

Summary
The espins are actin-bundling proteins of brush border microvilli and Sertoli cell-spermatid junctions. We have determined that espins are also present in hair cell stereocilia and have uncovered a connection between the espin gene and jerker, a recessive mutation that causes hair cell degeneration, deafness and vestibular dysfunction. The espin gene maps to the same region of mouse chromosome 4 as jerker. The tissues of jerker mice do not accumulate espin proteins, but contain normal levels of espin mRNAs. The espin gene of jerker mice has a frameshift mutation that affects the espin C-terminal actin-bundling module. These data suggest that jerker mice are, in effect, espin null and that the jerker phenotype results from a mutation in the espin gene.
Introduction
Cells that act as sensory receptors employ elaborate specializations to transduce environmental cues. Within the auditory and vestibular systems, hair cells use stereocilia as mechanoelectrical signal transducers to detect sound and motion (Roberts et al., 1988; Pickles and Corey, 1992; Eatock et al., 1998). Each hair cell stereocilium is a highly specialized fingerlike projection of the apical plasma membrane that contains a parallel bundle of actin filaments at its core (Tilney et al., 1992; Bartles, 2000). The stereocilium tapers near its base, so that only the central actin filaments of the core actin bundle extend a rootlet into the apical cytoplasm of the hair cell to encounter the dense actin filament meshwork known as the cuticular plate (Tilney et al., 1992). Stereocilia exist on the apical surface of the hair cell in highly organized collections: stereocilia of similar length are arranged in rows, and rows of different lengths produce staircase assemblies that adopt fixed orientations with respect to anatomical landmarks within the inner ear (Lim, 1986; Tilney et al., 1992). Mechanical linkages – tip links – become established between a stereocilium and its next tallest neighbor in an adjacent row. When the tuft of stereocilia is deflected in the direction of the tallest row, the stretching of tip links elicits an increase in the permeability of ion channels in the stereocilium plasma membrane and causes hair cell depolarization (Roberts et al., 1988; Pickles and Corey, 1992; Eatock et al., 1998). The available evidence suggests that the core actin bundle acts as a scaffold that provides mechanical support for the stereocilium, in effect, determining its position, orientation and dimensions and possibly also influencing its stiffness and ability to pivot about its base (Tilney et al., 1992).
The crosslinking of actin filaments to form a parallel actin bundle is mediated by actin-bundling proteins (Matsudaira, 1991; Puius et al., 1998; Bartles, 2000). The formation of parallel actin bundles in structures as diverse as the neurosensory bristles of Drosophila pupae and brush border microvilli appears to involve the sequential action of multiple actin-bundling proteins (reviewed by Bartles, 2000). Although the actin-bundling protein fimbrin is present in hair cell stereocilia (Flock et al., 1982; Sobin and Flock, 1983; Tilney et al., 1989), the identity of other actin-bundling proteins suspected to work in conjunction with fimbrin (Tilney et al., 1989) has remained a mystery.
Over the last several years, we have characterized a new family of actin-bundling proteins known as the espins (Bartles, 2000). Two espin isoforms have been identified: the ~110-kD espin of Sertoli cell-spermatid junctions (ectoplasmic specializations; Bartles et al., 1996; Chen et al., 1999) and the ~30-kD small espin of brush border microvilli in the intestine and kidney (Bartles et al., 1998). Encoded by a single gene (Chen et al., 1999), espin and small espin share a 167-amino acid C-terminal peptide that includes a 116-amino acid C-terminal actin-bundling module that is necessary and sufficient for actin bundle formation in vitro, but they contain different N termini (Bartles et al., 1998). Unlike many actin-bundling proteins, the espins bind actin filaments with high affinity and their actin-bundling activities are not inhibited by Ca2+ (Bartles et al., 1998; Chen et al., 1999).
A great deal has been learned from the identification of mutations that cause hearing impairment or vestibular dysfunction in mice (Steel, 1995; Steel et al., 1997; Probst and Camper, 1999). A significant number of these mutations have been mapped to genes that encode proteins of the hair cell actin cytoskeleton (e.g., Gibson et al., 1995; Avraham et al., 1995; Probst et al., 1998). However, a number of deafness mutations have yet to be assigned to a particular gene (Steel, 1995). One such mutation is jerker, a spontaneous autosomal recessive mutation, first reported nearly 60 years ago (Grüneberg et al., 1941). Cochlear development appears relatively normal in jerker mice until the early postnatal period, at which time the hair cells begin to show evidence of a progressive degeneration (Deol, 1954; Steel and Bock, 1983; Sjöström and Anniko, 1990a and b, 1992a and b). The degeneration is confined initially to the stereocilia and cuticular plate: stereocilia appear to shorten, lose their stiffness or merge, and the cuticular plate appears to protrude, fold or disintegrate. These changes begin in earnest around postnatal day 11, coincident with the onset of auditory function (Ehret, 1977; Steel and Bock, 1993), but have been noted in an occasional hair cell as early as postnatal day 1. After degeneration of their stereocilia and cuticular plate, hair cells appear to shrink and disappear so that after ~3 months all that remains of the organ of Corti is a thin layer of dedifferentiated cells resting between collapsed pillar cells on the basilar membrane. In the vestibular system of jerker mice, the macula utriculi and macula sacculi exhibit a similar, although slower, hair cell degeneration and loss, whereas the hair cells of the cristae ampullares appear to be less affected. Measurements of auditory brainstem response indicate that jerker mice are totally deaf from postnatal day 12 onward (Steel and Bock, 1983; Sjöström and Anniko, 1990b). In addition to deafness, jerker mice display other features of the typical shaker-waltzer behavior, namely hyperactivity, circling movements, and head tossing (Grüneberg et al., 1941), which are presumed to reflect abnormalities within the vestibular system (Anniko et al., 1989; Sjöström and Anniko, 1990a).
