

Abstract
The electrocardiographic PR interval reflects atrial and atrioventricular nodal conduction, disturbances of which increase risk of atrial fibrillation (AF). To identify underlying common genetic variation, we meta-analyzed genome-wide association results for PR interval from seven community-based studies of European-ancestry individuals in the CHARGE consortium: AGES, ARIC, CHS, FHS, KORA, Rotterdam Study, and SardiNIA (N=28,517). Statistically significant loci (P<5×10-8) were tested for association with AF (N=5,741 cases). We identified nine loci associated with PR interval. At chromosome 3p22.2, we observed two independent associations in voltage gated sodium channel genes SCN10A and SCN5A, while six loci were near cardiac developmental genes CAV1/CAV2, NKX2-5 (CSX1), SOX5, WNT11, MEIS1, and TBX5/TBX3. Another signal was at ARHGAP24, a locus without known relevance to the heart. Five of the nine loci, SCN5A, SCN10A, NKX2-5, CAV1/CAV2, and SOX5, were also associated with AF (P<0.0056). Common genetic variation, particularly in ion channel and developmental genes, contributes significantly to atrial and atrioventricular conduction and to AF risk.
Search Terms: genome-wide association study, quantitative trait, PR interval, PQ interval, developmental genes, voltage gated sodium channel, atrial fibrillation
In myocardial excitation, the delay between the excitation of the atria and ventricles is determined by the sum of atrial and atrioventricular nodal conduction. This delay, measured in milliseconds, is reflected on the standard 12-lead electrocardiogram (ECG) by the PR interval or PQ interval. The PR interval has a substantial heritable component, with heritability estimates ranging between 30 and 50% (1,2,3,4).
Atrial fibrillation (AF) is the most common sustained arrhythmia and is independently associated with increased risk of stroke, heart failure, dementia, and death (5). AF prevalence increases markedly with age, to nearly 9% in those 80-89 years of age, and is estimated to triple by the year 2050. (6). Common genetic risk factors for AF (7) include variants on chromosome 4q25 near the PITX2 gene (8), in 16q22.3 near the ZFHX3 (ATBF1) gene (9), in 1q21 in the KCNN3 gene (10) and the K897T variant in the KCNH2 gene on 7q36.1 (11).
The PR interval is an intermediate phenotype for AF, as alterations in atrial action potential duration and in atrioventricular conduction influence both PR interval and AF risk (12). Longitudinal data from the Framingham Heart Study (FHS) and the Atherosclerosis Risk in Communities Study (ARIC) demonstrate that PR interval prolongation is a predictor of increased AF risk (13, 14). In addition, PR interval prolongation has been shown in FHS to be an independent predictor in a multifactorial risk score for AF predisposition (15).
We undertook a meta-analysis of GWAS to investigate the genetic determinants of the PR interval and their relationship to AF risk. Our goal was to identify genes that can provide insights into atrial disease and lead to novel opportunities for AF prevention and therapy.
We studied individuals of European descent from seven community based studies: the Age, Gene/Environment Susceptibility-Reykjavik Study (AGES) (16), ARIC (17), the Cardiovascular Health Study (CHS) (18), FHS (19), the Kooperative Gesundheitsforschung in der Region Augsburg Study (KORA) (20), the Rotterdam Study (RS) (21), and the SardiNIA study (3) (Table 1 and Online Methods). Phenotypic data including resting 12-lead electrocardiography, height, weight, systolic blood pressure, and medication use were collected using standardized protocols in all studies. Exclusion criteria and covariates are described in Supplementary Table 1.
Table 1.
Characteristics of participants in the seven community cohorts included in the meta-analysis of genome-wide association studies of PR. Exclusion criteria is given in Supplementary Table 1. “Participants after exclusion with genome-wide genotypes available” gives the N used in the analysis. Standard deviation (SD) of PR interval is given after adjustment for all covariates. fSamples are from the three generations of the Framingham Heart Study (G1=685, G2=3178, G3=3773).
