

Abstract
Efforts to characterize the human transcriptome have largely been limited to blood, urine, and tissue analyses (i.e., normally sterile materials). We report here an extraction protocol using commercially available reagents to obtain high-yield, reverse-transcribable RNA from human stool. Quantitative reverse transcriptase polymerase chain reactions demonstrated minimal intra-specimen but considerable intra-subject variability over time of transcripts for interleukin-6 (IL-6), IL-8, epidermal growth factor (EGF), calprotectin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). This technique now expands opportunities to use the human fecal transcriptome to characterize gastrointestinal pathophysiology.
Keywords: stool, transcriptome, RNA, nucleic acid purification, newborn
Introduction
The human transcriptome, defined as the totality of messenger RNA transcripts made by a set of cells or tissues, holds considerable promise for use in studies of human physiology in health and disease. Studies of the human transcriptome have largely been confined to blood cells (1), urine (2), and soft tissue (3). Efforts to characterize transcripts in heterogeneous substances such as stool have been limited to attempts to detect specific transcripts relating to gastrointestinal malignancy in adults (4, 5), and bacterial virulence genes in human pathogens shed by patients and bovine hosts (6, 7). Attempts to study mRNA in other gastrointestinal substances, such as saliva, have also been variably successful (8, 9).
Published techniques to isolate RNA from human stool have used methods and reagents that are not commercially available (6, 10), or that do not consistently remove polymerase chain reaction (PCR) inhibitors (5). PCR inhibition, presents a particular challenge when attempting to analyze nucleic acid in stool or environmental samples. The problem appears to be that reagents used in downstream reactions, such as polymerases in PCR, are inhibited by substances carried over from the stool (11). Kang, et al. (12) recently compared several techniques to isolate RNA from murine stool, and concluded that a combination of bead-beating and silica column extraction is the best method to examine pooled samples from multiple time points.
Here we describe our efforts to purify mRNA from human stool that can then be used for reverse transcription and quantification. We extend the work of Kang, et al. (12) by reporting the degrees of intra-specimen, and intra-host variability of mRNA, presence and viability of selected host transcripts, and the utility of the silica-based method to remove potential inhibitors of downstream PCR.
Methods and Materials
Subjects and Specimen Handling
Stool was obtained weekly from three infants born after 27, 27, and 29 weeks estimated gestations. All stools were collected with a sterile, disposable wood spatulas (Cardinal Health, Dublin, OH, USA) from diapers after the stools were freshly passed, and then placed into a 125 mL sterile specimen container (Cardinal Health, Dublin, OH, USA) and stored at 4°C until transported to the laboratory within 4 hours of collection. Immediately after receipt in the laboratory, non-homogenized specimens were divided into three ~ 100 mg aliquots, which were then immediately flash frozen in separate 1.2 mL Nalgene screw top vials (Thermo Fisher Scientific, Inc., Hampton, NH, USA) by immersion in an ethanol-dry ice bath, which were then stored at −80°C until further analysis. RNA was extracted between one and four weeks after the samples were frozen. Three samples from three different time points for each of the three patients were chosen to assess intra-specimen, and intra-individual variability in mRNA content over time. For positive controls, we used either total RNA from adult human leukocytes (gift of David Hunstad, M.D.) extracted from whole human blood using the Qiagen Mini Kit, according to the manufacturer's instructions, or total RNA isolated from E. coli 055:H7, strain TB182A (13) stock cultures using a Qiagen RNeasy spin column according to the manufacturer's instructions. All human subjects research was approved by The Human Research Protection Office of Washington University School of Medicine.
RNA Extraction
A 100 mg portion of frozen stool was placed into a 1.5 mL Eppendorf tube to which 600 μL of room temperature phosphate buffered saline (pH 7.4) had been added. This tube was then vortexed until the contents were well suspended. 100 μL of this suspension was aspirated using a 1 mL blue pipette tip, and transferred to a fresh 1.5 mL Eppendorf tube. One mL of RNA Bee (Tel-Test, Inc. Friendswood, TX) was added to this suspension. Immediately thereafter, we added 200 mg of acid washed glass beads and performed “bead beating” with a Fast Prep FP120 (Thermo Savant) for 45 seconds at a speed setting of 6.5 in three different pulses, with two-minute intervals between pulsations. We then added 200 μL of chloroform to this suspension, and mixed the samples by inverting them several times before placing them on ice for 15 minutes. The samples were then centrifuged (13,000 g, 15 minutes). The clear, colorless upper (aqueous) phase was easily differentiated from the blue-green lower (organic) phase above a pellet of glass beads and stool debris. The aqueous phase (usually 500-700 μL) was aspirated and placed into a fresh 1.5 mL Eppendorf tube; the organic phase and pellets were discarded.