In this report, we demonstrate that espin actin-bundling proteins are present in hair cell stereocilia and supply multiple lines of evidence linking the jerker mutation to the espin gene.
Results
Espins Are Present in the Stereocilia of Hair Cells in the Cochlea and Vestibular System
An affinity purified, highly specific rabbit polyclonal antibody directed against the 167-residue C-terminal peptide shared among known espin isoforms was found to label hair cell stereocilia in a variety of species. This result is illustrated for mouse cochlear (Figure 1A) and rat vestibular hair cells (Figures 1B and C) in cryostat sections of decalcified preparations from adult animals and for hair cells in whole mounts of isolated cochlear sensory epithelium from 13-day chicken embryos (Figures 2A and B). Consistent with the localization of espins to parallel actin bundles in other cell types (Bartles et al., 1996, 1998), the stereocilia showed considerably more labeling than the bodies of the hair cells (Figures 1A and C) and the actin filament meshwork of the cuticular plate (cf. Figures 2B and C). Intense specific labeling of hair cell stereocilia was also observed in 9- and 21-day-old mice, 5-day-old chickens, day-15 chicken embryos, and 5-day-old gerbils (data not shown).
Figure 1. Affinity Purified Espin Antibody Labels Stereocilia on Cochlear and Vestibular Hair Cells in Cryosections of Decalcified Specimens from Adult Mouse and Rat (Immunoperoxidase Method).

(A) Mouse cochlea. The collections of stereocilia on the single row of inner hair cells (asterisks over hair cell bodies) are sectioned roughly longitudinally, whereas those on the three rows of outer hair cells (arrowheads) are sectioned tranversely at different levels near their base. Bar, 10 μm.
(B) Rat vestibular system, low magnification view. The collections of stereocilia on the patches of hair cells that constitute the macula utriculi (mu), macula sacculi (ms) and the cristae ampullares of the anterior (ca) and lateral (cl) semicircular canals are shown. (Asterisk, posterior semicircular canal). Bar, 300 μm.
(C) Rat vestibular system, higher magnification view of the macula utriculi. Bar, 20 μm.
Figure 2. Affinity Purified Espin Antibody Labels Hair Cell Stereocilia in Whole Mounts of Isolated Cochlear Sensory Epithelium from a 13-day Chicken Embryo (Confocal Immunofluorescence Method).
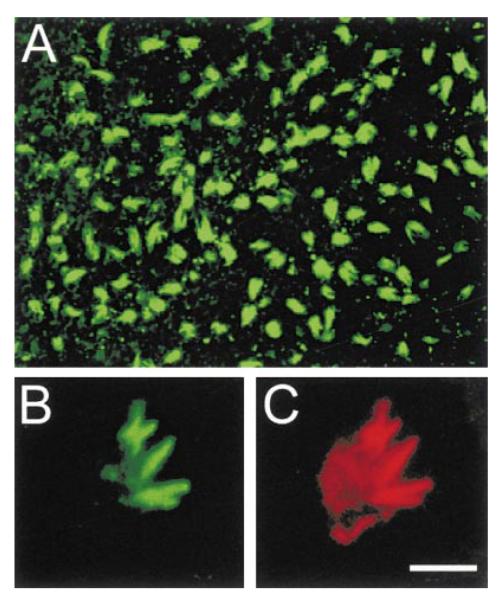
(A) Low magnification view from above the epithelium. Bar in (C), 28 μm.
(B) Higher magnification view showing several clumps of labeled stereocilia emanating from the apex of a single hair cell. Bar in (C), 5 μm.
(C) Double labeling of the same hair cell shown in (B) with rhodamine-phalloidin reveals the intensely labeled bundles of F-actin at the cores of the clumped stereocilia emanating from a less intensely labeled cuticular plate. Bar, 5 μm.
Mouse and chicken hair cells appeared to contain an espin protein intermediate in apparent molecular mass between espin (~110 kD) and small espin (~30 kD). Because of the small size of the inner ear, it was necessary to pool preparations from 5-6 animals to obtain enough material for western blotting. The affinity purified espin antibody directed against the 167-residue shared C-terminal peptide reacted with a major band of 45 kD and minor bands of 40 kD and 30 kD in SDS extracts of mouse temporal bone (Figure 3A, left). A faint band of 110 kD emerged with overexposure. The antibody also reacted with a 45-50-kD band in a more enriched preparation of hair cells: the isolated cochlear sensory epithelium of 5-day-old chicks (Figure 3A, right). This antibody does not react with actin, a major 43 kD protein. Although it is possible that the ~45-50-kD proteins result from the proteolysis of larger espin proteins, another actin-bundling protein, fimbrin, remained intact in these samples (Figure 3A, lanes F).
Figure 3. Detection of Inner Ear Espin Proteins and cDNAs by Western Blotting and Library Screening.

(A) Western blots of SDS extracts of mouse temporal bone homogenate and isolated chicken cochlear sensory epithelium showing that the affinity purified espin antibody reacts most strongly with proteins of ~45-50 kD (bracket at left) (PI, preimmune IgG control; IM, affinity purifed espin antibody). Fimbrin (lanes labeled F) remains largely intact at ~68-70 kD in these specimens (arrowhead).