| AGES | ARIC | CHS | FHS | KORA F3 | KORA S4 | Rotterdam | SardiNIA | |
|---|---|---|---|---|---|---|---|---|
| N - participants before exclusion | 3,219 | 11,478 | 2,084 | 12,174f | 1,644 | 1,100 | 5,271 | 4,305 |
| N - participants after exclusion with genome-wide genotypes available | 2,471 | 6,486 | 1,769 | 7,636f | 1,427 | 927 | 3,710 | 4,091 |
| Sex, Men, n (%) | 922 (37.3) | 2,977 (45.9) | 773 (43.7) | 3,511 (46.0) | 728 (51.0) | 447 (48.2) | 1,541 (39.8) | 1,782 (43.5) |
| Age, years, mean | 76.1 ± 5.4 | 53.9 ± 5.7 | 72.9 ± 5.4 | 39.9 ± 10.3 | 61.6 ± 9.9 | 56.2 ± 7.1 | 67.8 ± 8.5 | 42.5 ± 17.1 |
| PR interval, ms, mean | 171.4 ± 27.7 | 162.7 ± 23.7 | 167.9 ± 28.5 | 152.1 ± 21.9 | 163.3 ± 23.3 | 164.6 ± 21.2 | 165.9 ± 24.1 | 154.2 ± 26.7 |
| RR interval, ms, mean ± SD | 925.9 ± 156.5 | 915.7 ± 132.4 | 949.1 ± 155.5 | 905.7 ± 174.8 | 963.5 ± 160.6 | 930.7 ± 139.2 | 861.9 ± 137.4 | 921.2 ± 152.2 |
| BMI, kg/m2, mean ± SD | 27.1 ± 4.4 | 26.7 ± 4.7 | 26.5 ± 4.5 | 26.1 ± 4.9 | 28.0 ± 4.4 | 27.8 ± 4.5 | 26.1 ± 3.5 | 25.2 ± 4.6 |
| Height, cm, mean ± SD | 166.4 ± 9.1 | 168.8 ± 9.4 | 164.9 ± 9.6 | 169.1 ± 9.5 | 167.1 ± 9.1 | 167.4 ± 8.8 | 167.3 ± 9.3 | 160.1 ±8.9 |
| Systolic BP, mmHg, mean ± SD | 143 ± 20 | 117.2 ± 16.3 | 137 ± 21 | 120 ± 15 | 134 ± 20 | 132 ± 19 | 139 ± 22 | 125 ± 18 |
| Beta-Blockers (%) | 780 (31.6) | Excluded | 176 (10.0) | Excluded | 283 (19.8) | 99 (10.7) | Excluded | Excluded |
| Diuretics (%) | 733 (29.7) | 562 (8.7) | 434 (24.5) | ND | 273 (19.1) | 76 (8.2) | 292 (7.5) | 45 (1.1) |
| Calcium Antagonists* (%) | 131 (5.3) | Excluded | 78 (4.4) | Excluded | ND | ND | Excluded | Excluded |
| SD of PR residual after adjustment for covariates, ms | 25.9 | 23.0 | 26.9 | 20.5 | 21.3 | 20.1 | 23.0 | 25.5 |
Study participants were genotyped using a variety of genome-wide SNP arrays. To facilitate comparison of results across studies, we imputed to the 2.5 million HapMap SNPs (22). A recent review supports the validity of combining results across statistical and genotyping platforms (23). Genotyping details, SNP quality control filters, and imputation methods for each study are summarized in Supplementary Table 2.
After exclusions, 28,517 individuals were available for study. The association of each SNP with the PR interval was adjusted for age, sex, RR interval, height, body mass index (BMI), systolic blood pressure, and study site in studies with multiple recruitment sites. Studies adjusted for or excluded individuals using drugs known to alter the PR interval including beta-blockers, diuretics and non-dihydropyridine calcium antagonists.
Due to restrictions imposed by Institutional Review Boards at several of the study sites on the sharing of individual genetic data, it was not possible to perform analyses based on combined individual-level data. Therefore, we conducted inverse variance-weighted fixed-effects meta-analysis of the beta estimates from linear regression of PR interval. The coefficients, generated for each SNP, estimate the difference in PR interval per additional copy of the minor allele, adjusted for the covariates in the model. The genome-wide significance threshold was 5×10-8.
To determine if there was an association between the PR-associated loci and AF risk, we meta-analyzed results from 4 studies of AF in subjects of European descent. The first was a meta-analytic study of 896 prevalent AF cases and 15,768 referents from the CHARGE cohorts (9). The second was a meta-analytic study of 2,517 incident AF cases and 21,337 referents from the CHARGE cohorts. The third and fourth were independent case-control studies of prevalent AF: the German Competence Network on Atrial Fibrillation (AFNET, 2,145 cases and 4,073 controls) (10); and the Cleveland Clinic AF study (CCAF, 183 cases and 164 controls) (24) (Table 3 and Online Methods). We performed an inverse-variance weighted meta-analysis of the logistic-regression results from the prevalent AF studies and the proportional hazards results from the incident AF study. The Bonferroni adjusted significance threshold was P = 0.05/9 = 0.0056.
Table 3.
Characteristics of participants included in the meta-analysis of the association of the nine significant PR loci with atrial fibrillation. Overall the CHARGE prevalent AF sample included 896 cases, the CHARGE incident AF sample included 2,517 cases and the case-control sample included 2,328 cases aThe n given is the number of participants in the CHARGE cohorts and the number of cases plus controls in case control studies, bAge was defined as age at DNA collection. BMI: body mass index; NA: age at onset of prevalent AF not available for CHS and RS.