Ethanol (1:1 volume, 70%) was added to each sample. The resultant mixture was applied to an RNeasy spin column (Qiagen), which was then centrifuged, and washed twice with RPE buffer, per the manufacturer's instructions. Bound RNA was eluted with 50 μL of RNase-free water. DNase digestion was not performed. Our early work demonstrated no difference in RNA yield or effect on RT-PCR after DNase digestion, so we eliminated this step from our current protocol. When multiple samples were purified simultaneously, a QiaVac 24 Plus (Qiagen) was used to wash the corresponding silica membranes in parallel.
The time to simultaneously purify aqueous RNA from five to 20 samples from frozen samples was approximately two hours.
To assess effects of temperature and time from production to freezing on RNA yield, three aliquots of one stool sample were exposed to different pre-extraction intervals (0, 1, 2, 4, 12, and 24 hours) at 20°C or 4°C, prior to freezing. RNA was isolated and analyzed as with the other samples.
Assessments of RNA Yield and Quality
Specimens were first analyzed on a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.). RNA concentration was expressed as a function of the mass of the stool from which it was purified. Subsequently, a portion of each purified sample was diluted to ~5 ng/μL, and 1 μL of this diluted RNA solution was placed into an Agilent 5200 Bioanalyzer (Agilent Technologies), and quantified in duplicate. The electropherogram was examined and the RNA integrity number (RIN) was computed for each sample using the supplied software (14).
Quantitative Reverse Transcription PCR
We used quantitative reverse transcriptase PCR (qRT-PCR) to measure the ratios of various host mRNAs. Primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), interleukin-6 (IL-6), IL-8, S100A8 (a calprotectin subunit), and epidermal growth factor (EGF) were used in separate reactions, and in triplicate, for all samples. All primers were obtained from Applied Biosystems, Inc. (Product numbers: Hs99999905_m1 [GAPDH], Hs99999032_m1 [IL-6], Hs00171403_m1 [IL-8], Hs00374264_g1 [S100A8], Hs01099990_m1 [EGF], respectively). These particular mRNAs were chosen for study because we postulated that these transcripts might be present in the stools of normal or developing (GAPDH, EGF) or inflamed (IL-6, IL-8, S100A8) intestines. Each primer was designed to straddle splice sites to avoid amplifying residual DNA. Copy number standards were prepared according to the manufacturer's instructions. Ten μL reactions were prepared using 3 μL of RNA (concentrations adjusted to ~200 ng RNA per reaction), 5 μL TaqMan 2X Environmental Master Mix, 0.5 μL 20X primer mix, 0.1 μL SuperScript III Reverse Transcriptase, 0.2 μL RNase inhibitor, and 1.2 μL RNase-free H2O (all reagents from Applied Biosystems, Inc.). Amplifications were performed for 40 cycles in a 7500 Fast Real Time PCR System (Applied Biosystems, Inc.). Copy numbers for each transcript in each sample were calculated using 7500 Fast Real-Time PCR System Sequence Detection Software v. 1.3.1 (Applied Biosystems, Inc.). Positive controls consisted of purified human RNA from blood and negative controls consisted of RNase-free H2O. Positive controls amplified effectively with each primer, and negative controls did not amplify above threshold values for any primer pair. To assure PCR products of correct size were obtained, and to reiterate that RNA was indeed amplified rather than residual genomic DNA, a single aliquot from each sample was run on a 2% agarose gel. Each amplicon's size was estimated by an appropriately-sized marker lane run in parallel. All PCR products matched the expected amplification size in the documentation for each primer used.