(B) Stick-figure diagram highlighting the relationships between rat espin, rat small espin and the peptides encoded by the longest espin cDNAs obtained from rat and chicken cochlear cDNA libraries (PR, proline-rich peptide; ABM, 116-amino shared C-terminal actin-bundling module). The N-termini of the peptides encoded by the cochlear cDNAs are shown as dotted lines to signify that the cDNAs either are (chicken) or may be (rat) partial at their 5′ ends. None of the cochlear espin cDNAs encoded the two small peptides (shaded) that are unique to small espin (Bartles et al., 1998; Chen et al., 1999). The affinity purified antibodies used in this study are directed against the 167-residue shared C-terminal peptide of rat espin (amino acids 671-837) or a 262-residue peptide from a more central region of rat espin (amino acids 459-720) (thick lines above stick-figure of rat espin). The asterisk designates the position of the point mutation detected in jerker mice, which elicits a frameshift after R808 of mouse espin.
Espin cDNAs were detected in cochlear cDNA libraries. Four overlapping partial cDNAs were isolated from a chicken cochlear library. These cDNAs encoded espin fragments that included the shared C-terminal actin-bundling module and extended upstream to encompass the potential P-loop (three clones) or, in the case of a single longer cDNA (GenBank accession number AF239885), the C-terminal proline rich peptide (Figure 3B). A rat cochlear library yielded two cDNAs that were identical to rat espin cDNA (GenBank accession number U46007), except they began at nucleotide 1204 and, thus, encoded a protein that spanned from just upstream of the N-terminal proline-rich peptide to the C-terminus (Figure 3B). None of the cDNAs obtained from the cochlear libraries encoded the two small peptides unique to small espin (shaded in Figure 3B). It is presently unclear whether the cDNAs obtained from the rat cochlear library are full length or partial. These cDNAs begin at the 5′-end of an exon (GenBank accession number AF134858) and encode espin proteins of the expected size (~47 kD; Figure 3A). Although neither of the two in-frame ATG codons found at the 5′ end are in a Kozak consensus sequence (Kozak, 1987), cochlear espin cDNAs with additional upstream sequence were not identified in these cDNA libraries or in multiple attempts at 5′-RACE-PCR.
The Mouse Espin Gene Maps to the Same Region of Chromosome 4 As the Jerker Mutation, and the Tissues of the Jerker Mouse Are Missing Espin Protein, but Not Espin mRNA
An ~18-kb mouse espin genomic DNA fragment (Chen et al., 1999) was used to map the chromosomal location of the single espin gene (symbol Espn) to cytogenetic banding position E1 on chromosome 4 in the mouse [Mouse Genome Database (MGD) Accession number J:53411]. Of the large number of known and candidate deafness genes mapped by linkage studies in the mouse (Steel, 1995), one that shows primary defects at the level of the stereocilia and maps to this location is the jerker mutation (80.1 cM; Mouse Genome Informatics Accession ID: MGI:96636), first reported by Grüneberg et al. (1941).
Jerker mice showed prominent defects in espin protein expression in multiple tissues. The affinity purified espin antibody directed against the 167-residue shared C-terminal peptide reacted with bands expected for espin (~110 kD) and small espin (~30 kD) in the testis and kidney homogenates of jerker heterozygotes, but the levels of these proteins were less than those detected in age-matched CBA wild-type control mice (Figures 4A and C). More importantly, the testis and kidney homogenates of jerker homozygotes showed no specific antibody labeling in the positions expected for espin and small espin (Figures 4A and C). This was the case even upon further overexposure (data not shown). A similar result was obtained when the testis homogenate blots were labeled using a different affinity purified espin antibody directed against a 262-residue peptide (amino acids 459-720; Figure 3B) from a more central region of the espin protein (Figure 4B). This antibody detected a minor cross-reacting doublet at ~70 kD (Figure 4B, bracket), but these bands were also present in samples from the CBA wild-type control mice. Although major bands indicative of an accumulation of truncated or mutated espin proteins were not detected in jerker specimens, a faint triplet of specifically labeled bands at ~110 kD in the testis homogenates of jerker homozygotes emerged upon overexposure of the blot in Figure 4B (data not shown). The combined intensity of these three bands was estimated by laser densitometry to be ~3% of that observed for the 110-kD espin band of jerker heterozygotes.
Figure 4. Deficiency of Espin Protein, but Presence of Espin mRNA, in the Testis and Kidney of Adult Jerker Homozygotes.

(A-C) Western blots of homogenates prepared from testes or kidneys of duplicate CBA/CaJ wild-type mice (CBA), jerker heterozygotes (+/je), or jerker homozygotes (je/je) labeled with affinity purified espin antibody. Note the absence of specific labeling in the molecular mass regions expected for espin [~110 kD (A and B), arrowheads] or small espin [~30 kD (C), arrowhead] in jerker homozygotes. In (A) and (C), the affinity purifed espin antibody was directed against the 167-residue shared C-terminal peptide of the rat espins [ESPIN(671-837)]. In (B), the affinity purified espin antibody was directed against a 262-residue peptide from a more central region of rat espin [ESPIN(459-720)]. Bracket in B, ~70-kD doublet that shows weak cross-reaction with the antibody directed against the more central region of espin, but is also present in the samples from the CBA wild-type mice.
(D and E) RT-PCR analysis of total RNA isolated from the testis (D) or kidney of (E) of two jerker heterozygotes (+/je) and two jerker homozygotes (je/je) using primers that amplify a 288-bp fragment from the 3′ end of the coding region of the espins showing cDNA products of roughly comparable levels and sizes.