| Baseline Characteristics | CHARGE studies: Prevalent AF Analysis | CHARGE studies: Incident AF Analysis | Case Control Studies: Prevalent AF Analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AGES | CHS | FHS | RS | AGES | ARIC | CHS | FHS | RS | AFNET | CCAF | |
| Participants | |||||||||||
| na | 2,959 | 3,267 | 4,464 | 5,974 | 2,718 | 8,086 | 3,201 | 4,184 | 5,665 | 6,218 | 347 |
| Sex, men, n (%) |
1,154 (39.0) |
1,278 (39.1) |
2,004 (44.9) |
2,427 (40.6) |
1,011 (37.2) |
3,814 (47.2) |
1,241 (38.8) |
1,830 (43.7) |
2,282 (40.3) |
3,569 (57.4) |
202 (58.2) |
| Ageb, years, mean (SD) | 76.5 (5.5) | 72.3 (5.4) | 65.5 (12.7) | 69.4 (9.1) | 76.3 (5.5) | 57.0 (5.7) | 72.2 (5.3) | 64.7 (12.6) | 69.1 (9.0) | 52.7 (14.0) | 56.4 (8.2) |
| Ageb, years, min-max | 66-95 | 65-98 | 30-100 | 55-99 | 66-95 | 46-70 | 65-98 | 30-100 | 55-99 | 14-93 | 20-88 |
| Hypertension, n (%) |
2,260 (79.8) |
1,711 (52.4) |
2,263 (50.8) |
1,997 (33.4) |
2,145 (78.9) |
2192 (27.1) |
1,677 (52.4) |
2,062 (49.4) |
1,866 (32.9) |
1,902 (30.6) |
151 (43.5) |
| Cases | |||||||||||
| Atriial fibrillarion cases | 241 | 66 | 280 | 309 | 138 | 731 | 763 | 343 | 542 | 2,145 | 183 |
| Age at AF onset, mean (SD) | 76.9 (6.0) | NA | 70.6 (10.6) | NA | 80.6 (6.0) | 67.0 (6.7) | 81.2 (6.0) | 77.4 (10.5) | 77.7 (7.7) | 59.7 (11.2) | 46.0 (11.0) |
The study was performed in accordance with the Helsinki declarations and was approved by the local medical ethics and institutional review boards. All participants gave signed informed consent to use their DNA for genetic analyses.
The distribution of results from the meta-analysis of PR GWAS is summarized in Figure 1. The Q-Q plot in Figure 2 shows a clear excess of extreme p-values. Overall, nine loci showed independent association signals with P<5×10-8. We determined the genomic control factor (λ) for the linear regression analysis of PR interval to be 1.076 and report overall analysis results unadjusted for this λ value (25). We did not observe evidence of heterogeneity in effect sizes for any of the nine loci (I2-statistic, all p>0.05, Table 2).
Figure 1.
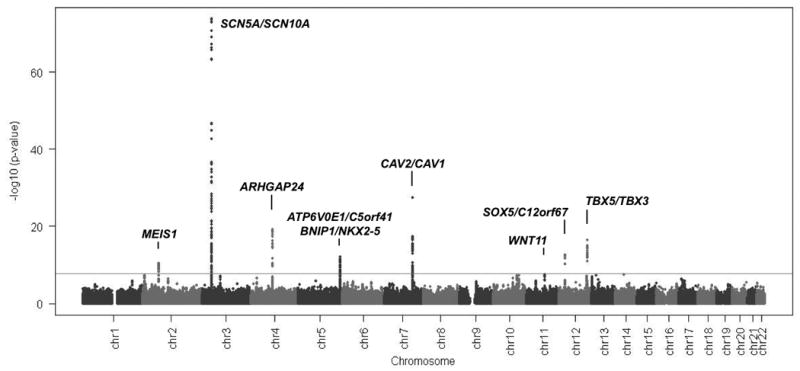
Manhattan Plot of genome-wide association analyses. Genome-wide association results were combined across all studies by inverse variance weighting. The blue line marks the threshold for genome-wide significance (P= 5×10-8). Coordinates are given in NCBI build 36.
Figure 2.
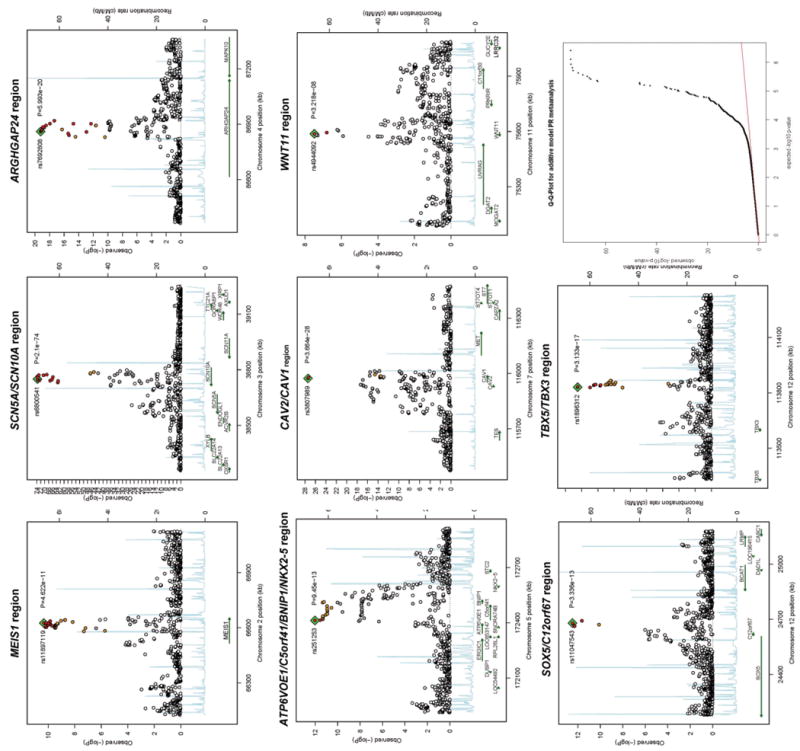
Association results at each significant locus. Associated loci are displayed in genomic order from left to right: MEIS1, SCN5A/SCN10A region, ARHGAP24, NKX2-5 region, CAV1/CAV2 region, WNT11, SOX5 region and TBX5/TBX3 region. Each panel spans ±500 kb around each SNP and has known gene transcripts annotated at the bottom. The SNPs are colored according to their degree of linkage disequilibrium (r2) with the leading variant highlighted with a blue square and displayed by name and achieved significance level in the meta-analysis. The lower right panel shows the QQ plot of the meta-analysis findings with a genomic control factor (λ) of 1.076.
