

Abstract
The aim of the study was to evaluate the frequency and to perform phenotypic and genotypic characterization of familial Parkinsonism and early onset Parkinson's disease (EOPD) in a Brazilian movement disorder unit. We performed a standardized clinical assessment of patients followed by sequencing of PRKN, PINK1, SNCA and LRRK2. During the period of study (January through December, 2006) we examined 575 consecutive patients of whom 226 (39.3%) met the diagnosis of Parkinsonism and idiopathic Parkinson's disease (IPD) was diagnosed in 202 of the latter. Of the IPD cases, 45 (22.3%) had EOPD. The age at onset in the EOPD cases (n=45) was 34.8±5.4 years (mean ±standard deviation). The age at onset in familial the late-onset PD patients (n=8) was 52.3±12.2 years. In the early-onset cases, we identified five known mutations in PRKN, two single heterozygous and three compound heterozygous (P153R, T240M, 255Adel, W54R, V3I); in addition we identified one novel mutation in PINK1 (homozygous deletion of exon 7). In the familial cases (late onset), one patient had a novel LRRK2 variant, Q923H, but no SNCA mutations were identified. We have demonstrated that EOPD accounts for a high frequency of IPD cases in our tertiary referral center. PRKN was the most commonly mutated gene but we also identified a novel mutation in PINK1 and a novel variant in LRRK2.
Keywords: LRRK2, PINK1, PRKN, SNCA, Brazil
Introduction
Idiopathic Parkinson's disease (IPD) is the most frequent cause of Parkinsonism, being diagnosed in up to 86% of cases1. Early onset PD (EOPD), defined by age of onset between 20 and 40 years of age2, accounts for 4-10% of all patients with PD. Recent epidemiologic studies have indicated a more readily evident genetic component in EOPD in contrast to late-onset IPD3,4. Several familial forms of PD have been described and mutation in the genes SNCA, PRKN, PARK7, PINK1, LRRK2 and GBA have been associated with genetic forms of PD 5-11. There is clear evidence that etiologic factors may vary depending on the geographic and ethnic background of the studied population; in particular the prevalence of monogenic forms of PD and specific disease causing mutations can vary considerably between populations12. In the current study we describe the frequency of PD, including EOPD and familial forms of PD in cases ascertained within a movement disorders clinic in Brazil; furthermore we examined EOPD and cases reporting a positive family history of parkinsonism for mutations in SNCA, PRKN, PINK1 and LRRK2.
Methods
During the year of 2006 we screened all consecutive patients seen at the Movement Disorders Clinic of the Federal University of Minas Gerais, a tertiary referral center located in the southeast of Brazil, covering an area with a population of 20 million people. All patients with parkinsonism, defined by the presence of bradykinesia and at least one of the following: rigidity, tremor or postural instability, entered the study. IPD was diagnosed according to the UK Brain Bank criteria13. Patients were classified as EOPD if the age at onset of parkinsonism was ≤ 40 years and ≥ 20 years14. Careful pedigree evaluation and a detailed neurological exam including the application of the UPDRS were performed for each patient15. We have calculated the rate of progression of motor disability using the formula: median of UPDRS motor score (when patient is on) within a year minus the basal motor score. Psychiatric problems were diagnosed using DSM- IV criteria16. Patients were recruited with their prior written informed consent and ethical committee approval. DNA was extracted from peripheral lymphocytes according to routine procedures. We tested PRKN, PINK1, SNCA and LRRK2 in the late onset PD familial group (n=8) and PRKN and PINK1 for the EOPD group (n=45). Amplification of the protein coding exons of PRKN, PINK1, SNCA and LRRK2 was performed by polymerase chain reaction in a total volume of 15μl containing 20ng of genomic DNA, 5pmol of forward and reverse primers, 5U of FastStart TaqDNA polymerase (Roche) containing all of the required buffers and dNTPs (details available upon request). Thermal cycling was performed using a standard 60-50 touchdown PCR program. Following confirmation of amplification on a 2% agarose gel, PCR products were purified by filtration on 96 well plates (Millipore). The product was then used as template in a dye-terminator sequencing reaction performed as per the manufacturer's protocol (BigDye® Terminator v3.1, Applied Biosystems, Foster City, CA). Each product was sequenced in both forward and reverse directions with the primers used for initial amplification. The resulting products were filter purified as above and run on an ABI3730XL; all data was analyzed using Sequencher (GeneCodes Corporation). To assay for genetic dosage alterations, quantitative duplex PCR of genomic DNA samples was performed on the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA). Protein coding exons of PRKN, PINK1 and exon 7 of SNCA were each co-amplified with βglobin, which served as an endogenous standard. PCR was carried out with TaqMan Universal PCR Master Mix using 25 ng genomic DNA, 900 nmol/L primers, and 250 nmol/L probes in a total reaction volume of 20 μL (Applied Biosystems). Primers and probes as well as PCR conditions are as previously published17,18. The cycle in the log phase of PCR amplification at which a significant fluorescence threshold was reached (Ct) was used to quantify each amplimer. The dosage of each amplimer relative to the reference gene and normalized to control DNA was determined using the 2-ΔΔCt method. To be considered valid, the requirements were SDs < 0.16 and threshold values reached before cycle 28. A value was considered a heterozygous deletion between 0.4 and 0.6, normal between 0.8 and 1.2, a heterozygous duplication between 1.3 and 1.7, and a triplication or homozygous duplication between 1.8 and 2.2.
Results
During the year of 2006 we examined 575 patients, of whom 226 met the diagnosis of Parkinsonism. IPD was diagnosed in 202 with 45 EOPD cases. Eight late onset familial cases were included in this study. Age at onset of the EOPD group was 34.8 ± 5.4 years (mean ± standard deviation); eight reported a positive family history for IPD, of whom seven showed a pattern of inheritance consistent with autosomal recessive transmission; the remaining patient reported a family history suggestive of an autosomal dominant mode of inheritance. The age at onset of the late onset PD group was 52.3±12.2 years; four had a pattern of inheritance consistent with autosomal recessive transmission and four patients reported a family history suggestive of autosomal dominant disease.
For the early onset cases, we identified 13.3% of patients with mutations: five previously reported mutations in PRKN: P253R in a compound heterozygosity with exon 5 duplication; T240M in a single heterozygosity and in compound heterozygosity with 255Adel; 255Adel in a single heterozygosity; W54R in a compound heterozygosity with V3I and one novel mutation in PINK1, a homozygous deletion of exon 7 (Fig 1). Clinical data of these patients are found in Tables 1 and 2. For the familial late onset cases, only one patient (5.5%) had a coding alteration absent from controls in the screened genes: a novel LRRK2 variant (Q923H) (Fig 2).
Figure 1.
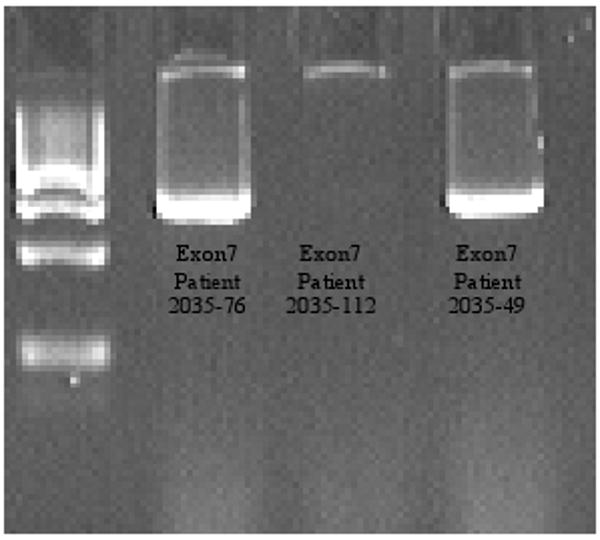
Confirmation of amplification on a 2% agarose gel. PINK1 exon 7 homozygous deletion (patient 2035-112)
Table 1.