The hair cells of jerker homozygotes were also found to contain reduced levels of espin protein. This result is illustrated in Figure 5 for vestibular hair cells in cryosections from jerker heterozygotes and homozygotes on postnatal day 16. Postnatal day 15 or 16 is when jerker homozygotes can first be reliably identified on the basis of their circling behavior, but it precedes the time when hair cell loss becomes pronounced within the vestibular system (Sjöström and Anniko, 1990a). When the sections were labeled with the affinity purified antibody directed against the more central portion of the espin protein (amino acids 459-720; Figure 3B), the stereocilia on the hair cells of the heterozygote showed strong specific labeling (Figure 5A). In contrast, the hair cells of the homozygote showed no labeling beyond a faint cytoplasmic background (Figure 5B) comparable to that observed in controls labeled with preimmune IgG instead of espin antibody (not shown). This absence of specific labeling also applied to the residual stereocilia on the vestibular hair cells of the homozygotes. Because these stereocilia were generally shorter, less numerous and unlabeled, they were often difficult to see using standard brightfield illumination (Figure 5B), but could be seen more clearly with higher magnification and contrast (Figure 5C). The absence of specific labeling on the hair cells of jerker homozygotes was also noted when using the affinity purified antibody directed against the 167-residue shared C-terminal peptide of the espins and for the small number of residual cochlear hair cells that could be identified in the sections from the jerker homozygotes (data not shown).
Figure 5. Reduced Levels of Espins in the Vestibular Hair Cells of Jerker Homozygotes on Postnatal Day 16.

(A and B) Immunoperoxidase labeling of cryosections of maculae utriculi from a jerker heterozygote (A) and homozygote (B) using the affinity purified antibody directed against the 262-residue peptide from a more central region of the rat espin protein (amino acids 459-720). Bar in B, 20 μm.
(C) Higher magnification view of the region between the arrows in (B) shown at higher contrast to better reveal the residual stereocilia. Bar, 10 μm.
Roughly equal levels of espin mRNAs – of normal apparent size – were detected in the tissues of jerker homozygotes and heterozygotes. This is illustrated for testis RNA (Figure 4D) and kidney RNA (Figure 4E) by RT-PCR using primers that amplified a 288-bp fragment from the 3′ end of the coding region of the espin and small espin mRNAs. Comparable results were obtained by RT-PCR using primers that amplified overlapping fragments along the entire coding sequences of the espin and small espin mRNAs (data not shown). In each instance, the identity of the fragment was confirmed by DNA sequence analysis (see below).
The Espin Gene of Jerker Mice Contains a Frameshift Mutation That Alters the Sequence of the Shared C-Terminal Actin-Bundling Module
The coding portion of the espin gene of jerker mice was found to contain a single point mutation, a frameshift mutation that affected the shared C-terminal actin-bundling module of the espins. The mutation was first noted upon DNA sequence analysis of espin and small espin cDNAs produced by RT-PCR analysis of testis and kidney RNA from jerker homozygotes and heterozygotes. Mouse espin cDNA (GenBank accession number AF239886) encoded a protein that was 96% identical to rat espin, but its sequence differed slightly from that inferred previously (Chen et al., 1999) in that it included two additional small exons (p and s, Figure 6A) not present in rat espin cDNA. Compared to the wild-type espin sequence (Figure 6B), the espin cDNA in the jerker homozygotes (Figure 6C) was missing a G at position 2426 in the coding sequence. The deletion of this nucleotide would cause a frameshift mutation that would profoundly alter the sequence of approximately the C-terminal half of the 116-amino acid shared C-terminal actin-bundling module (Figures 6B-E and asterisk in Figure 3B). The result would be a truncated protein, 24 amino acids shorter than the wild-type protein, that contained increased numbers of arginines and serines, decreased numbers of glutamates, and a single cysteine residue within its C terminus (Figures 6D and E). No other frameshift mutations or premature stop codons were detected within the coding portion of the espin cDNA from jerker homozygotes.
Figure 6. Organization of the Mouse Espin Gene and the Mutation Found in Jerker Mice.

(A) Revised map of part of the mouse espin gene highlighting the relative sizes and positions of the exons used to construct small espin (t-z) and the portion of espin downstream of the ankyrin-like repeats (n-s, u, and w-z) (PR, exons that encode the proline-rich peptides of espin; ABM, the exons that encode the 116-amino acid shared C-terminal actin-bundling module). Exons t and v, which are specific to small espin, have been shaded.
(B and C) Automated DNA sequence analysis of mouse espin and small espin cDNAs and exon x of the mouse espin gene showing that, compared to the wild-type sequences (B), jerker homozygotes (C) are missing a G at the position corresponding to nucleotide 2426 in the coding sequence of mouse espin cDNA. This deletion causes a frameshift mutation that changes the sequence of the shared C-terminal actin-bundling module and is expected to produce a protein that is 24 amino acids shorter because a stop codon is reached at a position corresponding to nucleotide 2543 instead of 2614 in the coding sequence of mouse espin cDNA. The espin and small espin cDNAs and exon x of the espin gene in jerker heterozygotes contain a mixture of the wild-type and mutated sequences.
(D and E) Comparisons of the amino acid sequences at the C-termini of mouse wild-type (D) and mutated jerker (E) espins.
The sequence of the espin cDNA in the jerker heterozygotes was identical to that in the jerker homozygotes throughout the entire coding region, with one notable exception: the sequence became undecipherable – appearing as if more than one sequence was present – beginning precisely at the position of the missing G when approached by automated DNA sequencing from either direction. This difference in sequence at the position corresponding to nucleotide 2426 in the coding sequence of espin cDNA was also noted when examining the mouse small espin cDNAs produced by RT-PCR analysis of RNA isolated from the kidneys of jerker homozygotes and heterozygotes (data not shown). Outside of this difference, the coding sequences of the mouse small espin cDNAs from the jerker homozygotes and heterozygotes were identical and contained no additional frameshift mutations or premature stop codons.
