses a previously unrecognized steric clash with a close-by Ser266-α residue. These clashes eliminate an essential hydrogen-bonding network for stabilizing the loop conformation, resulting in the disordered Ph-loops. The present results suggest that the phosphoryl group-induced disordering of the Ph-loops is the underlying mechanism for inactivation of the PDC and the related BCKDC by phosphorylation. This mechanism fundamentally differs from the one previously described based on an ordered Ph-loop A in the structure of the phosphomimetic mutant S264E-α E1p (Seifert et al., 2007).
RESULTS AND DISCUSSION
Complete Phosphorylation of E1p Variants by PDK4
As described above, each α subunit of human E1p carries three phosphorylation sites (site 1: Ser264-α; site 2: Ser271-α and site 3: Ser203-α), resulting in six total phosphorylation sites in the E1p α2β2 heterotetramer. To obtain uniformly phosphorylated E1p preparations, E1p variants containing only phosphorylation site 1 (S1-E1p) or site 2 (S2-E1p) were produced, in which the remaining two sites were inactivated by alanine substitutions (Korotchkina and Patel, 1995). As described above, previous studies indicated that only 50% phosphorylation was achieved, when the S1-E1p or the S2-E1p single-site variant and various PDK isoforms were both docked to the E2p/E3BP core (Kolobova et al., 2001; Korotchkina and Patel, 2001b). These results are consistent with the earlier report of half-of-the-sites phosphorylation with wild-type E1p, where only three of the six phosphorylation sites in the E1p heterotetramer were modified (Sugden and Randle, 1978). Apparent non-equivalent-site phosphorylation was also observed with the E1b component of the BCKDC. It was suggested that the positional effects occur when both E1b and BCKD kinase are tethered to the 24-meric E2b core; as a result, only the phosphorylation site proximal to the kinase is phosphorylated (Li et al., 2007).
Human PDK4 is capable of robustly phosphorylating free E1p in the absence of the E2p/E3BP cores (Wynn et al., 2008). Free S1-E1p or S2-E1p was incubated for up to 120 min with [γ-32P]ATP and PDK4 with a molar ratio of E1p: PDK4 = 2: 1. The incorporation of [γ-32P] into the E1p-α subunit was analyzed by SDS-PAGE and phosphorimaging (Figure 2). Nearly 100% phosphorylation at sites 1 and 2 with the S1-E1p and S2-E1p mutants, respectively, was achieved within 20 min. The data indicate that half-site phosphorylation does not occur when these E1p variants are phosphorylated by PDK4 in the absence of the E2p/E3BP cores. The scaled-up fully phosphorylated S1-E1p (phospho-S1-E1p) was used in the following kinetic and crystallographic studies.
Figure 2. Overall Structure of the Wild-type E1p Heterotetramer.
A stereo diagram of the crystal structure of the wild-type E1p heterotetramer (α2β2) is shown as surface representation models with each of the four subunits represented by a different color. The two phosphorylation loops (Ph-loops) are shown as ribbon models with Ph-loop A in orange and Ph-loop B in yellow. The three phosphorylation sites in each α subunit are depicted by spheres and labeled according to the site numbers. The red triangle indicates the entrance of the active-site channel where substrates pyruvate and lip-LBD enter. The axis of 2-fold symmetry in the heterotetramer is indicated by a grey arrow.
Phosphorylation of S1-E1p Abolishes Activities for Reductive Acetylation and the Overall Reaction
Enzyme kinetic studies of wild-type E1p, S1-E1p and phospho-S1-E1p were carried out by measuring activities for the overall reaction (Reaction 1), E1p-catalyzed decarboxylation (Reaction 2) and E1p-catalyzed reductive acetylation (Reaction 3) (Table 1). In all the assays, cofactor ThDP is present at a saturating concentration (2 mM). With pyruvate as the variable substrate, S1-E1p exhibits slightly reduced kcat (17.8 s-1) for the overall reaction when reconstituted with E2p/E3BP and E3, compared with wild-type E1p (21 s-1). The Km value of S1-E1p for pyruvate (58 μM) is 40% higher than that of the wild type (41 μM). These results indicate that S1-E1p is similar to wild-type E1p with respect to kcat and Km values for pyruvate. As expected, phospho-S1-E1p nullifies the overall activity of the PDC, when reconstituted with E2p/E3BP and E3.
Table 1.
Kinetic Parameters for Wild-type E1p, S1-E1p and Phospho-S1-E1p
| Assays | kcat (s-1) | Km for pyruvate (mM) | kcat/Km (s-1mM-1) |
|---|---|---|---|
| Overall | |||
| WT E1p | 21 ± 1 | 41 ± 4 | 0.51 |
| Y89F-α E1p | 19 ± 3 | 78 ± 13 | 0.24 |
| S1-E1p | 18 ± 2 | 58 ± 5 | 0.31 |
| Phospho-S1-E1p | NM | NM | - |
| DCPIP | |||
| WT E1p | 0.26 ± 0.02 | 8.1 ± 0.5 | 0.032 |
| Y89F-α E1p | 2.7 ± 0.05 | 31 ± 5 | 0.087 |
| S1-E1p | 0.31 ± 0.02 | 9.5 ± 0.8 | 0.032 |
| Phospho-S1-E1p | 0.21 ± 0.01 | 281 ± 21 | 0.00075 |
| Reductive acetylation | |||
| WT E1p | 0.030 ± 0.002 | - | - |
| Y89F-α E1p | 0.028 ± 0.002 | ||
| S1-E1p | 0.028 ± 0.001 | - | - |
| Phospho-S1-E1p | NM | - | - |
Phosphorylation studies on S1-E1p or Ser264-α (site 1) were carred out using a double alanine mutant S271A-α (site 2) and S203A-α (site 3). NM: not measurable.
When E1p-catalyzed decarboxylation activity (Reaction 2) was assayed in the presence of the artificial electron acceptor 2,6-dichlorophenolindophenol (DCPIP), wild-type and S1-E1p again exhibited similar kcat and Km values for pyruvate (Table 1). With phospho-S1-E1p, the kcat value for pyruvate is decreased from the corresponding values for wild-type E1p and S1-E1p by 19% and 32%, respectively. Significantly, the Km value for pyruvate of phospho-S1-E1p (281 μM) is increased about 30-fold compared to those of the wild type E1p (8.1 μM) and S1-E1p (9.5 μM). This result indicates that phosphorylation drastically reduces the affinity of phosphorylated S1-E1p for the substrate pyruvate. In parallel experiments, E1p-catalyzed reductive acetylation (Reaction 3) was assayed by determining the rate of incorporation of the hydroxyacetyl moiety from [U-C14]pyruvate into the lipoylated L2 domain. Wild-type E1p and S1-E1p show essentially identical kcat values for reductive acetylation of 0.03 s-1 (Table 1). Notably, phosphorylation of S1-E1p reduces reductive acetylation activity to non-measurable levels.