Clinical Features of Patients with Mutations
| Patient | 2035-21 | 2035-23 | 2035-46 | 2035-49 | 2035-76 | 2035-112 | 2035-43 |
|---|---|---|---|---|---|---|---|
| Gene | PRKN | PRKN | PRKN | PRKN | PRKN | PINK1 | LRRK2 |
| Mutation | W54R | 255Adel | T240M | P253R | 255Adel | Ex7 del | Q923H |
| V3I | Dup exon5 | T240M | Ex7 del | ||||
| Gender | Male | Female | Male | Male | Male | Female | Female |
| Current Age | 54 | 52 | 38 | 46 | 64 | 43 | 51 |
| Age et Onset | 40 | 40 | 33 | 40 | 22 | 24 | 46 |
| First symptom | Bradikynesia | Lower limb tremor | Tremor | Bradikynesia | Tremor | Tremor | Tremor |
| Follow up (years) | 7 | 7 | 2 | 4 | 11 | 14 | 2 |
| Family History | Negative | Negative | negative | negative | negative | negative | Positive (father and one brother) |
| UPDRS –III (On) | 50 | 12 | 17 | 54 | 36 | 10 | 52 |
| Progression rate (on motor score/year) | +2.5/year | +0.3/year | +1/year | +4/year | +0.8/year | +0.5/year | +5/year |
| Type | Rigid-akinetic | Tremor-dominant | Tremor-dominant | Rigid-akinetic | Tremor-dominant | Tremor-dominant | Rigid-akinetic |
| Neuroimaging | Normal CT scan | Normal MRI | Normal MRI | Normal CT scan | Normal CT scan | Normal MRI | Normal MRI |
| Time for ldopa use (from the first symptom) | 5 years | * | * | 1 year | 15 years | 6 years | 4 years |
| Time to develop dyskinesias | 2 years | ------ | ------- | 4 years | 1 year | 7 years | -------- |
| Dyskinesias – (UPDRS score) | 1-1-2-0 | 0 | 0 | 1-0-0-0 | 1-2-2-1 | 1-0-0-0 | 0 |
| Surgery | No | No | No | No | Palidotomy | No | No |
| Psychiatric Symptoms | None | Generalized Anxiety Disorder | Depression | Anxiety | None | Anxiety | Depression |
Patients not treated with Ldopa
Table 2.
Clinical Comparison Between EOPD With and Without mutations
| With mutation | Without mutation | |
|---|---|---|
| N | 6 | 39 |
| Age at onset | 33.1±7.6 | 35±5.4 |
| Male × female | 4 × 2 | 20 × 19 |
| Familiar history | all negative | 8 |
| Response to ldopa | ||
| Haven't been using | 15.4 % | 33.3% |
| Good response | 76.9 % | 66.6% |
| Intolerance | 7.7 % | ---- |
| Type | ||
| Rigid-akinetic | 59% | 33.3% |
| Tremor dominant | 41% | 66.6% |
| Dyskinesias | 66.6% | 76.9% |
Figure 2.
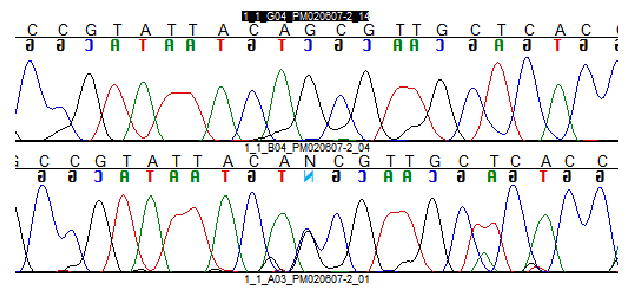
Novel mutation in LRRK2 - Q923
Discussion
In a previous study at our clinic, IPD was identified in 70% of 338 patients followed up from 1993 to 1995. During 2006, we have diagnosed 89.5% of all 226 consecutive patients with Parkinsonism. In our series, 22.3% of all IPD cases met criteria for EOPD. This is certainly more than expected for the general population, but, as is well recognized, in a tertiary center atypical patients are often over represented19. Nevertheless, this EOPD frequency is not very different than what was found in a community-based study, where it accounted for 17% of all parkinsonian patients20. Bower and colleagues found an incidence of EOPD of 0·8 cases per 100,000 per year in those aged 0–29 years, rising to 3·0 per 100,000 per year in those aged 30–49 years21. In a Korean series of EOPD, 5% of 94 patients had PRKN mutations (all in compound heterozygous state) and none had PINK1 mutations22. The frequency of those mutations within an Italian early onset group of patients was 8% (5% simple heterozygous mutations, 1% compound heterozygous and 1% homozygous)23. In a recent study, Clark et al, showed a PRKN mutation prevalence of 12.9% among 101 early onset cases; 10.9% carried simple heterozygous mutations, 1% homozygous and 1% compound heterozygous mutations24. The range of mutations in sporadic cases in previous studies varied between 9 and 18% with single mutations counting with 6.1 up to 12.8%25-27. According to Klein et al., mutation of PRKN occurs in 20% of European early onset cases, while