Table 2.
Genome-wide significant association findings for PR interval obtained at nine independent loci. In each locus at least one marker exceeds the genome-wide significant threshold of P<5 × 10-8. Betas estimate the difference in PR interval per one additional copy of the minor allele, adjusted for the covariates in the model. Column “Method” indicates whether a SNP was directly genotyped on one of the array platforms (G) or whether its genotype was imputed in all of the samples (I). The RSQR and the MAF are averages weighted by study size. RSQR (sometimes also termed OEvar) denotes the average of the observed by expected variance ratio of any SNP which indicates deviation from Hardy-Weinberg equilibrium and quality of imputation. All these SNPs met QC criteria in each study as outlined in Supplementary Tables 2 and 3. Effect size (beta) is reported in milliseconds (ms) per one copy of the minor allele. We observed no heterogeneity in effect size estimates between studies and report the I2-statistic results as the ratio of between-study to overall variance in betas.
| Locus | SNP | chr | Position (build 36) | Gene related position | minor/major allele | Meth od | RSQR (OEvar) | Freq coded (minor) allele | beta (ms) | SE (ms) | Association -P value | Heterogenei ty – I2 statistic | Heterogenei ty – P value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MEIS1 | rs11897119 | 2 | 66,625,504 | Intron 8 | C/T | G | 1.008 | 0.389 | 1.3624 | 0.207 | 4.62 × 10-11 | 0 | 0.803 |
| SCN5A | rs11708996 | 3 | 38,608,927 | Intron 14 | C/G | I | 0.927 | 0.149 | 3.0403 | 0.2886 | 6.00 × 10-26 | 0.045 | 0.396 |
| SCN10A | rs6800541 | 3 | 38,749,836 | Intron 14 | C/T | G | 0.995 | 0.404 | 3.7687 | 0.2065 | 2.10 × 10-74 | 0.490 | 0.056 |
| ARHGAP24 | rs7692808 | 4 | 86,860,173 | Intron 2 | A/G | I | 0.984 | 0.306 | -2.0146 | 0.2203 | 5.99 × 10-20 | 0 | 0.711 |
| NKX2-5 | rs251253 | 5 | 172,412,942 | 3kb 5′ of C5orf41 | C/T | G | 0.971 | 0.399 | -1.4924 | 0.2091 | 9.45 × 10-13 | 0 | 0.926 |
| CAV1/CAV2 | rs3807989 | 7 | 115,973,477 | Intron 2 of CAV1 | A/G | G | 0.970 | 0.395 | 2.2959 | 0.2086 | 3.66 × 10-28 | 0 | 0.626 |
| WNT11 | rs4944092 | 11 | 75,587,267 | Intron 1 | G/A | G | 1.006 | 0.321 | -1.1916 | 0.2155 | 3.22 × 10-08 | 0 | 0.549 |
| SOX5 | rs11047543 | 12 | 24,679,606 | 51 kb 5′ of C12orf67 | A/G | I | 0.983 | 0.147 | -2.0907 | 0.2872 | 3.34 × 10-13 | 0 | 0.912 |
| TBX5/TBX3 | rs1896312 | 12 | 113,830,807 | 226 kb 5′ of TBX3 | C/T | I | 0.936 | 0.279 | 1.9505 | 0.2311 | 3.13 × 10-17 | 0 | 0.768 |
The strongest genome-wide association signal for PR interval was in chromosomal region 3p22.2. In this region we detected two association signals, one covering SCN10A (rs6800541, P = 2.1×10-74) and the other SCN5A (rs11708996, P = 6.0×10-26) (Figure 2 and Table 2). These variants are in low LD (r2 = 0.031). In a meta-analysis of linear regression results from models including both SNPs, these SNPs remained independently associated with PR interval (rs6800541, P= 9.7×10-82; rs11708996, P = 1.1×10-33), suggesting they represent independent association signals.
SCN10A encodes the voltage gated sodium channel Nav1.8, essential for cold perception in afferent nociceptive fibers of sensory dorsal root ganglia (26). Nav1.8 is expressed in the peripheral sensory nervous system but has not been identified in the heart (27). In SCN10A, two common nonsynonymous SNPs are in high to moderate linkage disequilibrium with the sentinel SNP: rs6795970 (V1073A, r2=0.933) and rs12632942 (L1092P, r2=0.220) making both SNPs good candidates to mechanistically explain the strongest PR interval association identified in the human genome.