The above results represent the first kinetic parameters obtained with the fully phosphorylated E1p protein. The data are similar to those obtained with the pseudo-phosphorylated S264E-α E1p mutant (Seifert et al., 2007). The 30-fold increase in Km for pyruvate with phospho-S1-E1p (Table 1) is consistent with the 20-fold increase in the apparent dissociation constant (Kd) for binding of the pyruvate analogue methylacetylphosphonate (MAP) to the S264E-α E1p variant, compared to wild-type E1p (Seifert et al., 2007). Both S264E-α and S264Q-α E1p variants were essentially devoid of activity for reductive acetylation, also similar to phospho-S1-E1p (Table 1). In the study of Seifert et al., it was suggested that the presence of a bulky and/or negatively charged group alone precludes binding of the lipoamide moiety of LBD to the E1p active site. However, the structural basis for aborted reductive acetylation in phospho-S1-E1p remains unknown.
The S1-E1p Structure Is Unperturbed Compared To Wild-type E1p
To provide the structural basis for inactivation of the PDC by phosphorylation, we determined five different crystal structures of the human E1p heterotetramer: 1) wild-type E1p (containing Mn-ThDP), 2) S1-E1p (containing Mg-ThDP), 3) phospho-S1-E1p (containing Mn-ThDP), 4) ThDP-free (apo) phospho-S1-E1p, and 5) apo Y89F-α mutant E1p (Table 2). The last two structures (4 and 5) are described in Supplemental Data.
Table 2.
Data collection and refinement statistics
| WT E1p (Mn-ThDP) | S1-E1p (Mg-ThDP) | Phospho-S1-E1p (apo) | Phospho-S1-E1p (Mn-ThDP) | Y89F-α E1p (apo)e | |
|---|---|---|---|---|---|
| PDB code | 3EXE | 3EXF | 3EXG | 3EXH | 3EXI |
| Data collectiona | |||||
| Space group | P21 | P21 | P1 | C2 | P3221 |
| No. of E1p heterotetramers in the asymmetric unit | 2 | 2 | 8 | 2 | 0.5 (one heterodimer) |
| Unit cell | |||||
| a (Å) | 103.57 | 103.39 | 119.28 | 154.38 | 257.06 |
| b (Å) | 129.62 | 129.67 | 128.29 | 154.38 | 115.61 |
| c (Å) | 124.30 | 144.95 | 228.41 | 82.95 | 127.59 |
| α (°) | 90 | 90 | 90.14 | 90 | 90 |
| β (°) | 92.48 | 109.16 | 90.05 | 90 | 113.64 |
| γ (°) | 90 | 90 | 90.02 | 120 | 90 |
| Wavelength (Å) | 0.98 | 0.98 | 0.98 | 1.54 (Cu-Kα) | 0.98 |
| Resolution (Å) | 1.98 | 3.0 | 3.0 | 2.45 | 2.20 |
| Unique reflections | 225065 | 72201 | 263005 | 123888 | 57582 |
| Completeness (%) | 99.6 (99.0) | 99.8 (98.2) | 98.4 (98.0) | 98.1 (96.6) | 99.5 (99.5) |
| R-merge (%)b | 10.7 (55.1) | 14.6 (56.0) | 8.0 (53.5) | 15.5 (69.3) | 3.4 (39.9) |
| <I>/<σ(I)> | 13.3 (2.3) | 10.7 (2.6) | 10.3 (1.7) | 14.2 (2.8) | 33.0 (2.0) |
| Multiplicity | 4.9 (4.8) | 3.7 (3.4) | 2.1 (1.9) | 6.7 (6.5) | 6.6 (6.5) |
| Refinementa | |||||
| No. reflections (Work/Test) | 213690/11272 | 68497/3624 | 249289/13221 | 117572/6182 | 55098/1511 |
| No. atoms (Mean B value (Å2)) | |||||
| Protein | 21379 (17.7) | 21386 (21.6) | 83323 (55.7) | 5106 (49.2) | 21081 (18.8) |
| Solvents | 2316 (26.5) | 9 (6.9) | 0 | 318 (51.0) | 792 (19.0) |
| ThDP | 104 (11.4) | 104 (25.4) | 0 | 0 | 104 (17.7) |
| Metal and other | 32 (32.4) | 8 (32.8) | 16 (75.3) | 3 (65.7) | 58 (41.3) |
| R-work (%)c | 15.8 (20.1) | 18.4 (23.9) | 18.9 (29.5) | 16.5 (23.4) | 18.7 (28.1) |
| R-free (%)c | 20.6 (26.9) | 26.2 (35.8) | 25.3 (37.4) | 21.0 (28.5) | 22.5 (34.8) |
| R.m.s.d | |||||
| Bond length (Å) | 0.021 | 0.021 | 0.019 | 0.020 | 0.020 |
| Bond angle (°) | 1.761 | 2.044 | 1.883 | 1.733 | 1.842 |
| Ramachandran plot | |||||
| Most favored (%) | 96.5 | 90.2 | 92.1 | 96.1 | 96.3 |
| Allowed (%) | 2.2 | 6.8 | 5.7 | 2.8 | 2.5 |
| Disallowed (%) | 1.3 | 3.0 d | 2.2 d | 1.1 | 1.2 |
Values in parentheses refer to data in the highest resolution shell unless otherwise indicated.
R-merge = Σhkl Σj | Ij - <I> | / Σhkl Σj Ij, where <I> is the mean intensity of j observations from a reflection hkl and its symmetry equivalents.
R-work = Σhkl| |Fobs| − k|Fcalc| | / Σhkl |Fobs|. R-free = R-work for 5 % of reflections that were omitted from refinement.
The higher numbers result from the lower resolution (3.0 Å) of these structures than the others.
This crystal contains a complex of Y89F-α E1p with SBD, but SBD is not visible and excluded from the model.
The overall structure of S1-E1p is compared with the present wild-type E1p (Mn-ThDP) structure and two previously determined human E1p structures: wild-type E1p (Mg-ThDP) (PDB code: 1NI4) (Ciszak et al., 2003) and S264E-α mutant E1p (Mg-ThDP) (2OZL) (Ciszak et al., 2003; Seifert et al., 2007). The r.m.s. deviations between the S1-E1p structure and the other three are less than 0.3 Å for over 1300 equivalent Cα atoms in the E1p heterotetramer, indicating that the structure of S1-E1p is essentially identical to that of the wild type. Moreover, the conformations of the conserved Ph-loop A containing phosphorylation sites 1 and 2 (residues 259-α – 282-α) and Ph-loop B containing phosphorylation site 3 (residues 198-α – 205-α) in the E1p-α subunit are also unchanged in these structures (Figures 3A, S1A, S2A, and S2B). These data indicate that mutations to alanine at the phosphorylation sites 2 and 3 do not perturb the E1p structure as also supported by the activity data (Table 1).
Figure 3. Structures of the Phosphorylation loops in Wild-type and Phospho-S1-E1p.