PINK1 mutations occur in 2% to 7%23. Our data are also in agreement with such studies (PRKN, 11.1% and PINK1, 2.2% of cases) in EOPD. Single heterozygous mutations accounted with 4.4% in PRKN and 6.6% were compound heterozygous mutations. One patient hat a homozygous mutation in PINK1. Our findings suggest that despite the genetic heterogeneity of the contemporary Brazilian population, a result of mixture of people of European (mostly Portuguese but also Italian, German and other nationalities), African and Native origins, the frequency of these genetic mutations does not differ substantially from populations from other geographic areas. Surprisingly, since mutations in PRKN are highly correlated with lower age of onset and positive family history23, none of the familial early onset cases had mutation of the screened genes. One possible explanation for this finding is the small sample size. The clinical picture of patients with and without mutations was similar in the studied population. Two patients carrying single heterozygous mutations of PRKN did not differ from the three compound heterozygous subjects or from those patients without mutations. Whether these single heterozygous mutations play a role in the disease process remains to be determined. Two previous studies showed that single heterozygous mutations in PRKN are as common as in controls28,29. This was not confirmed in a third sudy30. Some individuals carrying a heterozygous mutation in PRKN or PINK1, considered as “asymptomatic patients” had some minor but unequivocal parkinsonian features31-33. PET scan studies of PRKN heterozygous mutant carriers showed reduction of striatal F-Dopa uptake34. Haploinsufficiency, a dominant negative effect or a risk factor associated to an environmental or another genetic cause have been used as hypotheses supporting the role of a single recessive mutation in promoting parkinsonism in these patients33,35,36. Since the frequency of PRKN mutations is higher in the EOPD group, it is possible that mutations in this gene might diminish the age at onset.
We did not find mutations or gene dosage alterations in SNCA. This confirms the findings of several other authors that SNCA mutation is a rare cause of parkinsonism, regardless of the age of onset of the patients37,38. LRRK2 mutations are more common, one study with 504 PD cases (of whom were 245 EOPD cases) and 341 controls showed a LRRK2 mutation frequency of ∼1% in sporadic cases and ∼5% in familial cases39; although as these studies primarily screen a single exon/mutation, they are likely an underestimation of the true prevalence. We tested all exons of LRRK2 only in familial cases (n=8) and we identified variant Q923H located in exon 21 in a typical PD case with a clear autosomal dominant inheritance. The new variant was not found in 192 healthy controls. Cross species comparison reveals a reasonable conservation of this residue. Although these findings support a pathogenic effect of this novel mutation, this result must be interpreted with caution. We were not able to study disease segregation in this family since the patients parents were deceased and thus we feel it remains to be determined if this variant is pathogenic. We did not identify the common G2019S mutation in our series, although given the relatively small sample series this is perhaps not surprising.
Prevalence studies of known genes in a larger number of patients are needed to provide a better view about the role of these mutations in the Brazilian population, whether it influences clinical features and age at onset.
Acknowledgments
We thank the patients for taking part in this study. This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services. The NIH project number associated with this work is Z01 AG000957-05.