We normalized the IL-6, IL-8, S100A8, and EGF transcripts to a constitutively expressed transcript, GAPDH. Transcript to GAPDH ratios were compared within the same sample, within the same patient, and among different patients to establish the extent of intra-specimen, inter-individual, and time variability. Additionally, the number of GAPDH transcripts per milligram of stool was computed and compared across three patients at three time points.
Each data point was obtained in five separate qRT-PCR runs. The standard deviation among these 5 measurements was determined for each point. Differences between time points, between patients, and between transcripts were computed using the two-tailed Wilcoxon test on SPSS PACW Statistics v. 17.1.
Assessment for PCR Inhibition
Several published methods of RNA extraction (4-7, 12) failed to remove PCR inhibitors from our samples (data not shown), so we paid particular attention to this problem in our comparative analysis. We homogenized stool in the lysis buffer described above, followed by silica column purification (4). We also bead beat samples, then purified them further via silica column, or guanidinium and phenol chloroform extraction, both described by Kang, et al. (12). We performed qRT-PCR as described above, but used primers for β-actin and 16S rRNA genes (from bacteria) for each purification method. To each PCR reaction, 3 μL of either total E. coli RNA or human leukocyte total RNA was combined with 2 μL of stool isolate purified by one of the above methods. After amplification, each sample was compared on 1% agarose gels for presence of an amplification band, to assess whether or not there were PCR inhibitors. This experiment was repeated with dilutions of the stool isolate of 1:2, 1:5, and 1:10 with nuclease-free H2O, but with otherwise identical reactions. RNA integrity number was not assessed for these samples.
Results
RNA Yield and Quality
The median concentration of RNA isolated by sequential bead-beating, RNA-Bee treatment, and silica column purification, was 132 μg RNA/g of stool (range 25.5- 254). The RIN for samples processed within 4 hours of collection (29 samples total) had a median value of 8.6 (range 7.9 – 8.9), indicating high quality RNA (14). For aliquots exposed to different temperatures, those held at 0, 1, 2, 4, 12, and 24 hours (two samples at each time / temperature point) and 4°C retained final RNA concentrations of 265.3, 243.0, 263.5, 212.3, 220.0, and 187.8 μg/g stool, respectively, while those held at 20°C had final concentrations of 219.5, 225.5, 200.3, 206.8, 32.5, and 18.5 μg/g stool, respectively (Fig. 1). These results affirm previous findings that storage and freezing before four hours have elapsed are desirable to preserve RNA integrity in stool(4).
Figure 1. RNA yield from variably stored specimens prior to freezing.

Final concentration of RNA in 50 μL of H2O from ~ 20 mg of stool incubated at 4°C (solid line) and 20°C (room temperature, dashed line) are provided.
Intra-specimen variability
To compare between and within samples, we computed the ratio of each transcript to the amount of GAPDH mRNA in the same sample. When comparing multiple aliquots from the same stool sample, we found the relative quantity of various transcripts to be equivalent for the three separate mRNA isolations within each sample (Fig. 2). All intra-sample values were not significantly different (all p-values > 0.05), with the exception of the first aliquot analyzed using EGF on Patient 2 (p-value = 0.045).
Figure 2. Intraspecimen uniformity of transcripts.

The relative concentrations of 3 different transcripts, in 3 different aliquots of the same sample, in 3 different patients show minimal intra-specimen variability. Error bars indicate standard deviation within the 5 RT-PCR reactions for each sample.
Inter-patient and age-dependent variability
In contrast to the minimal intra-sample variability, we noted considerable differences in the relative quantities of various transcripts between different patients and within the same patient at different time points. No pattern is apparent among the transcripts assayed in this study (Fig. 3). There were variable statistically significant differences between time points among all transcripts and patients. Intra-subject, different day measurements for all transcripts were quite discordant (Table 1). The median ratios for each transcript between subjects was also quite variable (Table 2).
Figure 3. Day – dependent variability among transcripts and patients.

The relative concentrations of 3 different transcripts, at 3 different time points, in 3 different patients show considerable inter-patient, inter-time, and inter-transcript variability. Note differences in scale used to depict the three transcript ratios.
Table 1.