The neighboring SCN5A gene encodes Nav1.5, the major cardiac sodium channel, with mutations resulting in Brugada Syndrome, long-QT Syndrome, dilated cardiomyopathy, cardiac conduction disease, idiopathic ventricular fibrillation and AF (28). SCN5A sentinel SNP rs11708996 is in weak LD with rs1805124 (H558R, r2=0.034) as well as with two non-coding variants, rs12053903 (r2=0.030) and rs11129795 (r2=0.058), recently reported to be associated with QT interval (29, 30). This suggests that the PR interval and QT interval modifying effects are distinct.
Six PR interval associations were identified in or near genes involved in human cardiac development (Figure 2 and Table 2). NKX2-5 (rs251253 P= 9.5×10-13) is the homolog of the Drosophila tinman gene and encodes the cardiac specific homeobox transcription factor Nkx2.5 (Csx). Mutations can cause atrium septum defect (ASD) with conduction defects (OMIM #108900), tetralogy of Fallot (OMIM #187500), and high degree AV block (31). In the NKX2-5 gene region the association extends over 200Kb and includes three other genes BNIP1, C5orf41, and ATP6V0E1. The signal at the TBX5/TBX3 locus is 230kb downstream of the paralog TBX5 and TBX3 genes (rs1896312, P= 3.1×10-17). Both encode T-box containing transcription factors important for cardiac conduction system formation in the developing heart (32). TBX5 is required for the patterning and maturation of the murine atrioventricular and bundle branch conduction system (33). Deletion of TBX5 results in longer PR intervals in mice (34). Mutations are seen in Holt-Oram syndrome (OMIM #142900) with atrial and ventricular septal defects, conduction disease, and occasionally AF (35). TBX3 controls formation of the sinus node and imposes pacemaker function on atrial cells (36). Mutations cause ulnar-mammary syndrome (OMIM #181450) with limb, mammary, tooth, genital and cardiac abnormalities (37).
The CAV1 and CAV2 genes (rs3807989, P = 3.7×10-28) encode caveolins necessary for the development of caveolae involved in signal transduction (38). CAV1 is expressed in atrial myocytes. Mice deficient in Cav1 develop dilated cardiomyopathy and pulmonary hypertension (39). SOX5 (rs11047543, P = 3.3×10-13) and the nearby C12orf67 encode transcription factors. SOX5 knockout mice die with heart failure marked by hepatic congestion and peripheral edema (40). MEIS1 (rs11897119, P = 4.6×10-11) encodes a homeobox transcription factor implicated in cardiac, hematopoietic and neural development. MEIS1 deficient mice have malformed cardiac outflow tracts with overriding aorta and ventricular septal defect (41).
WNT11 (rs4944092, P = 3.2×10-8) encodes a signaling protein inducing cardiogenesis in Xenopus and in mice by noncanonical WNT signaling (42). The nearest gene to a signal on chromosome 4 (rs7692808, P = 6.0×10-20) is ARHGAP24, which encodes a Rho-GTPase-activating protein and key angiogenic regulator involved in cell polarity, cell morphology, and cytoskeletal organisation (43), but without known relevance to the heart.
Of the nine identified PR loci, five were associated with AF risk (p<0.0056). These were at SCN10A (rs6800541, P=1.5 × 10-4) and SCN5A (rs11708996, P=7.0 × 10-4), as well as at three regions harboring developmental genes, NKX2-5 (P=2.3 × 10-3), CAV1/CAV2 (P=2.2 × 10-5), and SOX5 (P=2.1 × 10-4). In all instances the minor alleles were associated with a decrease in AF risk, irrespective of the direction of their association with PR interval (Table 4). Protective ratios against AF were between 0.93 and 0.88 for the minor alleles.
Table 4.
Meta-analytic results for the association of the nine significant PR loci with atrial fibrillation. Meta-analyzed were results from a meta-analysis of prevalent AF conducted in the CHARGE cohorts, results from a meta-analysis of incident AF conducted in the CHARGE cohorts, results from the AFNET case-control study and results from the CCAF case-control study. The Bonferroni adjusted table-wide significance threshold is P= 0.05/9= 5.6E-03, the p(adjusted) column reports the significance thresholds after adjustment for 9 tests. For all five SNPs we identified as associated with AF the minor allele decreased the risk of AF irrespective of the direction of its effect on PR interval.
