

Abstract
A diverse array of nitrosoalkenes derived from both acyclic and cyclic ketones, as well as aldehydes, via the Denmark protocol using α-chloro-O-TBS-oximes can be trapped efficiently in situ by a wide variety of potassium ester enolates to afford conjugate addition products in good yields.
Keywords: Conjugate additions, Oximes, Nitrosoalkenes
Vinylnitroso compounds are highly reactive, generally unstable species which have only found sporadic use in organic synthesis.1 There are presently two procedures most commonly used to generate nitrosoalkenes (Scheme 1). The most widely applied method involves base-promoted 1,4-elimination of an α-halo oxime 1 to produce the vinylnitroso species 3. These transient intermediates are known to undergo rapid conjugate additions with a variety of hetero and carbon nucleophiles in a Michael-type reaction to produce adducts 4 in good yields. When forming the vinylnitroso species via this process it is common to utilize at least two equivalents of a nucleophile, one of which acts as the base for the initial elimination step. Such a procedure, however, is inefficient when using valuable nucleophiles.
Scheme 1.
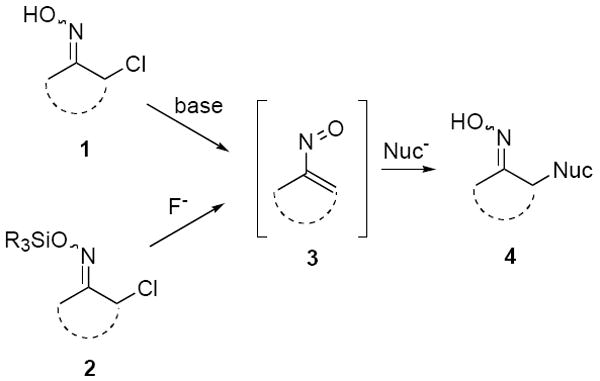
A second, less widely used method for nitrosoalkene generation developed by Denmark, et al. relies on treatment of an O-silyl-α-halo oxime 2 with a fluoride source to form 3.2 Several scattered examples have appeared describing the production of vinylnitroso compounds via this procedure in the presence of a nitrogen or oxygen heteronucleophile to afford the corresponding conjugate addition products.3 In addition, two reports exist of the generation and intermolecular trapping of carbon nucleophiles starting from silyl-α-halo oximes like 2.4 Recently we have used the Denmark procedure to effect the first examples of intramolecular conjugate additions of enolates to vinylnitroso compounds.5
In view of our interest in exploring the potential of nitrosoalkenes as enolonium ion equivalents in organic synthesis,7 we have studied effecting intermolecular conjugate additions of a number of vinylnitroso compounds formed by the Denmark strategy with a wide variety of ester enolates. It should be noted that vinylnitroso compounds derived from cyclic ketones6 as well as aldehydes4a are still relatively rare and therefore we have opted to explore reactions involving such systems to probe the scope of the methodology.
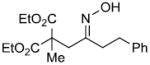
We have developed a general experimental procedure to effect this transformation as shown in Scheme 2 and have explored the scope and limitations of this methodology. Thus, an ester derivative 5 (1.2 equiv) is first converted into its potassium enolate with potassium hexamethyldisilazide in THF at -78 °C. Addition of an α-chloro-O-TBS-oxime 6 (1.0 equiv) to the enolate solution is followed by slow addition of tetrabutylammonium fluoride solution in THF (1.2 equiv). The mixture is then slowly warmed to 0 °C, and after two hours the reaction is worked up to yield alkylation product 7. A number of examples of this process along with chromatographically isolated yields of oxime products 7 are listed in Table 1. In most cases a single oxime geometric isomer is formed, assumed to have the more stable (E)-configuration, although occasionally oxime mixtures are produced (see Table 1). We were pleased to find that in general the alkylation procedure works well with a variety of α-chloro-O-TBS-oxime substrates including those derived from cyclic ketones and aldehydes. Moreover, it was gratifying to see that with the aldehyde-derived nitrosoalkene in entries n and o it is possible to produce adjacent quaternary carbon centers.
Scheme 2.
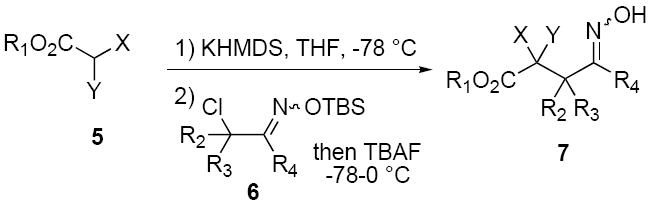
Table 1.