Comparison of p-values computed between multiple patients, multiple time points, and multiple transcripts.
| S100A8 | IL-8 | EGF | ||
|---|---|---|---|---|
| Time Points | p-Values | |||
| Patient 1 | 1 and 2 | 0.033 | 0.018 | 0.092 |
| 2 and 3 | <0.001 | 0.006 | 0.126 | |
| 1 and 3 | 0.001 | 0.435 | 0.938 | |
| Patient 2 | 1 and 2 | 0.071 | 0.181 | 0.096 |
| 2 and 3 | 0.152 | 0.785 | 0.112 | |
| 1 and 3 | 0.074 | 0.003 | 0.061 | |
| Patient 3 | 1 and 2 | 0.082 | 0.301 | 0.009 |
| 2 and 3 | 0.806 | 0.049 | 0.008 | |
| 1 and 3 | <0.001 | 0.088 | 0.718 |
Table 2.
Comparison of p-values computed between the average value of each transcript among multiple patients.
| S100A8 | IL-8 | EGF | |
|---|---|---|---|
| Patients | p-Values | ||
| 1 and 2 | 0.151 | 0.041 | 0.146 |
| 2 and 3 | 0.769 | 0.141 | 0.789 |
| 1 and 3 | 0.133 | 0.879 | 0.205 |
Variation in the average ratio obtained across all patients between different transcripts was high. There was no significant difference in ratios seen between IL-8 and EGF (p = 0.189), but SA100A8 ratios to IL-8 and EGF were significantly different (p = 0.043 and 0.034, respectively).
The concentration of GAPDH per mass of stool ranged from 47-31064 copies detected per mg of stool (Fig. 4). There was notable intra-specimen difference in this concentration.
Figure 4. Change over time in number of GAPDH transcripts per mg of stool.
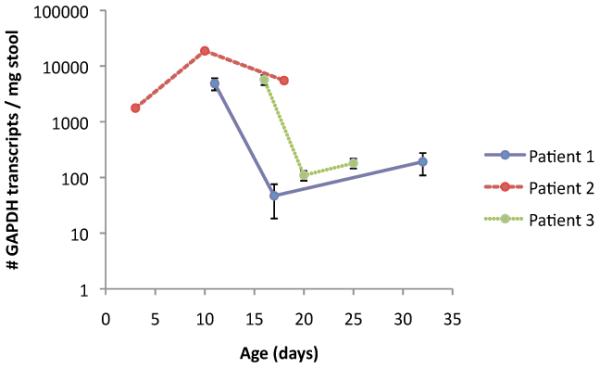
The absolute concentration of GAPDH mRNA transcripts found in stool for 3 different patients at 3 different time points. Note the logarithmic scale.
IL-6 mRNA was not detected in any of our samples.
PCR Inhibition
Agarose gel electrophoresis demonstrated no detectable β-actin or 16S RNA in the three methods we compared. Control RNA alone (Fig. 5, lanes 1 and 2) and control RNA combined with stool isolate obtained via sequential bead-beating, phenol-chloroform extraction, and silica column purification (lanes 3 and 4) were amplified with both β-actin and 16S primers. Control RNA to which a stool isolate obtained with lysis buffer homogenization and silica column purification (lanes 5 and 6), bead-beating followed by silica column extraction, (lanes 7 and 8), or phenol-chloroform extraction with ethanol precipitation (lanes 9 and 10) was not amplified with β-actin or 16S primers. Diluted samples also inhibited PCR: all but the 1:10 dilutions using bead-beating followed by silica column extraction failed to amplify either 16S or β-actin (data not shown). Lack of amplification in these samples, which had been spiked with control primable RNA, suggests the presence of PCR inhibitors.
Figure 5. Presence of PCR inhibition after different stool RNA isolation techniques.