The structures of Ph-loops in E1p-α subunits are shown as ribbon models against other parts of the protein in surface representation. Ph-loop A (residues from 259-α to 282-α) is in orange, and Ph-loop B (from 198-α to 205-α) in yellow. The three phosphorylation sites are indicated by spheres and labeled according to the site numbers. The bound ThDP is shown as a ball-and-stick model. (A) Fully ordered Ph-loops in wild-type E1p. (B) Wild-type-like ordered Ph-loops in phospho-S1-E1p with bound Mn-ThDP. Two of the four E1p-α subunits in the asymmetric unit (depicted as 2/4 in the figure caption) exhibit this conformation, which is maintained through interactions with a symmetry-related molecule (cf. Figure S3B). (C) Completely disordered Ph-loops in phospho-S1-E1p containing the bound Mn-ThDP. One of the four E1p-α subunit in the asymmetric unit (1/4) has this conformation. No symmetry-related molecule is present near the Ph-loops (cf. Figure S3A). (D) Variant ordered conformation of Ph-loop A and partially ordered Ph-loop B in the remaining E1p-α subunits (1/4). This conformation is also maintained by interactions with a symmetry-related molecule (cf. Figure S3C).
Phosphorylation of S1-E1p Causes Disordering of the Ph-loops Harboring the Three Phosphorylation Sites
To examine the effect of ThDP binding on the conformation of Ph-loops in E1p, it was necessary to produce the apo (without the bound-ThDP) form of E1p. Attempts to obtain “completely” apo wild-type E1p by exhaustive dialysis in the presence of EDTA were not successful, since its non-phosphorylated form has high binding affinity for Mg-ThDP (Kd = 0.11 μM) (Table 3). In the wild-type E1p structure, Tyr89-α directly interacts with the diphosphate group of the bound ThDP, but not with either Ph-loop A or Ph-loop B (data not shown). Therefore, we generated the Y89F-α mutant E1p, which exhibits 450-fold weaker binding affinity for ThDP than the wild type (Table 3). At saturating ThDP concentrations, this mutant E1p shows essentially wild-type activity levels in the overall reaction as well as E1p-catalyzed decaroxylation and reductive acetylation (Table 1). The Y89F-α E1p structure shows no electron density for ThDP, Ph-loop A and Ph-loop B, confirming the presence of an apo form (Figure S1B). By contrast, in the wild-type holo-E1p structure the Ph-loops are well ordered with the bound ThDP (Figure 3A). Thus, the apo Y89F-α E1p structure supports the thesis that the bound ThDP induces a disorder-to-order transition of the Ph-loops, analogous to that observed in the related E1b component of BCKDC (Li et al., 2004; Nakai et al., 2004) (see Supplementary Data for more details).
Table 3.
Dissociation constants (Kd) for the binding of cofactor ThDP to wild-type E1p, S1-E1p and phospho-S1-E1p in the presence of magnesium ions
| Protein | Kd (μM) | Maximum quenching (%) |
|---|---|---|
| Wild-type E1p | 0.11 ± 0.01 | 76.1 ± 2.1 |
| Y89F-α E1p | 50.2 ± 4.6 | 67.5 ± 2.3 |
| S1-E1p | 0.29 ± 0.02 | 68.4 ± 2.5 |
| Phospho-S1-E1p | 13.1 ± 1.1 | 73.1 ± 1.9 |
Dissociation constants are determined by fluorescence quenching as described under “Experimental Procedures”.
Crystals of phospho-S1-E1p were initially produced in the presence of Mg-ThDP, but they did not contain the cofactor and are thus also referred to as the apo form. In this structure, the Ph-loops are completely or partially disordered (see Supplementary Data). This result is in good agreement with 100-fold weaker binding of phospho-S1-E1p for Mg-ThDP compared to the wild type (Table 3), establishing the important role of the Ph-loops for ThDP binding (see below). On the other hand, the bound ThDP is essential for ordering the Ph-loops as deduced from the Y89F-α E1p structure (Figure S1B). It is, therefore, impossible to discern whether the phosphorylation of site 1, the absence of a bound ThDP or a combination of the two induces the disordering of the Ph-loops in the apo phospho-S1-E1p structure. Since ThDP binds to phospho-S1-E1p with markedly higher affinity in the presence of Mn2+ than Mg2+ (Figure S3), phospho-S1-E1p was later crystallized at a saturating concentration of Mn-ThDP.
Phospho-S1-E1p with bound Mn-ThDP crystallized with the symmetry of space group C2 with two heterotetramers in the asymmetric unit. The overall structure of phospho-S1-E1p is very similar to that of the wild type (r.m.s.d = 0.296 Å for 1379 equivalent Cα atoms in the heterotetramer) and S1-E1p (r.m.s.d = 0.268 Å for 1299 Cα atoms). However, the Ph-loops are significantly different. Among the four E1p-α subunits in the asymmetric unit, three different conformations of the Ph-loops are observed. In one of the E1p-α subunits, there is no discernable electron density for either Ph-loop A or Ph-loop B, indicating that these loops are disordered (Figure 3C). It must be pointed out, however, that the electron density for the bound Mn-ThDP is well defined in all E1p heterotetramers. Therefore, the bound cofactor does not seem to be able to order the phosphorylated Ph-loops. There are no crystal-packing contacts near both Ph-loops (Figure S4A). Taken together, the disordered conformation appears to be an intrinsic property of the phosphorylated Ph-loops.
Unexpectedly, the Ph-loops of two other E1p-α subunits adopt wild-type-like conformations with electron densities of the phosphoryl group on Ser264 (site 1) clearly visible (Figures 3B, 4D, and S2C). These E1p-α subunits belong to different heterotetramers in the asymmetric unit and are related by a 2-fold non-crystallographic symmetry axis (Figure S4B). The α-helix in the ordered Ph-loop A in each E1p-α subunit interacts with the neighboring heterotetramer, which appears to maintain the Ph-loops in the wild-type ordered conformation. In the fourth molecule, the Ph-loop A conformation is very similar to the conformation with an extended α-helix observed in the apo phospho-S1-E1p structure (see Supplemental Data and Figures 3D, S1C, and S2D). In this structure, the entire backbone of the Ph-loop A has well defined electron density, but the phosphoryl group on Ser264-α is disordered. This variant Ph-loop A conformation is also stabilized through interactions with a symmetry-related molecule as observed in the corresponding apo structure (Figure S4C). The physiological significance of this variant Ph-loop A conformation is presently unclear.
Figure 4. Hydrogen-Bond Networks Involving Phosphorylation Site 1.