| Nearest gene | SNP | Chr | pos build36 | PR prolonging allele | Frequency – PR prolonging allele | OR for AF – PR prolonging Allele | OR for AF – 95% CI lower bound | OR for AF – 95% CI upper bound | P (unadjusted) | P (adjusted) | Effect of PR prolonging allele towards AF risk | Effect of minor allele towards AF risk |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MEIS1 | rs11897119 | 2 | 66,625,504 | C | 0.39 | 1.01 | 0.97 | 1.06 | 0.65 | - | ns | ns |
| SCN5A | rs11708996 | 3 | 38,608,927 | C | 0.15 | 0.90 | 0.84 | 0.96 | 7.0E-04 | 6.30E-03 | Decreased | Decreased |
| SCN10A | rs6800541 | 3 | 38,749,836 | C | 0.40 | 0.92 | 0.88 | 0.96 | 1.5E-04 | 1.35E-03 | Decreased | Decreased |
| ARHGAP24 | rs7692808 | 4 | 86,860,173 | G* | 0.69 | 1.01 | 0.97 | 1.06 | 0.56 | - | ns | ns |
| NKX2-5 | rs251253 | 5 | 172,412,942 | T* | 0.61 | 1.07 | 1.03 | 1.12 | 2.3E-03 | 2.07E-02 | Increased | Decreased |
| CAV1/CAV2 | rs3807989 | 7 | 115,973,477 | A | 0.40 | 0.91 | 0.87 | 0.95 | 2.2E-05 | 1.98E-04 | Decreased | Decreased |
| WNT11 | rs4944092 | 11 | 75,587,267 | A* | 0.67 | 0.94 | 0.90 | 0.99 | 0.01 | - | ns | ns |
| SOX5 | rs11047543 | 12 | 24,679,606 | G* | 0.85 | 1.13 | 1.06 | 1.20 | 2.1E-04 | 1.89E-03 | Increased | Decreased |
| TBX5/TBX3 | rs1896312 | 12 | 113,830,807 | C | 0.30 | 0.99 | 0.95 | 1.04 | 0.72 | - | ns. | ns. |
The observation that SNPs associated with both PR interval and AF risk did not exhibit consistent directions of effect may initially seem counterintuitive. However, PR interval is an amalgamated measure of atrial and atrioventricular nodal conduction, which independently affect AF risk. PR intervals at both high and low extremes may be associated with an increase in AF risk. Indeed, existing data from humans and animal models suggest that the effects of genetic variants on atrial repolarization and action potential duration, and their relationship with atrial arrhythmias, are complex (11). In analogy to the QT interval duration, where both long and short QT intervals are associated with increased ventricular tachycardia risk, assuming a linear association model for AF with PR interval and its underlying genetic variants may not capture the complexity of these relations.
Our study was subject to a number of potential limitations. False positive associations from multiple testing is a limitation of any GWAS, so we used a well-accepted genome-wide association significance threshold equivalent to a Bonferroni correction for 1 million independent tests to reduce false positive findings (44). Population stratification is also a concern, so we only included study subjects of European descent. The low genomic control inflation factor suggested there was no strong influence of population stratification on our results.
Our study also did not examine patterns of haplotype association. Thus complex haplotype associations may not have been captured. However, genome-wide meta-analysis of haplotypes is currently not feasible, and, in common with other GWAS, our use of imputation to the HapMap leverages available linkage disequilibrium information.
The identification of SCN10A was unexpected, as Nav1.8 was previously not thought to play a role in cardiac electrophysiology. In addition, SCN10A was the only locus where two common nonsynonymous variants were in high LD with a sentinel SNP and thus are likely causal candidates. The second key finding was that the majority of association signals we identified were in cardiac developmental genes including two, NKX2-5 and TBX5, in which mutations are known to cause well-defined cardiac malformations involving the atrial septum and the atrioventricular junction. The biological mechanisms by which the identified variants influence PR interval and AF remain speculative, and detailed functional investigation will be required to determine the potential contribution of each genomic region.
Supplementary Material
Acknowledgments
We gratefully acknowledge all of the participants in the studies.
AGES: NIH N01-AG-12100, NIA and NIH Intramural Research Programs, Hjartavernd (Icelandic Heart Association), Althingi (the Icelandic Parliament), NHLBI, NEI, and NIDCD.
ARIC: NHLBI N01-HC-55015, N01-HC-55016, N01-HC-55018 through N01-HC-55022, R01-HL-087641, R01-HL-59367 and R01-HL-086694, and R01-HL-054512, NHGRI U01-HG004402, NIH HHSN268200625226C, and the Donald W. Reynolds Cardiovascular Clinical Research Center. Infrastructure was supported by NIH UL1-RR025005.
CCAF: NHLBI R01-HL090620 and P50-HL077107, Intramural funding from the Heart and Vascular Institute, Department of Cardiovascular Medicine, Cleveland Clinic.
CHS: NHLBI N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01-HC-15103, N01-HC-55222, N01-HC-75150, N01-HC-45133, U01-HL-080295 R01-HL-087652, R01-HL-088456, NCRR M01-RR-00425, NIDDK DK063491, NINDS, the Cedars-Sinai Board of Governors Chair in Medical Genetics.
FHS: NIH N01-HC-25195, HL-076784, AG-028321, N01-HC25195, HL-080025 and 6R01-NS-17950, NHLBI N01-HC-25195, BU School of Medicine and Boston Medical Center (LINGA-II), the Robert Dawson Evans Endowment, the Doris Duke Charitable Foundation, the SHARe project, DFG Fellowship SCHN 1149/1-1, Affymetrix contract for genotyping services (N02-HL-6-4278) and Pfizer.
KORA/AFNET: We thank Benno Pütz, Michael Putz and Guido Fischer for their contributions to genotyping and imputation. BMBF NGFN 01-GS-0499, 01-GR-0103, 01-GR-0803, AFNET 01-GI-0204 01-GS-0838, the Leducq Foundation 07-CVD 03, LMU FöFoLe 557/569, the LMU Excellence Initiative, MC Health as part of LMUinnovativ, the HMGU and the State of Bavaria.