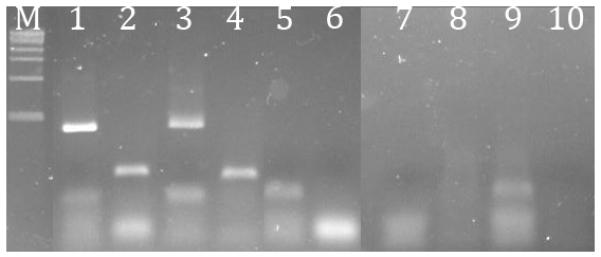
1% agarose gel electrophoresis of RT-PCR products shows inhibition of PCR by stool isolates using different purification techniques. M = DNA marker. Lanes 1 and 2 are the positive control using E. coli RNA with 16S rRNA gene primers primer or human leukocyte RNA with β-actin primer, respectively. Lanes 3 and 4 are the positive controls sequentially treated with bead-beating, RNA-Bee extraction, and silica column extraction, with 16S or β-actin primers, respectively. Lanes 5 and 6 are positive controls sequentially treated with lysis buffer homogenization and silica column extraction, with 16S or β-actin primers, respectively. Lanes 7 and 8 are positive controls sequentially treated with bead-beating and column extraction, with 16S or β-actin primers, respectively. Lanes 9 and 10 are positive controls sequentially treated with phenol:chloroform extraction and ethanol precipitation, with 16S or β-actin primers respectively.
Discussion
Human RNA can be isolated from stool quickly, easily, and efficiently using commercially available materials. Furthermore, the composition of recoverable mRNA in an individual sample is consistent, but strikingly variable between patients and time points. The quantity of GAPDH across several samples and time points varies considerably, while ratios of GAPDH to specific transcripts are more stable and reproducible. Changes in GAPDH over time and among patients may itself be a useful determination, possibly as a surrogate for epithelial or inflammatory cell turnover in the host, although changes in the relative quantity of bacterial to host cell mass are also likely to be considerable sources of variation. Thus, mRNAs in human stool appear to be a dynamic population, but also a surprisingly promising analyte that might be used to gain insight into human gastrointestinal epithelial biology.
We also demonstrated that many methods used to isolate stool RNA do not remove inhibitors of PCR. We have addressed this by adopting an approach that utilizes as pure a sample as possible and practical. Kang et al. (12) state that silica column purification can be added at the end of their preferred method if further purity is desired. We agree, but extend their findings by formulation of a three-step approach of bead-beating, followed by phenol-chloroform extraction, followed by silica column purification to offer the most consistent production of pure, reverse-transcribable and amplifiable RNA.
As demonstrated by Ahmed, et al(4), freezing or purifying samples within 4 hours of collection is essential to maintain RNA integrity. This is reflected largely by the fall in RNA yield that occurs with longer incubation times at room temperature or at 4°C, but the rate of degradation appears to be lower at the latter temperature.
We chose primers for some of the genes that would be most likely to be expressed in an inflamed intestine, so it is not surprising there is no clear pattern in children without obvious gut pathology. Variable amounts of EGF were also observed, which is also expected, because its precise role in early infancy is still poorly understood. In future investigations we will focus on broader transcript profile, and in diverse human hosts.
One potential limitation of our study is our study population. By choosing infant stools in a confined environment (our neonatal ICU), from patients who are also premature, we are unable to assess how applicable the technique will be to subjects in other settings. Moreover, these children consumed breast milk, formula, or both, but no plant matter, which might contain PCR inhibitors (15). Another limitation is that much of our analysis was limited to eukaryotic transcripts. We plan to undertake a more comprehensive analysis of the stool microbial transcriptome to help ascertain if the characteristics of fecal prokaryotic transcripts are similar to what we describe for eukaryotic transcripts. Finally, inter-subject comparisons were difficult for different transcripts, in view of the different gestational ages, and days of life on which samples were obtained.
In summary, high-quality, measurable, human mRNA can be isolated from neonatal stool. The intra-specimen variability of this analyte is low, while variabilities over time and between subjects are high. PCR inhibition can be minimized by a three-step, but simple, approach. Our refinement of RNA extraction from stool is rapid, high-yield, economical, reproducible, and potentially scalable. This protocol appears to overcome some of the technical challenges in previously published methods. The fecal transcriptome offers an unexplored opportunity to interrogate gene expression in normal development and under pathological conditions.