References
- 1.Rajput AH, Offord KP, Beard CM, Kurland LT. Epidemiology of parkinsonism: incidence, classification, and mortality. Ann Neurol. 1984 Sep;16(3):278–82. doi: 10.1002/ana.410160303. [DOI] [PubMed] [Google Scholar]
- 2.Quinn N, Critchley P, Marsden CD. Young onset Parkinson's disease. Mov Disord. 1987;2(2):73–91. doi: 10.1002/mds.870020201. [DOI] [PubMed] [Google Scholar]
- 3.Payami H, Zareparsi S, James D, Nutt J. Familial aggregation of Parkinson disease: a comparative study of early-onset and late-onset disease. Arch Neurol. 2002 May;59(5):848–50. doi: 10.1001/archneur.59.5.848. [DOI] [PubMed] [Google Scholar]
- 4.Sveinbjornsdottir S, Hicks AA, Jonsson T, Petursson H, Gugmundsson G, Frigge ML, Kong A, Gulcher JR, Stefansson K. Familial aggregation of Parkinson's disease in Iceland. N Engl J Med. 2000 Dec 14;343(24):1765–70. doi: 10.1056/NEJM200012143432404. [DOI] [PubMed] [Google Scholar]
- 5.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997 Jun 27;276(5321):2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 6.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998 Apr 9;392(6676):605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 7.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 8.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004 May 21;304(5674):1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 9.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004 Nov 18;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 10.Zimprich A, Müller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, Stoessl J, Uitti RJ, Pfeiffer RF, Trenkwalder C, Homann N, Ott E, Wenzel K, Asmus F, Hardy J, Wszolek Z, Gasser T. The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the diseasecontaining interval. Am J Hum Genet. 2004 Jan;74(1):11–9. doi: 10.1086/380647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med. 2004;351:1972–1977. doi: 10.1056/NEJMoa033277. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann Neurol. 2006 Oct;60(4):389–98. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- 13.Daniel SE, Lees AJ. Parkinson's Disease Society Brain Bank, London: overview and research. J Neural Transm Suppl. 1993;39:165–72. [PubMed] [Google Scholar]
- 14.Quinn N, Critchley P, Marsden CD. Young onset Parkinson's disease. Mov Disord. 1987;2:73–91. doi: 10.1002/mds.870020201. [DOI] [PubMed] [Google Scholar]
- 15.Fahn S, Elton RL, the UPDRS development Committee . Unified Parkinson disease rating scale. In: Fahn S, Marsden CD, Calne DB, editors. Recent developments in Parkinson's disease. Floral Park, NJ: Macmillan; 1987. pp. 293–304. [Google Scholar]
- 16.American Psychiatric Association. The Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition Text Revision. Washington DC: American Psychiatric Association; 2000. 943p. [Google Scholar]
- 17.Hedrich K, Kann M, Lanthaler AJ, et al. The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset parkinsonism. Hum Mol Genet. 2001;10(16):1649–56. doi: 10.1093/hmg/10.16.1649. [DOI] [PubMed] [Google Scholar]
- 18.Johnson J, Hague SM, Hanson M, et al. SNCA multiplication is not a common cause of Parkinson disease or dementia with Lewy bodies. Neurology. 2004;63(3):554–6. doi: 10.1212/01.wnl.0000133401.09043.44. [DOI] [PubMed] [Google Scholar]
- 19.Cardoso F, Camargos ST, Silva GA., Junior Etiology of parkinsonism in a Brazilian movement disorders clinic. Arq Neuropsiquiatr. 1998 Jun;56(2):171–5. doi: 10.1590/s0004-282x1998000200001. [DOI] [PubMed] [Google Scholar]
- 20.Dekker MC, van Switen JC, Houwing-Duistermaat JJ, et al. A clinical-generic study of Parkinson's disease in a genetically isolated community. J Neurol. 2003;250(9):1056–1062. doi: 10.1007/s00415-003-0151-z. [DOI] [PubMed] [Google Scholar]
- 21.Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology. 1999;52:1214–20. doi: 10.1212/wnl.52.6.1214. [DOI] [PubMed] [Google Scholar]