Intermolecular Michael additions of carbon nucleophiles to nitrosoalkenes
| entry | ester derivative | nitrosoalkene precursor | product | yield |
|---|---|---|---|---|
| a |  |
 |
95%a | |
| b | 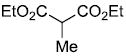 |
|
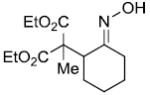 |
84% |
| c | 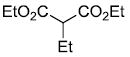 |
|
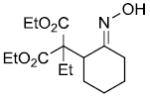 |
69% |
| d | |
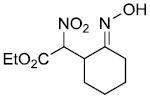 |
57%b,c | |
| e | 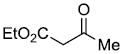 |
|
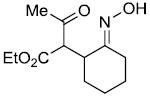 |
71%c |
| f | |
 |
95%c | |
| g | |
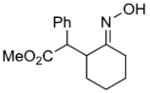 |
79%c | |
| h | 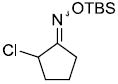 |
 |
85%d,e | |
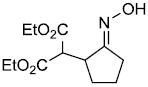
| i | |
|
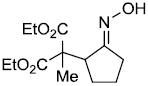 |
72% |
| j | |
|
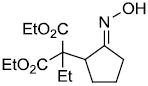 |
73% |
| k | |
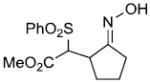 |
82%c | |
| l | |
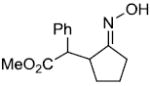 |
55%c,d | |
| m | 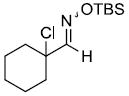 |
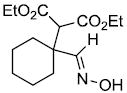 |
64%d,e | |
| n | |
|
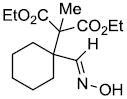 |
74% |
| o | |
|
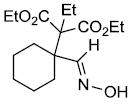 |
69%d |
| p | |
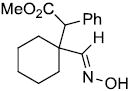 |
63% | |
| q | |
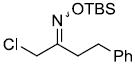 |
 |
88% |
| r | |
|
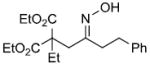 |
71% |
| s | |
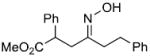 |
75% | |
| t | 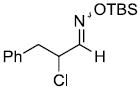 |
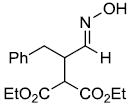 |
51%f | |
| u | |
|
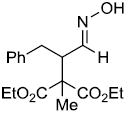 |
69% |
| v | |
|
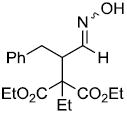 |
68%d,g |
| w | |
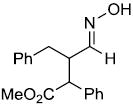 |
75%c,d | |
| x | 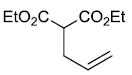 |
|
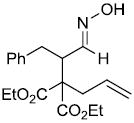 |
67%d |
Use of LiHMDS and NaHMDS gave yields of 91% and 94%, respectively.
No desired product was formed when using LiHMDS or NaHMDS.
An accurate stereochemical assignment could not be made since the products exist as a complex mixture of E/Z-isomers and/or diastereomers which were not separable by column chromatography.
2 eq of KHMDS and 2 eq of ester derivative were used.
The deprotonation step was performed at 0 °C to prevent freezing of the reaction mixture.
Use of LiHMDS and NaHMDS gave 34% and 51%, respectively.
E:Z ratio could not be determined.
One interesting observation which was made is that with some ester and α-chloro silyl oxime combinations, the nature of the base used for the enolization can affect the product yield. For example, using ethyl α-nitroacetate (entry d) KHMDS gave the desired alkylation product in 57% yield whereas with NaHMDS and LiHMDS none of the product was formed. With diethyl malonate and the aldehyde-derived substrate in entry t, KHMDS and NaHMDS gave similar product yields but LiHMDS gave a substantially reduced yield. On the other hand, using diethyl malonate and the cyclic ketone derived silyl oxime substrate in entry a, the yield of alkylation product is only slightly dependant upon the base: 95% with KHMDS, 94% with NaHMDS and 91% with LiHMDS. In a few of the examples in the table (entries h, l, m, o, v,w,x) it was found that there was a significant improvement in product yield if the amount of the ester potassium enolate is increased to 2 equiv.
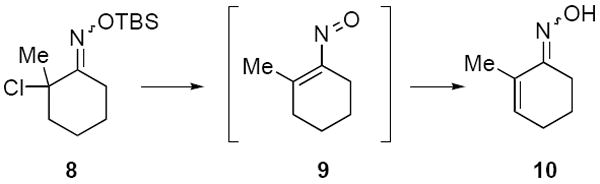
To our surprise, it was observed that enolates of 1,3-diketones and simple ketone enolates do not add to vinylnitroso compounds under these conditions. At present we cannot rationalize this failure since there are a number of examples in the literature of such Michael reactions of nitrosoalkenes generated from base elimination of simple α-halo oximes.1,8 In addition, all attempts to add ester enolates to the more highly substituted nitrosoalkene 9 formed from α-chloro-O-TBS-oxime 8 only led to the tautomerized α,β-unsaturated oxime 10 in varying yields (Scheme 3).
Scheme 3.