Acknowledgements
This research has been supported by NIH grants NIDDK T32 DK077653-17, 5UL1RR02499202, UH2 AIO83265, the Institute of Clinical and Translational Sciences (ICTS) at Washington University, and the Doris Duke Clinical Research Fellowship Program. We thank David Hunstad, M.D. for providing human leukocyte RNA samples for use as controls, Rebecca Rashid, Ph.D. for technical advice, and Elizabeth Wolf for administrative assistance.
Bibliography
- 1.Mohr S, Liew CC. The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol Med. 2007;13(10):422–32. doi: 10.1016/j.molmed.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Roos PH, Golka K, Hengstler JG. Predictive biomarkers and signatures in urinary bladder cancer. Curr Opin Mol Ther. 2008;10(3):243–50. [PubMed] [Google Scholar]
- 3.Farber CR, Lusis AJ. Integrating global gene expression analysis and genetics. Adv Genet. 2008;60:571–601. doi: 10.1016/S0065-2660(07)00420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed FE, Vos P, iJames S, Lysle DT, Allison RR, Flake G, et al. Transcriptomic molecular markers for screening human colon cancer in stool and tissue. Cancer Genomics Proteomics. 2007;4(1):1–20. [PubMed] [Google Scholar]
- 5.Ahmed FE, James SI, Lysle DT, Dobbs LJ, Jr., Johnke RM, Flake G, et al. Improved methods for extracting RNA from exfoliated human colonocytes in stool and RT-PCR analysis. Dig Dis Sci. 2004;49(1112):1889–98. doi: 10.1007/s10620-004-9589-9. [DOI] [PubMed] [Google Scholar]
- 6.Rashid RA, Tabata TA, Oatley MJ, Besser TE, Tarr PI, Moseley SL. Expression of putative virulence factors of Escherichia coli O157:H7 differs in bovine and human infections. Infect Immun. 2006;74(7):4142–8. doi: 10.1128/IAI.00299-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sjoling A, Qadri F, Nicklasson M, Begum YA, Wiklund G, Svennerholm AM. In vivo expression of the heat stable (estA) and heat labile (eltB) toxin genes of enterotoxigenic Escherichia coli (ETEC) Microbes Infect. 2006;8(1213):2797–802. doi: 10.1016/j.micinf.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Kumar SV, Hurteau GJ, Spivack SD. Validity of messenger RNA expression analyses of human saliva. Clin Cancer Res. 2006;12(17):5033–9. doi: 10.1158/1078-0432.CCR-06-0501. [DOI] [PubMed] [Google Scholar]
- 9.Setzer M, Juusola J, Ballantyne J. Recovery and stability of RNA in vaginal swabs and blood, semen, and saliva stains. J Forensic Sci. 2008;53(2):296–305. doi: 10.1111/j.1556-4029.2007.00652.x. [DOI] [PubMed] [Google Scholar]
- 10.Alexander RJ, Raicht RF. Purification of total RNA from human stool samples. Dig Dis Sci. 1998;43(12):2652–8. doi: 10.1023/a:1026699126899. [DOI] [PubMed] [Google Scholar]
- 11.Abu Al-Soud W, Radstrom P. Capacity of nine thermostable DNA polymerases To mediate DNA amplification in the presence of PCR-inhibiting samples. Appl Environ Microbiol. 1998;64(10):3748–53. doi: 10.1128/aem.64.10.3748-3753.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang S, Denman SE, Morrison M, Yu Z, McSweeney CS. An efficient RNA extraction method for estimating gut microbial diversity by polymerase chain reaction. Curr Microbiol. 2009;58(5):464–71. doi: 10.1007/s00284-008-9345-z. [DOI] [PubMed] [Google Scholar]
- 13.Bokete TN, O'Callahan CM, Clausen CR, Tang NM, Tran N, Moseley SL, et al. Shiga-like toxin-producing Escherichia coli in Seattle children: a prospective study. Gastroenterology. 1993;105(6):1724–31. doi: 10.1016/0016-5085(93)91069-t. [DOI] [PubMed] [Google Scholar]
- 14.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koonjul PK, Brandt WF, Farrant JM, Lindsey GG. Inclusion of polyvinylpyrrolidone in the polymerase chain reaction reverses the inhibitory effects of polyphenolic contamination of RNA. Nucleic Acids Res. 1999;27(3):915–6. doi: 10.1093/nar/27.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]