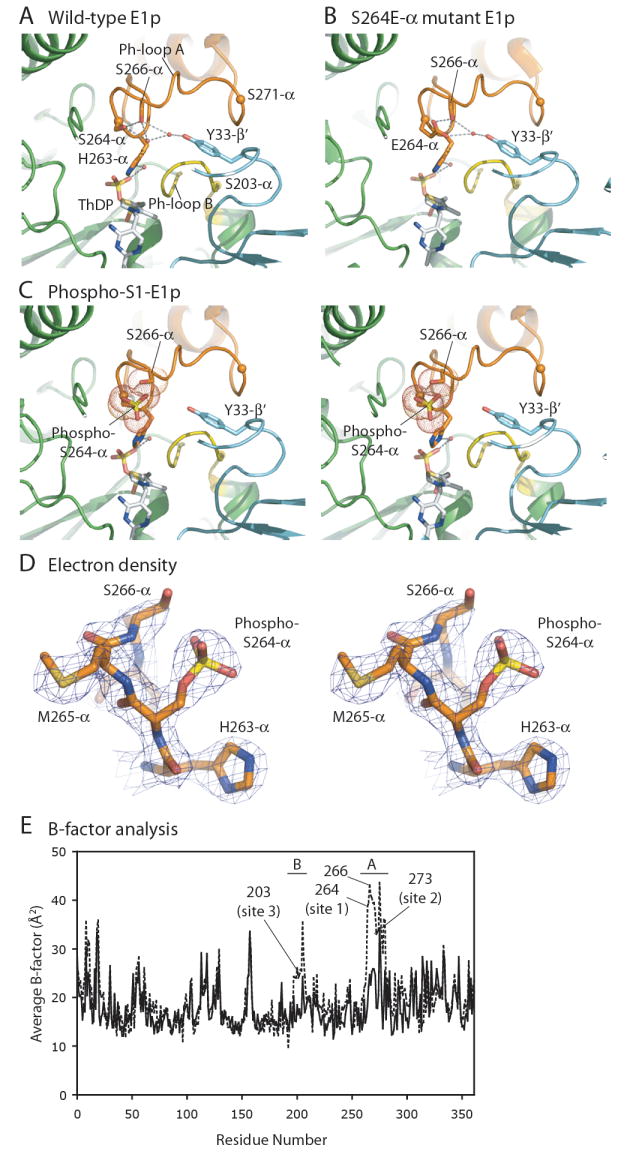
(A) The H-bond network connecting phosphorylation site 1 and Tyr33-β’ of the E1p-β’ subunit in the wild-type E1p structure. Ph-loop A is in orange, and Ph-loop B in yellow. The positions of the three phosphorylation sites are shown as spheres at the corresponding Cα atom positions. Water molecules are depicted as small red dots. H-bonds are indicated by grey dashed lines. (B) A similar H-bond network in the Ser264E-α mutant E1p structure (PDB code: 2OZL) (Seifert et al., 2007). (C) A stereo diagram showing absence of the H-bond network in phospho-S1-E1p containing bound Mn-ThDP. The phosphoryl group on Ser264-α (site 1) clashes with the side chain of the neighboring Ser266-α. Van der Waals radii of the phosphoryl group and side chains of both serine residues are shown as spheres of red dots. (D) A stereo figure of the 2Fo-Fc electron density map (contoured at 1σ) at phosphorylation site 1 with a stick representation of the refined model. (E) Average B-factor plots of the wild-type and phospho-S1-E1p structures. Average B-factors for individual residues in one of the four non-phosphorylated wild-type E1p-α subunits (solid line) and one of the two phospho-S1-E1p-α subunits with the wild-type-like “ordered” Ph-loops (dashed line) are plotted against the residue number. The residue ranges for Ph-loops A and B are indicated on top of the graph. Each of the three phosphorylation sites and residue Ser266-α are labeled.
Phosphorylation of Ser264-α Interferes with the Ordering of the Ph-loops
In our wild-type E1p structure, Ser264-α (site 1) and adjacent S266-α in Ph-loop A form a hydrogen (H)-bond network with two neighboring water molecules (Figure 4A). One of these water molecules further establishes an H-bond with Tyr33-β’ from the E1p-β’ subunit. Interestingly, a similar H-bond network is present in the S264E-α mutant E1p structure, comprising residues Glu264-α, Ser266-α and Tyr33-β’ that coordinate to a water molecule (Figure 4B) (Seifert et al., 2006). Ph-loop A in this mutant E1p structure is apparently stabilized by a variant H-bond network involving the introduced Glu264-α residue. By contrast, no bridging water molecule between Ph-loop A and Tyr33-β’ is present in the phospho-S1-E1p structure, which contains the wild-type-like “ordered” Ph-loops stabilized by crystal packing (Figure 4C). Moreover, the phosphoryl group of Ser264-α occupies the position where the side-chain hydroxyl group of Ser266-α is located in the non-phosphorylated wild-type E1p structure. This conformation potentially imposes a steric clash between the bulky phosphoryl group and the side chain of Ser266-α (Figure 4C). As a result, the hydroxyl group of Ser266-α is rotationally disordered and exhibits poor electron density (Figure 4D). The average B-factors of individual residues in both Ph-loop A and Ph-loop B of phospho-S1-E1p are significantly higher than those of the equivalent regions in the non-phosphorylated wild type E1p-α subunit (Figure 4E). The data suggest that both phosphorylated Ph-loop A and non-phosphorylated Ph-loop B are locally unstable, particularly in the vicinity of Ser266-α. In the absence of crystal packing, phosphorylated Ph-loop A remains in the disordered conformation, as observed in the other phospho-S1-E1p-α subunit in the same crystal (Figure 3C). We therefore conclude that phosphorylation of Ser264α prevents the ordering of both Ph-loops in the phospho-S1-E1p structure.
Concerted Disordering of Ph-loops A and B Reduces ThDP Binding to Phospho-S1-E1p
The disordering of Ph-loop B is tightly coupled to that of Ph-loop A. This conjecture is based on the observations that Ph-loop B is well defined when Ph-loop A is in the ordered wild-type conformation in the crystal structures (Figures 3A, 3B and S1A). When Ph-loop A is disordered or in the variant partially ordered conformation, Ph-loop B also assumes similar conformations (Figures 3C, 3D, S1B, and S1C). Ph-loop B extensively interacts with Ph-loop A through direct and indirect H-bonds (Figure 5). Alterations in Ph-loop A conformations apparently disrupt these interactions, producing reciprocal effects on the Ph-loop B conformation.
Figure 5. Extensive Interactions Between the Two Ph-loops in the Wild-Type E1p Structure.

The stereo diagram illustrates extensive interactions between Ph-loop A (orange) and Ph-loop B (yellow) in the non-phosphorylated wild-type E1p structure. The Ph-loops are shown as stick models. Side chains of some residues are omitted for clarity. The three phosphorylation sites are S264-α (site 1), S271-α (site 2), and S203-α (site 3). Water molecules are shown as small red spheres and the manganese atom as a pink sphere. H-bonds are indicated by grey lines.
In the wild-type holo-E1p structure, the side chains of Arg259-α and His263-α from Ph-loop A serves as key anchors for the bound ThDP by forming indirect H-bonds with a phosphate of the bound ThDP through the same water molecule (Figure 5). The corresponding residues Arg287-α and His291-α in the E1b component of BCKDC have been shown to be indispensable anchoring residues for ThDP binding (Li et al., 2004; Wynn et al., 2003). In the Ph-loop B of wild-type E1p, the main-chain carbonyl group of Tyr198-α provides a ligand to the cation ion essential for ThDP binding (Figure 5). Thus, the disordering of both Ph-loops A and Ph-loop B in phosphorylated E1p removes these important anchors for the bound ThDP, resulting in weaker binding affinity for this cofactor.