The frameshift mutation noted in the espin and small espin cDNAs was also evident in the espin gene of jerker mice. Exon x of the espin gene (Figure 6A) was amplified from the genomic DNA of jerker heterozytgotes and homozygotes by high-fidelity PCR. When the PCR products obtained from the heterozygote were subcloned and sequenced individually, a mixture of mutated and wild-type sequences was obtained. Two-thirds of the clones (12/18) displayed a sequence like that of the wild-type espins, which included the G at position 2426 (Figure 6B), whereas one-third of the clones (6/18) were missing the G (Figure 6C), just like the espin and small espin cDNAs obtained from the jerker homozygote. As expected, all eight of the exon x clones obtained from the jerker homozygote were missing the G.
Discussion
The Jerker Phenotype As the Result of a Mutation in the Espin Gene
The data reported above provide strong support for the conclusion that the hair cell degeneration, deafness and behavioral abnormalities that are the hallmarks of the jerker phenotype are the result of a mutation in the gene that encodes the espin actin-bundling proteins. First, the espin gene maps to the same location of mouse chromosome 4 as the jerker mutation. Second, espin proteins are localized to hair cell stereocilia, the mechanoelectrical signal transducers of the auditory and vestibular systems and the primary sites of degeneration noted in jerker mice. Third, the espin gene of jerker mice has a frameshift mutation that alters the sequence of the C-terminal actin-bundling module shared among all known espin isoforms, including the espins of hair cells. Fourth, although jerker mice appear to have normal levels of espin mRNAs, espin proteins do not accumulate in multiple tissues of jerker mice, including hair cells. This latter result confirms that jerker mice have a defect at the level of their espin proteins and suggests that the frameshift mutation adversely affects the stability of the espins in all cells that ordinarily express the proteins. In light of these findings, we propose that the espin gene be added to the list of deafness genes that encode proteins of the hair cell actin cytoskeleton. A rat homolog of jerker has been identified and mapped to the syntenic region of rat chromosome 5 (Truett et al., 1996). Knowledge of the position of a putative human espin gene (chromosome 1p36.11-36.31; GenBank accession numbers AL031848 and AF134401) can now be used to help identify humans that carry homologous deafness mutations.
The Possible Role of Espins in Hair Cell Stereocilia
Espin and small espin have been hypothesized to act as high affinity, Ca2+-insensitive actin-bundling proteins of ectoplasmic specialization junctional plaques and brush border microvilli (Bartles et al., 1996, 1998; Chen et al., 1999; Bartles, 2000). By analogy, the espin proteins detected in hair cells may be the heretofore unidentified actin-bundling proteins postulated to work in conjunction with fimbrin to cross-link the actin filaments in the parallel actin bundle found at the core of the hair cell stereocilium (Tilney et al., 1989). Consistent with this view, the hair cell espins are concentrated within stereocilia and contain the C-terminal actin-bundling module that is necessary and sufficient for actin-bundling activity in vitro (Bartles et al., 1998), in addition to many other recognizable features of the espins. Further experiments will be required to determine whether the ~45-50-kD proteins detected in inner ear extracts represent a novel isoform, identical to Sertoli cell espin but with a shorter N-terminus, or whether they are derived from espin by proteolysis.
The deficiency of espins in jerker mice appears to have a greater impact on stereocilium stability than stereocilium assembly. To the extent that they have been examined, the morphological aspects of hair cell stereociliogenesis appear remarkably normal in jerker homozygotes until postnatal day 11 (Deol, 1954; Steel and Bock, 1983; Sjöström and Anniko, 1990a and b, 1992a and b). Thus, it would appear that most of the currently recognized steps involved in the positioning and growth of stereocilia (Lim and Anniko, 1985; Tilney et al. 1992; Kaltenbach et al., 1994) can occur in jerker homozygotes, even though espins are present at exceedingly low levels and contain a frameshift mutation in their shared actin-bundling module. The degeneration of stereocilia and cuticular plate noted in jerker homozygotes occurs late in stereociliogenesis, beginning on postnatal day 11 (Deol, 1954; Steel and Bock, 1983; Sjöström and Anniko, 1990a and b, 1992a and b), coincident with the onset of auditory function (Ehret, 1977; Steel and Bock, 1983). Unlike the other known actin-bundling protein of hair cell stereocilia, fimbrin, the espins bind to F-actin with high affinity, and their actin-bundling activities are not blocked by Ca2+ (Bartles et al., 1996, 1998; Chen et al., 1999). Mechanoelectrical signal transduction by hair cells involves increases in Ca2+ influx across the plasma membrane of the stereocilium (Ohmori, 1988; Denk et al., 1995; Lumpkin and Hudspeth, 1998). Therefore, one possibility is that hair cell espin cross-links stabilize the core actin bundle of the stereocilium against local increases in the concentration of Ca2+ that occur upon activation of the hair cell. Thus, like the neurosensory bristles of Drosophila pupae (Tilney et al., 1998), hair cell stereocilia appear to represent structures in which multiple actin-bundling proteins (fimbrin and an espin) function sequentially. A role in stabilizing parallel actin bundles was postulated previously for small espin because it is the last of three actin-bundling proteins (villin, fimbrin, and small espin) to accumulate in microvilli during assembly of the enterocyte brush border (Bartles et al., 1998).