Rotterdam Study: We thank Pascal Arp, Mila Jhamai, Dr Michael Moorhouse, Marijn Verkerk and Sander Bervoets for their help in creating the database, Karol Estrada for his help with the analyses and Maxim Struchalin for contributions to genotype imputation. NWO 175.010.2005.011, 911.03.012 and 050-060-810, RIDE, NGI, ZonMw, Netherlands Hartstichting, Ministry of Education Culture and Science, Ministry of Health Welfare and Sports; the European Commission; Erasmus Medical Center, Erasmus University Rotterdam and the Municipality of Rotterdam
SardiNIA: We thank Angelo Scuteri and Marco Orrù for longstanding continuous support of the project and for phenotype characterization. NIA NO1-AG-1-2109, 263-MA-410953, NIH and NIA Intramural Research Programs, NHGRI and NHLBI.
Role of the Sponsor: None of the funding organizations had any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; or preparation, review, or approval of the manuscript.
Extended acknowledgements section: A more detailed acknowledgement section can be found in the supplementary material.
Footnotes
Conflict of Interest Statement: Aravinda Chakravarti is a paid member of the Scientific Advisory Board of Affymetrix, a role that is managed by the Committee on Conflict of Interest of the Johns Hopkins University School of Medicine.
Methods: Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Note: Supplementary information is available on the Nature Genetics website.
URL: Information about the CHARGE consortium is available at: http://depts.washington.edu/chargeco/wiki/Main_Page
Author Contributions: Study concept and design: AP, DEA, AVS, NS, JIR, AH, BHCS, CMVD, GE, AC, KLL, VG, PTE, SS, SK, JCMW, EJB, SRH; Acquisition of data: AP, CVN, DEA, KVT, MFS, JIR, FR, JAK, BHCS, AGU, BMB, WS, CG, CNC, TJW, MKC, JDS, DRVW, SSN, GBE, AC, EZS, SP, JCMW, AA, SRH; Analysis and interpretations of data: AP, CVN, KDM, DEA, AVS, MM, NS, GCV, ML, JIR, CNC, TJW, RSV, TA, SSN, GBE, AC, EZS, KLL, SP, VG, EJB, SRH; Drafting of the manuscript: AP, DEA, NS, PTE, SK, EJB, SRH; Critical revision of the manuscript: CVN, KDM, MGL, AVS, KVT, MM, MFS, GCV, WHLK, AK, JC, JCB, BMP, KR, JIR, FR, AH, JAK, BHCS, AGU, CMVD, BMB, CG, SAL, CNC, TJW, JWM, RBS, MKC, JB, JDS, DRVW, RSV, GE, LJL, TBH, EL, DS, MU, GRA, BMM, EB, EZS, KLL, HEW, TM, DL, VG, SS, JCMW, AA; Statistical analysis: AP, CVN, DEA, MGL, AVS, MM, GCV, ML, WHLK, JCB, KR, TA, KLL; Obtained funding: AP, MFS, BMP, JIR, FR, AH, AGU, MKC, JDS, RSV, GE, DS, MU, GRA, EB, AC, HEW, TM, DL, VG, JCMW, SRH; Study supervision: JIR, FR, AH, BHCS, AGU, CMVD, GE, AC, VG, JCMW, SRH; The following authors had full data access and take responsibility for analysis: AP, CVN, MM, JIR, AC, KLL, SRH.
References
- 1.Havlik RJ, Garrison RJ, Fabsitz R, Feinleib M. Variability of heart rate, P-R, QRS and Q-T durations in twins. J Electrocardiol. 1980;13:45–48. doi: 10.1016/s0022-0736(80)80008-2. [DOI] [PubMed] [Google Scholar]
- 2.Hanson B, Tuna N, Bouchard T, Heston L, Eckert E, Lykken D, Segal N, Rich S. Genetic factors in the electrocardiogram and heart rate of twins reared apart and together. Am J Cardiol. 1989;63:606–609. doi: 10.1016/0002-9149(89)90907-7. [DOI] [PubMed] [Google Scholar]
- 3.Pilia G, Chen WM, Scuteri A, Orrú M, Albai G, Dei M, Lai S, Usala G, Lai M, Loi P, Mameli C, Vacca L, Deiana M, Olla N, Masala M, Cao A, Najjar SS, Terracciano A, Nedorezov T, Sharov A, Zonderman AB, Abecasis GR, Costa P, Lakatta E, Schlessinger D. Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet. 2006;2:e132. doi: 10.1371/journal.pgen.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton-Cheh C, Guo CY, Wang TJ, et al. Genome Wide Association Study of Electrocardiographic and Heart Rate Variability Traits: the Framingham Heart Study. BMC Med Genet. 2007;8(1):S7. doi: 10.1186/1471-2350-8-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benjamin EJ, Chen PS, Bild DE, Mascette AM, Albert CM, Alonso A, Calkins H, Connolly SJ, Curtis AB, Darbar D, Ellinor PT, Go AS, Goldschlager NF, Heckbert SR, Jalife J, Kerr CR, Levy D, Lloyd-Jones DM, Massie BM, Nattel S, Olgin JE, Packer DL, Po SS, Tsang TS, Van Wagoner DR, Waldo AL, Wyse DG. Prevention of atrial fibrillation: report from a national heart, lung, and blood institute workshop. Circulation. 2009;119:606–618. doi: 10.1161/CIRCULATIONAHA.108.825380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heeringa J, van der Kuip DA, Hofman A, Kors JA, van Herpen G, Stricker BH, Stijnen T, Lip GY, Witteman JC. Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur Heart J. 2006;27:949–953. doi: 10.1093/eurheartj/ehi825. [DOI] [PubMed] [Google Scholar]