To further investigate these interactions, binding affinities of wild-type E1p, S1-E1p and phospho-S1-E1p for cofactor ThDP in the presence of Mg2+ were measured by the quenching of tryptophan fluorescence from residue Trp135-β’ of the E1p-β’ subunit upon ThDP binding (Table 3) (Ali et al., 1995). Both wild-type and S1-E1p exhibit high affinities for ThDP with dissociation constants (Kd) of 0.11 μM and 0.29 μM, respectively. Phosphorylation of S1-E1p increases the Kd for ThDP immensely by more than 100-fold over wild-type E1p to 13.1 μM. The data indicate that the disruption of interactions between the anchoring residues and ThDP by phosphorylation via the disordered Ph-loops leads to the decreased binding affinity of E1p for ThDP. In contrast, the introduction of a glutamate or glutamine to phosphorylation site 1 of E1p (S264E-α and S264Q-α E1p variants) did not alter the affinity of the E1p mutants for ThDP, compared with wild-type E1p at 0.47 μM (Seifert et al., 2007). The inability of the amino acid substitution to induce the disordering of the Ph-loops in the S264E-α E1p structure may explain the wild-type binding affinities exhibited by these E1p mutants.
No Detectable L2 Binding to the Phosphorylated S1-E1p Protein
Isothermal titration calorimetry (ITC) measurements were utilized to characterize the binding of lipoylated L2 domain to wild-type and phosphorylated E1p proteins in the presence of 200 μM ThDP (Figure 6). Analysis of the data shows that both the wild-type and non-phosphorylated S1-E1p proteins bind L2 with the same affinity (Kd = 1.3 μM). By contrast, no heat changes were detected for phosphorylated S1-E1p, consistent with the absence of L2 binding. The data provide evidence that the absence of reductive acetylation (Reaction 3) activity for ThDP in phosphorylated S1-E1p results from the inability of the substrate L2 to bind to E1p.
Figure 6. Binding of the Lipoylated-L2 Domain to E1p Determined by Isothermal Titration Calorimetry.
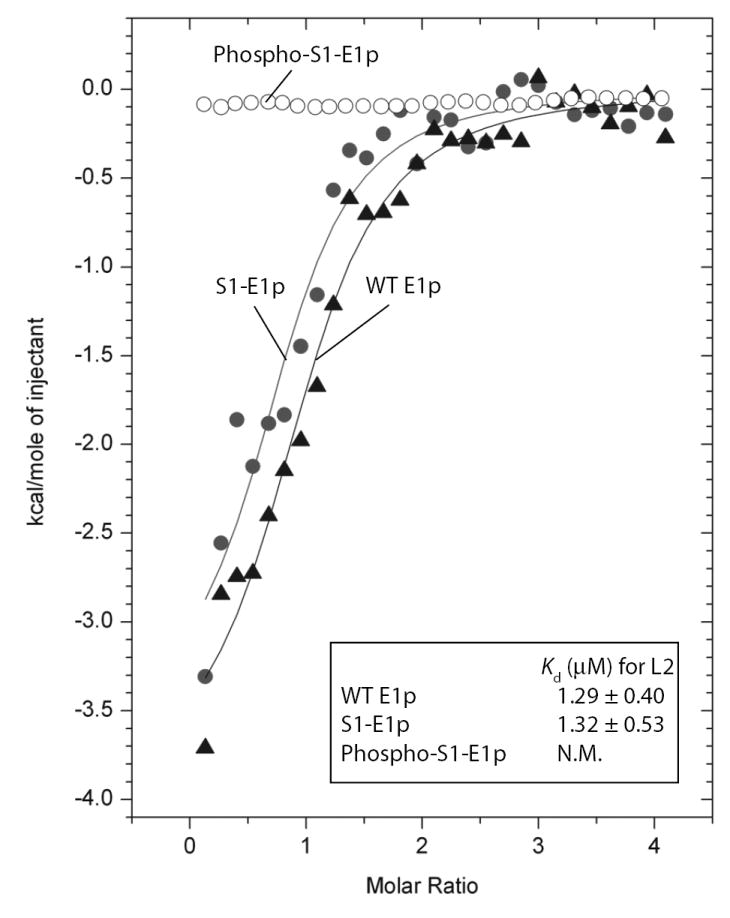
Wild-type E1p, S1-E1p or phospho-S1-E1p protein (35 μM, heterodimer) was titrated with 0.65 mM lip-L2 (residues 128-265 in E2p) as described in the Experimental Procedures. The binding isotherms were fit using ORIGIN v. 7.0 software. For wild-type E1p and S1-E1p, the dissociation constants Kd are averages of triplicates; for phospho-S1-E1p, there are no detectable heats.
Structural Mechanism for Inactivation of E1p by Phosphorylation
Based on the present biochemical and structural data, we propose the following structural mechanism by which phosphorylation of Ser264-α (site 1) inactivates the PDC. As shown in the structure of the Y89F-α E1p mutant, the Ph-loops assume the disordered conformation in the absence of the cofactor ThDP (Figures 7A, left panel and S1B). We suggest that in non-phosphorylated wild-type E1p, the Ph-loops are in equilibrium between the ordered and the disordered conformations. ThDP binding induces the ordering of Ph-loop A and Ph-loop B through: 1) anchoring interactions provided by the bound ThDP via His263-α and Arg259-α of Ph-loop A, 2) coordination between Tyr198-α of Ph-loop B and the bound magnesium or manganese ion, and 3) an H-bond network comprising Ser264-α, Ser266-α of Ph-loop A and Tyr33-β’ from the β’subunit (Figure 7A, middle panel). The presence of both ordered Ph-loops confers the essential determinants for binding of lip-LBD to E1p. With these ordered Ph-loop conformations, E1p is able to catalyze both oxidative decarboxylation (Reaction 2) and reductive acetylation (Reaction 3) (Figure 7A, right panel).
Figure 7. Mechanism of PDC Inactivation by Site 1 Phosphorylation in E1p.