The Failure to Accumulate Espin Protein in Jerker Mice
Even though the frameshift mutation detected jerker mice is near the end of the espin coding sequence, we were unable to convincingly demonstrate the presence of mutated espin proteins in the tissues of jerker homozygotes. This was the case when using two different affinity purified espin antibodies, including one directed against a peptide considerably N-terminal to the mutated portion. Judging from the intensity of the unidentified triplet at ~110-kD in the testis of jerker homozygotes revealed upon further overexposure of the blot in Figure 4B, we estimate that, at most, mutated espin proteins could be present at a level that is 1-2% of that observed in wild-type mice. Although it remains to be determined whether mutated espin proteins accumulate transiently in the cells of jerker mice, e.g., during a particular stage of development, our results raise the possibility that jerker mice are, in effect, espin null. As such, the jerker mouse promises to be of great value in our ongoing efforts to elucidate the roles of the espins in hair cell stereocilia and the parallel actin bundle-containing specializations of other cells. Since the espin deficiency is evident in multiple tissues of jerker mice, it is surprising that defects in the auditory and vestibular systems are the only aspects of the jerker phenotype that have been reported. The homozygous jerker males are not sterile, although in our colony we have noticed smaller litter sizes, as well as smaller body sizes for homozygous neonates. A detailed analysis of the effects of the jerker mutation on other espin-containing structures and cells is in progress.
The sizes and levels of espin and small espin mRNAs appeared to be same in the tissues of jerker heterozygotes and homozygotes. Moreover, no additional frameshift mutations or premature stop codons were detected upstream in the coding sequences of the jerker espin and small espin cDNAs. Therefore, we hypothesize that the frameshift mutation detected in the espin gene of jerker mice destabilizes espin proteins. Alternative hypotheses invoking second-site mutations in noncoding regions of the espin gene of jerker mice that might impair translation of the espin and small espin mRNAs seem unlikely in light of the spontaneous origin of the jerker mutation and the fact that the 5′ untranslated sequences of the espin and small espin mRNAs are derived from different exons. Although the frameshift mutation does not introduce an obvious PEST sequence, like those commonly found in rapidly degraded proteins (Rechsteiner and Rogers, 1996), it does change the amino acid sequence at the espin C terminus. On the basis of our previous deletion mutagenesis studies (Bartles et al., 1998), the mutation is expected to disrupt one of two actin-binding sites present within the 116-amino acid shared C-terminal actin-bundling module. The loss of this actin-binding site may alone be sufficient to destabilize the jerker espins. However, the frameshift mutation also substitutes novel peptide sequence with, among other properties, a high net positive charge (+14 versus +3 for the wild-type), a dramatically reduced propensity to form an α-helix and coiled-coil (see Bartles et al., 1998 for details), an increased number of serine residues (7 versus 1 for the wild-type), and a single cysteine near the C terminus. While the detailed mechanisms by which changes in the levels or structures of the espin proteins contribute to hair cell pathogenesis in the jerker mouse remain to be elucidated, the connection between the espin actin-bundling proteins and the jerker mutation provides the first insight into the molecular basis of a relatively uncharacterized hereditary deafness disorder.
Experimental Procedures
Immunolocalization and Western Blot Analysis
Antibodies to espin were prepared in rabbits by immunization with a maltose-binding protein fusion protein that included the C-terminal 379 amino acids of rat espin, espin(459-837) (Bartles et al., 1996) and affinity purified on columns of 6 x His-tagged espin(671-837) (Bartles et al., 1998) or espin(339-720) (Chen et al., 1999) coupled to cyanogen bromide-activated Sepharose 4B (Sigma Chemical Co., St. Louis, MO). Rabbit antiserum to fimbrin (R 164.2) was provided by Dr. Paul Matsudaira (Whitehead Institute for Biomedical Research, Cambridge, MA). For immunolocalization, mice (Jackson Labs, Bar Harbor, ME) and rats (Charles River Breeding Laboratories, Wilmington, MA) were fixed by transcardiac perfusion with 4% formaldehyde in 0.12 M phosphate buffer, pH 7.4. The temporal bone was removed and decalcified in 10% (w/v) EDTA in 0.9% NaCl, pH 8.0, and infiltrated with 30% sucrose for cryoprotection. Cryostat sections, 30-μm thick, were collected on gelatin-coated slides and subjected to immunoperoxidase labeling with affinity purified espin antibody or preimmune IgG using the ABC method (Vector Laboratories, Burlingame, CA). For western blotting, mouse temporal bones were pooled, crushed, homogenized and extracted with hot SDS gel sample buffer. The extracts were resolved in SDS-polyacrylamide gels under reducing conditions, transferred to nitrocellulose and labeled with affinity purified espin antibody or pre-immune IgG followed by 125I-Protein A (Bartles et al., 1996). Cochlear sensory epithelium was isolated from chicken embryos and chicks using the methods outlined by Tilney et al. (1989) and analyzed by western blotting or by confocal fluorescence microscopy after fixation with 4% formaldehyde, brief permeabilization with cold 0.5% (v/v) Triton X-100 and labeling with affinity purified espin antibody or pre-immune IgG, fluorescein-labeled goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) and rhodamine-phalloidin (Molecular Probes, Eugene, OR).