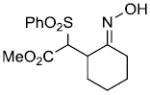
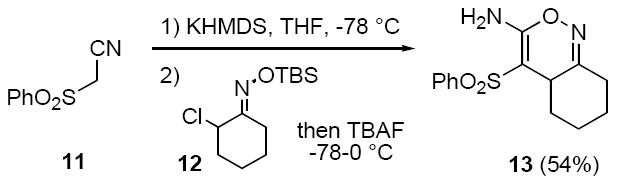
Finally, the potassium anion from α-phenylsulfonylacetonitrile (11) reacts with the nitrosoalkene from α-chloro-O-TBS-oxime 12 but produces adduct 13 where the oxime hydroxyl group has cyclized onto the initially formed cyano sulfone (Scheme 4). The moderate yield of 13 is probably due to its instability on silica gel chromatography.
Scheme 4.
In conclusion, we have described a general procedure whereby vinylnitroso compounds formed via the Denmark protocol from a diverse array of α-chloro-O-TBS-ketoximes and -aldoximes can be trapped in situ with a wide range of potassium ester enolates to give Michael-type adducts in good yields. We are currently exploring some extensions of the methodology and applications to the synthesis of complex molecules
General procedure for intermolecular Michael additions of carbon nucleophiles to in situ-generated nitrosoalkenes
To a -78 °C solution of ester derivative 5 (0.46 mmol) in THF (1 mL) was added KHMDS (917 μL, 0.5 M in PhMe, 0.46 mmol). The resulting solution was then stirred for 45 min at that temperature. The O-TBS-oxime 6 dissolved in THF (0.38 mmol in 0.3 mL of THF) was added slowly over 1 min, followed by the dropwise addition of TBAF (458 μL, 1.0 M in THF, 0.46 mmol) over 3 min. The resulting solution was immediately transferred to a 0 °C ice bath and stirred for an additional 2 h. The reaction mixture was diluted with conc. aqueous NH4Cl and EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue, which was purified by flash column chromatography on silica gel eluting with a mixture of ethyl acetate and hexanes. Isolated yields of conjugate addition products 7 are shown in Table 1.
Acknowledgments
We are grateful to the National Institutes of Health (9R56GM-087733) for financial support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For reviews of vinylnitroso compounds and lead references see: Gilchrist TL. Chem Soc Rev. 1983;11:53.Lyapkalo IM, Ioffe SL. Russ Chem Rev. 1998;67:467.
- 2.(a) Denmark SE, Dappen MS. J Org Chem. 1984;49:798. [Google Scholar]; (b) Denmark SE, Dappen MS, Sternberg JA. J Org Chem. 1984;49:4741. [Google Scholar]; (c) Denmark SE, Dappen MS, Sear NL, Jacobs RT. J Am Chem Soc. 1990;112:3466. [Google Scholar]
- 3.(a) Hassner A, Murthy K. Tetrahedron Lett. 1987;28:683. [Google Scholar]; (b) Padwa A, Chiacchio U, Dean DC, Schoffsatll AM, Hassner A, Murthy KSK. Tetrahedron Lett. 1988;29:4169. [Google Scholar]; (c) Hassner A, Maurya R, Mesko E. Tetrahedron Lett. 1988;29:5313. [Google Scholar]; (d) Hassner A, Murthy KSK, Padwa A, Bullock WH, Stull PD. J Org Chem. 1988;53:5063. [Google Scholar]; (e) Hassner A, Murthy KSK, Padwa A, Chiacchio U, Dean DC, Schoffstall AM. J Org Chem. 1989;54:5277. [Google Scholar]; (f) Hassner A, Maurya R, Friedman O, Gottlieb HE, Padwa A, Austin D. J Org Chem. 1993;58:4539. [Google Scholar]; (g) Trewartha G, Burrows JN, Barrett AGM. Tetrahedron Lett. 2005;46:3553. [Google Scholar]
- 4.(a) Hassner A, Maurya R. Tetrahedron Lett. 1989;30:5803. [Google Scholar]; (b) Kaiser A, Wiegrebe W. Monat Chem. 1998;129:937. [Google Scholar]
- 5.Korboukh I, Kumar P, Weinreb SM. J Am Chem Soc. 2007;129:10342. doi: 10.1021/ja074108r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.See for example: Ohno M, Torimitsu S, Naruse N, Okamoto M, Sakai I. Bull Chem Soc Jpn. 1966;39:1129.Trost BM, Barrett D. Tetrahedron. 1996;52:6903.Corey EJ, Melvin LS, Jr, Haslanger MF. Tetrahedron Lett. 1975:3117.
- 7.For some other examples of enolonium ion equivalents see: Fuchs PL. J Org Chem. 1976;41:2935.Wender PA, Erhardt JM, Letendre LJ. J Am Chem Soc. 1981;103 and references cited.
- 8.Oppolzer W, Battig K, Hudlicky T. Tetrahedron. 1981;37:4359. [Google Scholar]