- 22.Chung EJ, Ki CS, Lee WY, Kim IS, Kim JY. Clinical features and gene analysis in Korean patients with early-onset Parkinson disease. Arch Neurol. 2006 Aug;63(8):1170–4. doi: 10.1001/archneur.63.8.1170. [DOI] [PubMed] [Google Scholar]
- 23.Klein C, Djarmati A, Hedrich K, Schafer N, Scaglione C, Marchese R, Kock N, Schule B, Hiller A, Lohnau T, Winkler S, Wiegers K, Hering R, Bauer P, Riess O, Abbruzzese G, Martinelli P, Pramstaller PP. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur J Hum Genet. 2005 Sep;13(9):1086–93. doi: 10.1038/sj.ejhg.5201455. [DOI] [PubMed] [Google Scholar]
- 24.Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Case-Control Study of the Parkin Gene in Early-Onset Parkinson Disease. Arch Neurol Volume. 2006 April;63(4):548–552. doi: 10.1001/archneur.63.4.548. [DOI] [PubMed] [Google Scholar]
- 25.Periquet M, Latouche M, Lohmann E, et al. Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain. 2003;126:1271–1278. doi: 10.1093/brain/awg136. [DOI] [PubMed] [Google Scholar]
- 26.Kann M, Jacobs H, Mohrmann K, et al. Role of parkin mutations in 111 community based patients with early-onset parkinsonism. Ann Neurol. 2002;51:621–625. doi: 10.1002/ana.10179. [DOI] [PubMed] [Google Scholar]
- 27.Poorkaj P, Nutt JG, James D, et al. Parkin mutation analysis in clinic patients with early-onset Parkinson's disease. Am J Med Genet A. 2005;139A:56. doi: 10.1002/ajmg.a.30157. [DOI] [PubMed] [Google Scholar]
- 28.Lincoln SJ, Maraganore DM, Lesnick TG, et al. Parkin variants in North American Parkinson's disease: cases and controls. Mov Disord. 2003;18(11):1306–11. doi: 10.1002/mds.10601. [DOI] [PubMed] [Google Scholar]
- 29.Kay DM, Moran D, Moses L, et al. Heterozygous parkin point mutations are as common in control subjects as in Parkinson's patients. Ann Neurol. 2007;61(1):47–54. doi: 10.1002/ana.21039. [DOI] [PubMed] [Google Scholar]
- 30.Clark LN, Afridi S, Karlins E, et al. Case-control study of the parkin gene in early-onset Parkinson disease. Arch Neurol. 2006;63(4):548–52. doi: 10.1001/archneur.63.4.548. [DOI] [PubMed] [Google Scholar]
- 31.Hedrich K, Hagenah J, Djarmati A, et al. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch Neurol. 2006;63(6):833–8. doi: 10.1001/archneur.63.6.833. [DOI] [PubMed] [Google Scholar]
- 32.Hiller A, Hagenah JM, Djarmati A, et al. Phenotypic spectrum of PINK1-associated parkinsonism in 15 mutation carriers from 1 family. Mov Disord. 2007;22(1):145–7. doi: 10.1002/mds.21059. [DOI] [PubMed] [Google Scholar]
- 33.Pramstaller PP, Schlossmacher MG, Jacques TS, et al. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58(3):411–22. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- 34.Hilker R, Klein C, Ghaemi M, et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49(3):367–76. [PubMed] [Google Scholar]
- 35.Khan NL, Valente EM, Bentivoglio AR, et al. Clinical and subclinical dopaminergic dysfunction in PINK1-linked parkinsonism: an 18F-dopa PET study. Ann Neurol. 2002;52(6):849–53. doi: 10.1002/ana.10417. [DOI] [PubMed] [Google Scholar]
- 36.Sun M, Latourelle JC, Wooten GF, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006 Jun;63(6):826–32. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- 37.Johnson J, Hague SM, Hanson M, et al. SNCA multiplication is not a common cause of Parkinson disease or dementia with Lewy bodies. Neurology. 2004;63(3):554–6. doi: 10.1212/01.wnl.0000133401.09043.44. [DOI] [PubMed] [Google Scholar]
- 38.Klein C. Implications of Genetics on the Diagnosis and Care of Patients With Parkinson Disease. Arch Neurol. 2006 Mar;63(3):328–34. doi: 10.1001/archneur.63.3.328. [DOI] [PubMed] [Google Scholar]
- 39.Clark LN, Wang Y, Karlins E, et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. 2006;67:1786–1791. doi: 10.1212/01.wnl.0000244345.49809.36. [DOI] [PubMed] [Google Scholar]