Localization of the Espin Gene, Library Screening, PCR and Sequence Analysis
An ~18-kb mouse genomic DNA fragment (Chen et al., 1999) was used to determine the location of the espin gene on 4′,6-diamidino-2-phenylindole-labeled mitotic mouse chromosomes by fluorescence in-situ hybridization using a biotinylated probe (Heng et al., 1992; Heng and Tsui, 1993). Localization to a single pair of chromosomes was observed in 94 out of the 100 collections of mitotic figures examined. The position of the espin gene was determined on the basis of a detailed comparison to the banding patterns observed in photographs of labeled mitotic figures. Lambda phage libraries of chicken (provided by Dr. J. Carl Oberholtzer, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, Philadelphia, PA) and rat (provided by Dr. Melissa Hartley, Laboratory of Molecular Neurobiology, The Salk Institute, La Jolla, CA) cochlear cDNAs were screened by Southern blotting using 32P-labeled rat espin cDNA. Positive clones were subjected to automated DNA sequence analysis using Big Dye terminator and the model 377 sequencer (Applied Biosystems, Foster City, CA). RNA was isolated from mouse testis and kidney using Trizol (Life Technologies, Grand Island, NY) and reverse transcribed using M-MLV reverse transcriptase in the presence of RNAsin (Promega, Madison, WI). The resulting cDNAs were amplified by PCR using Taq DNA polymerase (Life Technologies) and mouse espin and small espin cDNA primers, purified by agarose gel electrophoresis and subjected to automated DNA sequence analysis. The sequences of the PCR products obtained from jerker homozygotes and heterozygotes were compared base-for-base in both directions along the entire coding sequences of mouse espin (testis) and small espin (kidney). Genomic DNA was isolated from mouse testis using DNAzol (Life Technologies). Selected exons of the mouse espin gene were amplified by high fidelity PCR using Deep Vent DNA polymerase (New England Biolabs, Beverly, MA) and subjected to automated sequence analysis with or without prior subcloning into the pBluescript vector (Stratagene, La Jolla, CA).
Acknowledgements
We thank Dr. Paul Matsudaira for the fimbrin antiserum; Dr. J. Carl Oberholtzer for the chicken cochlear cDNA library; Dr. Melissa Hartley for the rat cochlear cDNA library; Dr. David Z. Z. He for initial dissections of gerbil cochlea; Anli Li, Min Wang, Jodi Irwin and Melissa Burns for assistance with library screening and DNA sequence analysis; and Dr. Rex Chisholm, Dr. Carolyn Jahn, Dr. Donna Whitlon, Dr. Bin Chen, and Benjarat Changyaleket for valuable discussions. This work was supported by NIH grants DC04314 and HD35280, NIH Independent Scientist (K02) Award HD01210 and American Cancer Society grant #RPG-96-094-04-CSM awarded to JRB; NIH grant DC02764-05 awarded to EM; and NIH grant GM52857 awarded to LGT.
References
- Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of the inner ear hair cells. Nat. Genet. 1995;11:369–375. doi: 10.1038/ng1295-369. [DOI] [PubMed] [Google Scholar]
- Anniko M, Sjöström B, Webster D. The effects of auditory deprivation on morphological maturation of the ventral cochlear nucleus. Arch. Otorhinolaryngol. 1989;246:43–47. doi: 10.1007/BF00454133. [DOI] [PubMed] [Google Scholar]
- Bartles JR. Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 2000;12:72–78. doi: 10.1016/s0955-0674(99)00059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartles JR, Wierda A, Zheng L. Identification and characterization of espin, an actin-binding protein localized to the F-actin-rich junctional plaques of Sertoli cell ectoplasmic specializations. J. Cell Sci. 1996;109:1229–1239. doi: 10.1242/jcs.109.6.1229. [DOI] [PubMed] [Google Scholar]
- Bartles JR, Zheng L, Li A, Wierda A, Chen B. Small espin: a third actin-bundling protein and potential forked protein ortholog in brush border microvilli. J. Cell Biol. 1998;143:107–119. doi: 10.1083/jcb.143.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Li A, Wang D, Wang M, Zheng L, Bartles JR. Espin contains an additional actin-binding site in its N terminus and is a major actin-bundling protein of the Sertoli cell-spermatid ectoplasmic specialization junctional plaque. Mol. Biol. Cell. 1999;10:4327–4339. doi: 10.1091/mbc.10.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Holt JR, Shepherd GM, Corey DP. Calcium imaging of single stereocilia in hair cells: localization of transduction channels at both ends of tip links. Neuron. 1995;15:1311–1321. doi: 10.1016/0896-6273(95)90010-1. [DOI] [PubMed] [Google Scholar]
- Deol MS. The anomalies of the labyrinth of the mutants varitint-waddler, shaker-2 and jerker in the mouse. J. Genetics. 1954;52:562–588. [Google Scholar]
- Eatock RA, Rusch A, Lysakowski A, Saeki M. Hair cells in mammalian utricles. Otolaryngol. Head Neck Surg. 1998;119:172–181. doi: 10.1016/S0194-5998(98)70052-X. [DOI] [PubMed] [Google Scholar]
- Ehret G. Postnatal development in the acoustic system of the house mouse in light of developing masked thresholds. J. Acoust. Soc. Am. 1977;62:143–148. doi: 10.1121/1.381496. [DOI] [PubMed] [Google Scholar]
- Flock A, Bretscher A, Weber K. Immunohistochemical localization of several cytoskeletal proteins in inner ear sensory and supporting cells. Hear. Res. 1982;7:75–89. doi: 10.1016/0378-5955(82)90082-x. [DOI] [PubMed] [Google Scholar]