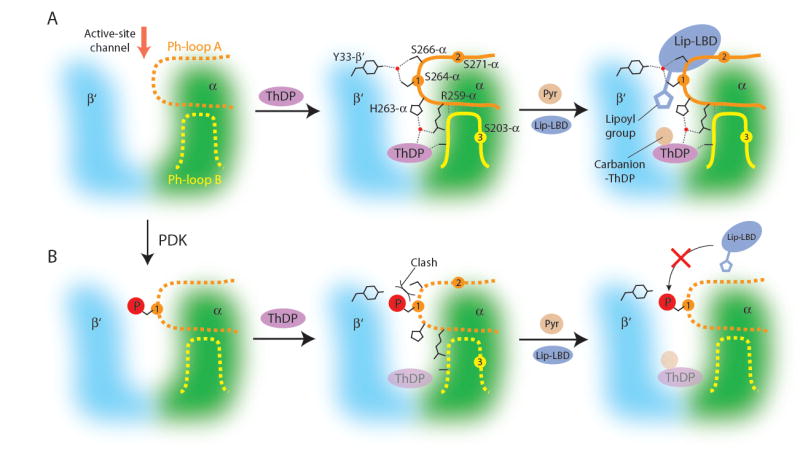
Reaction scheme for non-phosphorylated E1p. The E1p active site is formed by portions of the α subunit (green) and the β’ subunit (cyan). The red arrow depicts the entrance of the active-site channel for substrates pyruvate and lip-LBD. In apo E1p, Ph-loop A (orange) and Ph-loop B (yellow) are disordered (shown as dashed lines in the left panel). Binding of the cofactor ThDP (magenta) at the bottom of the active-site channel induces ordering of both Ph-loop A and Ph-loop B (middle panel). Phosphorylation sites are indicated by orange and yellow circles and numbered. Water molecules are represented by red dots. H-bonds are shown as dashed lines. These ordered Ph-loops mediate E1p-catalyzed decarboxylation of pyruvate and reductive acetylation of lip-LBD (right panel). (B) Inactivation mechanism for phosphorylated E1p. When Ph-loop A is disordered in apo E1p, Ser264-α (site 1) is phosphorylatable by PDK (from panel A left to panel B left). A steric clash between the phosphoryl group and adjacent Ser266-α prevents the ordering of Ph-loops (middle panel). In the absence of the ordered Ph-loops, ThDP binding to the E1p active site is significantly attenuated (shown as a transparent oval). Nonetheless, under saturating ThDP conditions, the E1p-catalyzed decarboxylation of pyruvate proceeds (right panel). However, the reductive acetylation of lip-LBD is abolished due to the loss of determinants on the disordered Ph-loops, which are essential for lip-LBD binding to the E1p active site. The abrogation of E1p-catalyzed reductive acetylation interrupts substrate channeling in the PDC, resulting in the inactivation of this catalytic machine.
The equilibrium between the disordered and the ordered loop conformations may also exist in the E1 active sites of other α-ketoacid dehydrogenase complexes, which are not regulated by phosphorylation/dephosphorylation. A case in point, the activation loop, which is equivalent to Ph-loop A, has been shown to assume both the disordered and the ordered conformations in the E1p component of the Bacillus stearothermophilius PDC (Frank et al., 2005; Frank et al., 2004). The premise is that these active-site loops are intrinsically flexible by the necessity of the many roles they play in catalysis, ThDP binding and interactions with other enzyme components.
When Ph-loop A is disordered in apo E1p, Ser264-α (site 1) is readily available for phosphorylation by PDK (Figure 7B, left panel). Upon phosphorylation, the bulky phosphoryl group could pose a steric clash with the side chain of Ser266-α (Figure 4). This clash not only eliminates the H-bond network involving the serine residue but also destabilizes the local conformation of Ph-loop A. These negative effects prevent the disorder-to-order transition of phosphorylated Ph-loop A, despite the presence of the bound ThDP, leaving this loop in the disordered conformation (Figure 7B, middle panel). Ph-loop B extensively interacts with Ph-loop A (Figure 5), therefore, the disordering of Ph-loop A is coupled to that of Ph-loop B (Figure 3C). The disordering of both Ph-loops synergistically decreases the binding affinity of E1p for ThDP, through flexibility of the cofactor-anchoring residues (Table 3). With reduced affinity for ThDP, phospho-S1-E1p still catalyzes the decarboxylation of pyruvate (Reaction 2), when assayed at a saturating cofactor concentration (Figure 7B, right panel) (Table 1). However, the reductive acetylation of lip-LBD (Reaction 3) by phospho-S1-E1p is nullified due to the displacement of the critical determinants for lip-LBD binding to the E1p active site (Figure 6). The interruption at the reductive acetylation step forestalls the ensuing reaction steps (Reactions 4 - 6) mediated by the E2p and E3 components, thereby abolishing the overall activity (Reaction 1) of the PDC.
Concluding Remarks
One of the remaining questions is whether the mechanism by which phosphorylation at site 2 or site 3 leads to inactivation of the PDC is similar to phosphorylation at site 1. Ser271-α (site 2) is involved in the H-bond network between Ph-loop A and Ph-loop B (Figure 5). In addition, Ser271-α is close to Tyr33-β’, which is involved in the neighboring H-bond network including Ser264-α (site 1) and Ser266-α (Figure 4). Phosphorylation of Ser271-α could disrupt both the H-bond networks promoting the disordering of both Ph-loops. On the other hand, Ser203-α (site 3) is surrounded by Tyr198-α, Glu205-α and several water molecules, which invariably participate in the H-bond network between Ph-loop A and Ph-loop B (Figure 5). Phosphorylation of site 3 could also revoke this H-bonding network and cause the disordering of both Ph-loops. However, the deactivating effects of phosphorylation at site 2 or site 3 do not appear to be as complete as those of site 1, since residual PDC activity is present in either the site-2-only or site-3-only phosphorylated E1p variants (Korotchkina and Patel, 2001b). The latter results and the slower rates of phosphorylation at site 2 and site 3 than site 1 (Korotchkina and Patel, 1995; Sale and Randle, 1981b) support the notion that site 1 is the major site for down-regulation of PDC activity by phosphorylation.
The disordering of the Ph-loops in E1p is similar to that occurring in the phosphorylated E1b component of BCKDC, which we described previously (Wynn et al., 2004). The structural mechanism for the disordering of the lone Ph-loop in E1b was not clear, since in the absence of crystal contacts, the entire loop was not visible in the electron density upon phosphorylation. However, Ser264-α (site 1) and the adjacent Ser266-α, which participate in the H-bond network to stabilize Ph-loop A in the current wild-type E1p structure, are conserved in the E1b component of BCKDC (Ser292-α and Ser294-α, respectively). The conformation of the Ph-loop in S292D-α E1b is maintained by a variant H-bond network produced by the side chains of the mutant aspartate and the juxtaposed Ser294-α (Wynn et al., 2004), equivalent to the H-bond network that stabilizes Ph-loop A in the pseudo-phosphorylated S264E-α E1p structure (Seifert et al., 2007). Based on the present results with E1p, a steric clash between the phosphorylated Ser292-α (site 1) and the side chain of Ser294-α would dislodge a putative H-bond network in E1b, presumably resulting in the disordered Ph-loop. Thus, the disordering of Ph-loops appears to be the conserved mechanism between mammalian α-ketoacid dehydrogenase complexes for inactivation by phosphorylation. Conversely, the disordering of Ph-loops upon phosphorylation is also necessary for reactivation through dephosphorylation by the cognate phosphatases. For example, the active site of pyruvate dehydrogenase phosphatase is located in the bottom of a cavity and appears to be unable to accommodate the ordered conformation of Ph-loops (Vassylyev and Symersky, 2007). These results, taken together, strongly suggest that the disordering of Ph-loops is the unifying design for the regulation of the mammalian PDC and BCKDC by reversible phosphorylation.