- Gibson R, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SD. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- Grüneberg H, Burnett JB, Snell GD. The origin of jerker, a new gene mutation of the house mouse, and linkage studies made with it. Proc. Natl. Acad. Sci. USA. 1941;27:562–565. doi: 10.1073/pnas.27.12.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng HHQ, Tsui L-C. Modes of DAPI banding and simultaneous in situ hybridization. Chromosoma. 1993;102:325–332. doi: 10.1007/BF00661275. [DOI] [PubMed] [Google Scholar]
- Heng HHQ, Squire J, Tsui L-C. High resolution mapping of mammalian genes by in situ hybridization to free chromatin. Proc. Natl. Acad. Sci. USA. 1992;89:9509–9513. doi: 10.1073/pnas.89.20.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach JA, Falzarano PR, Simpson TH. Postnatal development of the hamster cochlea. II. Growth and differentiation of stereocilia bundles. J. Comp. Neurol. 1994;350:187–198. doi: 10.1002/cne.903500204. [DOI] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5′ noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DJ. Functional structure of the organ of Corti: a review. Hear. Res. 1986;22:117–146. doi: 10.1016/0378-5955(86)90089-4. [DOI] [PubMed] [Google Scholar]
- Lim DJ, Anniko M. Developmental morphology of the mouse inner ear. A scanning electron microscopic observation. Acta Otolaryngol. Suppl. (Stockh.) 1985;422:1–69. [PubMed] [Google Scholar]
- Lumpkin EA, Hudspeth AJ. Regulation of free Ca2+ concentration in hair-cell stereocilia. J. Neurosci. 1998;18:6300–6318. doi: 10.1523/JNEUROSCI.18-16-06300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsudaira P. Modular organization of actin crosslinking proteins. Trends Biochem. Sci. 1991;16:87–92. doi: 10.1016/0968-0004(91)90039-x. [DOI] [PubMed] [Google Scholar]
- Ohmori H. Mechanical stimulation and Fura-2 fluorescence in the hair bundle of dissociated hair cells of the chick. J. Physiol. (Lond.) 1988;399:115–137. doi: 10.1113/jphysiol.1988.sp017071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles JO, Corey DP. Mechanoelectrical transduction by hair cells. Trends Neurosci. 1992;15:254–259. doi: 10.1016/0166-2236(92)90066-h. [DOI] [PubMed] [Google Scholar]
- Probst FJ, Camper SA. The role of mouse mutants in the identification of human hereditary hearing loss genes. Hear. Res. 1999;13:1–6. doi: 10.1016/s0378-5955(98)00231-7. [DOI] [PubMed] [Google Scholar]
- Probst FJ, Fridell RA, Raphael Y, Saunders TL, Wang A, Liang Y, Morell RJ, Touchman JW, Lyons RH, Noben-Trauth K, Friedman TB, Camper SA. Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science. 1998;280:1444–1447. doi: 10.1126/science.280.5368.1444. [DOI] [PubMed] [Google Scholar]
- Puius YA, Mahoney NM, Almo SC. The modular structure of actin-regulatory proteins. Curr. Opin. Cell Biol. 1998;10:23–34. doi: 10.1016/s0955-0674(98)80083-5. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- Roberts WM, Howard J, Hudspeth AJ. Hair cells: transduction, tuning, and transmission in the inner ear. Ann. Rev. Cell Biol. 1988;4:63–92. doi: 10.1146/annurev.cb.04.110188.000431. [DOI] [PubMed] [Google Scholar]
- Sjöström B, Anniko M. Morphologically specific vestibular hair cell degeneration in the jerker mutant mouse. Eur. Arch. Otorhinolaryngol. 1990a;247:51–55. doi: 10.1007/BF00240951. [DOI] [PubMed] [Google Scholar]
- Sjöström B, Anniko M. Variability in genetically induced age-related impairment of auditory brainstem response thresholds. Acta Otolaryngol. (Stockh.) 1990b;109:353–360. doi: 10.3109/00016489009125155. [DOI] [PubMed] [Google Scholar]
- Sjöström B, Anniko M. Cochlear structure and function in a recessive type of genetically induced inner ear degeneration. ORL J. Otorhinolaryngol. Relat. Spec. 1992a;54:220–228. doi: 10.1159/000276302. [DOI] [PubMed] [Google Scholar]
- Sjöström B, Anniko M. Genetically induced inner ear degeneration. A structural and functional study. Acta Otolaryngol. Suppl. (Stockh.) 1992b;493:141–146. [PubMed] [Google Scholar]
- Sobin A, Flock A. Immunohistochemical identification and localization of actin and fimbrin in vestibular hair cells in the normal guinea pig and in a strain of the waltzing guinea pig. Acta Otolaryngol. (Stockh) 1983;96:407–412. doi: 10.3109/00016488309132726. [DOI] [PubMed] [Google Scholar]
- Steel KP. Inherited hearing defects in mice. Ann. Rev. Genet. 1995;29:675–701. doi: 10.1146/annurev.ge.29.120195.003331. [DOI] [PubMed] [Google Scholar]
- Steel KP, Bock GR. Cochlear dysfunction in the jerker mouse. Behav. Neurosic. 1983;97:381–391. doi: 10.1037//0735-7044.97.3.381. [DOI] [PubMed] [Google Scholar]
- Steel KP, Mburu P, Gibson F, Walsh J, Varela A, Brown K, Self T, Mahony M, Fleming J, Pearce A, Harvey D, Cable J, Brown SD. Unravelling the genetics of deafness. Ann. Oto. Rhinol. Laryngol. Suppl. 1997;168:59–62. [PubMed] [Google Scholar]
- Tilney MS, Tilney LG, Stephens RE, Merte C, Drenckhahn D, Cotanche DA, Bretscher A. Preliminary characterization of the stereocilia and cuticular plate of hair cells in the chick cochlea. J. Cell Biol. 1989;109:1711–1723. doi: 10.1083/jcb.109.4.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Tilney MS, DeRosier DJ. Actin filaments, stereocilia and hair cells: How cells count and measure. Annu. Rev. Cell Biol. 1992;8:257–274. doi: 10.1146/annurev.cb.08.110192.001353. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Connelly PS, Vranich KA, Shaw MK, Guild GM. Why are two different crosslinkers necessary for actin bundle formation in vivo and what does each crosslink contribute? J. Cell Biol. 1998;143:121–133. doi: 10.1083/jcb.143.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truett GE, Jr., Walker JA, Brock JW. A rat homolog of the mouse deafness mutant jerker (je) Mamm. Genome. 1996;7:356–358. doi: 10.1007/s003359900103. [DOI] [PubMed] [Google Scholar]