EXPERIMENTAL PROCEDURES
Materials
Pyruvate, thiamine diphosphate (ThDP), and 2,6-dichlorophenolindophenol (DCPIP) were obtained from Sigma-Aldrich (St. Louis, MO). [γ32P]ATP and [2-14C]pyruvate were purchased from MP Biomedicals (Solon, OH). Expression plasmids for human wild-type E1p and E2p/E3BP (Harris et al., 1997) were gifts from Dr. Kirill Popov at University of Alabama at Birmingham. Construction, expression and purification of recombinant proteins are provided as Supplementary data.
Phosphorylation of E1p
Phosphorylation of S1-E1p or S2-E1p was carried out with SUMO-PDK4, and the degree of phosphorylation of E1p was estimated as described in Supplementary data.
To scale up production of the fully phosphorylated protein, S1-E1p was incubated with tag-free PDK4, and purified as described in Supplementary data.
Enzyme Assays
The activity assays for the overall reaction catalyzed by the PDC (Reaction 1), the E1p-catalyzed decarboxylation (Reaction 2), and the E1p-catalyzed reductive acetylation of the lipoylated L2 domain (Reaction 3) were conducted as described in Supplementary data.
Fluorescence Quenching for ThDP Binding
Steady-state tryptophan fluorescence quenching upon ThDP binding to the wild-type and mutant E1p proteins (87 nM, heterotetramer) was measured in the presence of either Mg2+ or Mn2+ (1 mM) as described previously (Wynn et al., 2003). Fluorescence was corrected for the inner-filter effect at high ThDP concentrations (Lakowicz et al., 1983).
L2-Binding Measurement by ITC
ITC measurements were performed in a VP-ITC microcalorimeter from MicroCal (Northampton, MA). Titrations were carried out in 50 mM potassium phosphate buffer (pH 7.5), 10 mM KCl, 200 μM ThDP, 2 mM MgCl2 and 5 mM β-mercaptoethanol at 20°C. In a typical measurement of lip-L2-E1p binding, 30 injections (at 3 min intervals) with 8 μl each of L2 (0.65 mM) were made into 1.8 ml of the E1p protein (35 μM, heterodimer) in the cell. Curve fitting and the derivation of thermodynamic parameters were carried out with ORIGIN v. 7.0 software package provided by MicroCal. The concentrations of human E1p proteins and human lip-L2 were determined using extinction coefficients ε278nm of 145.7 mM-1cm-1 (base on tetramer) and 11.2 mM-1cm-1, respectively.
Structure Determination
Crystallization and data collection are provided in Supplementary data. The E1p structures were determined by molecular replacement using the program Phaser (Read, 2001) of the CCP4 package (CCP4, 1994) with the structure of human E1p (PDB code: 1NI4) as the search model. After rigid-body refinement with REFMAC5 (Murshudov et al., 1997), the electron density map was improved using the program DM (Cowtan and Main, 1996). The protein models were manually modified using the program Coot (Emsley and Cowtan, 2004). After modeling of the protein part was complete, ThDP, magnesium or manganese ions and water molecules were added in subsequent refinement cycles. Data processing and refinement statistics are summarized in Table 2. Structural representations were created with the program PyMOL (DeLano Scientific LLC, Palo Alto, CA).
Supplementary Material
Supplemental Data including four supplemental figures can be found with this article online at http://www.structure.org/XXX.
Acknowledgments
We are indebted to Drs. Diana Tomchick and Chad Brautigam in the UT Southwestern Structural Biology Laboratory for help with synchrotron data collection. This work was supported by grants DK62306 and DK26758 from the National Institutes of Health and grant I-1286 from the Welch Foundation. The use of the Argonne National Laboratory Structural Biology Center beam lines at the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Energy Research under Contract No. W-31-109-ENG-38.
Footnotes
ACCESSION NUMBERS Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 3EXE (wild-type E1p), 3EXF (S1-E1p), 3EXG (apo phospho-S1-E1p), 3EXH (phospho-S1-E1p with Mn-ThDP), and 3EXI (Y89F-α E1p).
The authors declare no financial conflict of interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali MS, Shenoy BC, Eswaran D, Andersson LA, Roche TE, Patel MS. Identification of the tryptophan residue in the thiamin pyrophosphate binding site of mammalian pyruvate dehydrogenase. J Biol Chem. 1995;270:4570–4574. doi: 10.1074/jbc.270.9.4570. [DOI] [PubMed] [Google Scholar]
- Bao H, Kasten SA, Yan X, Hiromasa Y, Roche TE. Pyruvate Dehydrogenase Kinase Isoform 2 Activity Stimulated by Speeding Up the Rate of Dissociation of ADP. Biochemistry. 2004;43:13442–13451. doi: 10.1021/bi0494875. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. PNAS. 2007;104:9445–9450. doi: 10.1073/pnas.0611662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Ciszak EM, Korotchkina LG, Dominiak PM, Sidhu S, Patel MS. Structural basis for flip-flop action of thiamin pyrophosphate-dependent enzymes revealed by human pyruvate dehydrogenase. J Biol Chem. 2003;278:21240–21246. doi: 10.1074/jbc.M300339200. [DOI] [PubMed] [Google Scholar]
- Cowtan KD, Main P. Phase combination and cross validation in iterated density-modification calculations. Acta Cryst. 1996;D52:43–48. doi: 10.1107/S090744499500761X. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallographica Section D - Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Frank RA, Pratap JV, Pei XY, Perham RN, Luisi BF. The molecular origins of specificity in the assembly of a multienzyme complex. Structure (Camb) 2005;13:1119–1130. doi: 10.1016/j.str.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Frank RA, Titman CM, Pratap JV, Luisi BF, Perham RN. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science. 2004;306:872–876. doi: 10.1126/science.1101030. [DOI] [PubMed] [Google Scholar]
- Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul. 2002;42:249–259. doi: 10.1016/s0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- Harris RA, Bowker-Kinley MM, Wu P, Jeng J, Popov KM. Dihydrolipoamide dehydrogenase-binding protein of the human pyruvate dehydrogenase complex. DNA-derived amino acid sequence, expression, and reconstitution of the pyruvate dehydrogenase complex. J Biol Chem. 1997;272:19746–19751. doi: 10.1074/jbc.272.32.19746. [DOI] [PubMed] [Google Scholar]
- Holness MJ, Kraus A, Harris RA, Sugden MC. Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes. 2000;49:775–781. doi: 10.2337/diabetes.49.5.775. [DOI] [PubMed] [Google Scholar]
- Kato M, Chuang JL, Tso SC, Wynn RM, Chuang DT. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 2005;24:1763–1774. doi: 10.1038/sj.emboj.7600663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001;358:69–77. doi: 10.1042/0264-6021:3580069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem. 1995;270:14297–14304. doi: 10.1074/jbc.270.24.14297. [DOI] [PubMed] [Google Scholar]
- Korotchkina LG, Patel MS. Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J Biol Chem. 2001a;276:5731–5738. doi: 10.1074/jbc.M007558200. [DOI] [PubMed] [Google Scholar]
- Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem. 2001b;276:37223–37229. doi: 10.1074/jbc.M103069200. [DOI] [PubMed] [Google Scholar]
- Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 2004;53:899–910. doi: 10.2337/diabetes.53.4.899. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR, Maliwal BP, Cherek H, Balter A. Rotational freedom of tryptophan residues in proteins and peptides. Biochemistry. 1983;22:1741–1752. doi: 10.1021/bi00277a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Machius M, Chuang JL, Wynn RM, Chuang DT. The two active sites in human branched-chain alpha -ketoacid dehydrogenase operate independently without an obligatory alternating-site mechanism. J Biol Chem. 2007;282:11904–11913. doi: 10.1074/jbc.M610843200. [DOI] [PubMed] [Google Scholar]
- Li J, Wynn RM, Machius M, Chuang JL, Karthikeyan S, Tomchick DR, Chuang DT. Cross-talk between thiamin diphosphate binding and phosphorylation loop conformation in human branched-chain alpha-keto acid decarboxylase/dehydrogenase. J Biol Chem. 2004;279:32968–32978. doi: 10.1074/jbc.M403611200. [DOI] [PubMed] [Google Scholar]
- Linn TF, Pettit FH, Reed LJ. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci U S A. 1969;62:234–241. doi: 10.1073/pnas.62.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G, Vagin A, Dodson E. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakai T, Nakagawa N, Maoka N, Masui R, Kuramitsu S, Kamiya N. Ligand-induced conformational changes and a reaction intermediate in branched-chain 2-oxo acid dehydrogenase (E1) from Thermus thermophilus HB8, as revealed by X-ray crystallography. J Mol Biol. 2004;337:1011–1033. doi: 10.1016/j.jmb.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Pan JG, Mak TW. Metabolic Targeting as an Anticancer Strategy: Dawn of a New Era? Sci STKE. 2007;2007:e14. doi: 10.1126/stke.3812007pe14. [DOI] [PubMed] [Google Scholar]
- Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. Faseb J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- Perham RN. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem. 2000;69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- Popov KM, Hawes JW, Harris RA. Mitochondrial alpha-ketoacid dehydrogenase kinases: a new family of protein kinases. Adv Second Messenger Phosphoprotein Res. 1997;31:105–111. [PubMed] [Google Scholar]
- Read R. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallographica Section D. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Reed LJ. A Trail of Research from Lipoic Acid to alpha-Keto Acid Dehydrogenase Complexes. J Biol Chem. 2001;276:28329–28336. doi: 10.1074/jbc.R100026200. [DOI] [PubMed] [Google Scholar]
- Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci. 2007;64:830–849. doi: 10.1007/s00018-007-6380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche TE, Hiromasa Y, Turkan A, Gong X, Peng T, Yan X, Kasten SA, Bao H, Dong J. Essential roles of lipoyl domains in the activated function and control of pyruvate dehydrogenase kinases and phosphatase isoform 1. Eur J Biochem. 2003;270:1050–1056. doi: 10.1046/j.1432-1033.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- Sale GJ, Randle PJ. Analysis of site occupancies in [32P]phosphorylated pyruvate dehydrogenase complexes by aspartyl-prolyl cleavage of tryptic phosphopeptides. Eur J Biochem. 1981a;120:535–540. doi: 10.1111/j.1432-1033.1981.tb05733.x. [DOI] [PubMed] [Google Scholar]
- Sale GJ, Randle PJ. Occupancy of sites of phosphorylation in inactive rat heart pyruvate dehydrogenase phosphate in vivo. Biochem J. 1981b;193:935–946. doi: 10.1042/bj1930935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert F, Ciszak E, Korotchkina L, Golbik R, Spinka M, Dominiak P, Sidhu S, Brauer J, Patel MS, Tittmann K. Phosphorylation of serine 264 impedes active site accessibility in the E1 component of the human pyruvate dehydrogenase multienzyme complex. Biochemistry. 2007;46:6277–6287. doi: 10.1021/bi700083z. [DOI] [PubMed] [Google Scholar]
- Seifert F, Golbik R, Brauer J, Lilie H, Schroder-Tittmann K, Hinze E, Korotchkina LG, Patel MS, Tittmann K. Direct kinetic evidence for half-of-the-sites reactivity in the E1 component of the human pyruvate dehydrogenase multienzyme complex through alternating sites cofactor activation. Biochemistry. 2006;45:12775–12785. doi: 10.1021/bi061582l. [DOI] [PubMed] [Google Scholar]
- Sugden MC. PDC deletion: the way to a man’s heart disease. Am J Physiol Heart Circ Physiol. 2008;295:H917–H919. doi: 10.1152/ajpheart.00663.2008. [DOI] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003;284:E855–862. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Hutson NJ, Kerbey AL, Randle PJ. Phosphorylation of additional sites on pyruvate dehydrogenase inhibits its re-activation by pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1978;169:433–435. doi: 10.1042/bj1690433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden PH, Randle PJ. Regulation of pig heart pyruvate dehydrogenase by phosphorylation. Studies on the subunit and phosphorylation stoicheiometries. Biochem J. 1978;173:659–668. doi: 10.1042/bj1730659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague WM, Pettit FH, Yeaman SJ, Reed LJ. Function of phosphorylation sites on pyruvate dehydrogenase. Biochem Biophys Res Commun. 1979;87:244–252. doi: 10.1016/0006-291x(79)91672-3. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Symersky J. Crystal structure of pyruvate dehydrogenase phosphatase 1 and its functional implications. J Mol Biol. 2007;370:417–426. doi: 10.1016/j.jmb.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes. 1999;48:1593–1599. doi: 10.2337/diabetes.48.8.1593. [DOI] [PubMed] [Google Scholar]
- Wynn RM, Kato M, Chuang JL, Tso SC, Li J, Chuang DT. Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robust core-free basal activity. J Biol Chem. 2008;283:25305–25315. doi: 10.1074/jbc.M802249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn RM, Kato M, Machius M, Chuang JL, Li J, Tomchick DR, Chuang DT. Molecular mechanism for regulation of the human mitochondrial branched-chain alpha-ketoacid dehydrogenase complex by phosphorylation. Structure (Camb) 2004;12:2185–2196. doi: 10.1016/j.str.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Wynn RM, Machius M, Chuang JL, Li J, Tomchick DR, Chuang DT. Roles of His291-alpha and His146-beta’ in the reductive acylation reaction catalyzed by human branched-chain alpha-ketoacid dehydrogenase: refined phosphorylation loop structure in the active site. J Biol Chem. 2003;278:43402–43410. doi: 10.1074/jbc.M306204200. [DOI] [PubMed] [Google Scholar]
- Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, Dixon GH. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry. 1978;17:2364–2370. doi: 10.1021/bi00605a017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data including four supplemental figures can be found with this article online at http://www.structure.org/XXX.