- 7.Fox CS, Parise H, D'Agostino RB, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 8.Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, Jonasdottir A, Baker A, Thorleifsson G, Kristjansson K, Palsson A, Blondal T, Sulem P, Backman VM, Hardarson GA, Palsdottir E, Helgason A, Sigurjonsdottir R, Sverrisson JT, Kostulas K, Ng MC, Baum L, So WY, Wong KS, Chan JC, Furie KL, Greenberg SM, Sale M, Kelly P, MacRae CA, Smith EE, Rosand J, Hillert J, Ma RC, Ellinor PT, Thorgeirsson G, Gulcher JR, Kong A, Thorsteinsdottir U, Stefansson K. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 9.Benjamin EJ, Rice KM, Arking DE, Pfeufer A, van Noord C, Smith AV, Schnabel RB, Bis JC, Boerwinkle E, Sinner MF, Dehghan A, Lubitz SA, D'Agostino RB, Sr, Lumley T, Ehret GB, Heeringa J, Aspelund T, Newton-Cheh C, Larson MG, Marciante KD, Soliman EZ, Rivadeneira F, Wang TJ, Eiríksdottir G, Levy D, Psaty BM, Li M, Chamberlain AM, Hofman A, Vasan RS, Harris TB, Rotter JI, Kao WH, Agarwal SK, Stricker BH, Wang K, Launer LJ, Smith NL, Chakravarti A, Uitterlinden AG, Wolf PA, Sotoodehnia N, Köttgen A, van Duijn CM, Meitinger T, Mueller M, Perz S, Steinbeck G, Wichmann HE, Lunetta KL, Heckbert SR, Gudnason V, Alonso A, Kääb S, Ellinor PT, Witteman JC. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–881. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellinor PT, et al. Common Variants in KCNN3 are Associated with Lone Atrial Fibrillation. doi: 10.1038/ng.537. (unpublished observations) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinner MF, Pfeufer A, Akyol M, Beckmann BM, Hinterseer M, Wacker A, Perz S, Sauter W, Illig T, Näbauer M, Schmitt C, Wichmann HE, Schömig A, Steinbeck G, Meitinger T, Kääb S. The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: results from a systematic candidate gene-based analysis of KCNH2 (HERG) Eur Heart J. 2008;29:907–914. doi: 10.1093/eurheartj/ehm619. [DOI] [PubMed] [Google Scholar]
- 12.Olsson SB, Cotoi S, Varnauskas E. Monophasic action potential and sinus rhythm stability after conversion of atrial fibrillation. Acta medica Scand. 1971;190:381–387. doi: 10.1111/j.0954-6820.1971.tb07446.x. [DOI] [PubMed] [Google Scholar]
- 13.Soliman EZ, Prineas RJ, Case LD, Zhang ZM, Goff DC., Jr Ethnic distribution of ECG predictors of atrial fibrillation and its impact on understanding the ethnic distribution of ischemic stroke in the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2009;40:1204–1211. doi: 10.1161/STROKEAHA.108.534735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng S, Keyes MJ, Larson MG, McCabe EL, Newton-Cheh C, Levy D, Benjamin EJ, Vasan RS, Wang TJ. Long-term outcomes in individuals with prolonged PR interval or first-degree atrioventricular block. JAMA. 2009;301:2571–2577. doi: 10.1001/jama.2009.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schnabel RB, et al. Development of a Risk Score for Atrial Fibrillation (The Framingham Heart Study): a community-based cohort study. The Lancet. 2009;373:739–745. doi: 10.1016/S0140-6736(09)60443-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris TB, Launer LJ, Eiriksdottir G, Kjartansson O, Jonsson PV, Sigurdsson G, Thorgeirsson G, Aspelund T, Garcia ME, Cotch MF, Hoffman HJ, Gudnason V. Age, Gene/Environment Susceptibility-Reykjavik Study: multidisciplinary applied phenomics. Am J Epidemiol. 2007;165:1076–1087. doi: 10.1093/aje/kwk115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 18.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 19.Splansky GL, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol. 2007;165:1328–1335. doi: 10.1093/aje/kwm021. [DOI] [PubMed] [Google Scholar]
- 20.Wichmann HE, Gieger C, Illig T. KORA-gen–resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;671(1):S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 21.Hofman A, Breteler MM, van Duijn CM, Krestin GP, Pols HA, Stricker BH, Tiemeier H, Uitterlinden AG, Vingerling JR, Witteman JC. The Rotterdam Study: objectives and design update. Eur J Epidemiol. 2007;22:819–829. doi: 10.1007/s10654-007-9199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nothnagel M, Ellinghaus D, Schreiber S, Krawczak M, Franke A. A comprehensive evaluation of SNP genotype imputation. Hum Genet. 2009;125:163–171. doi: 10.1007/s00439-008-0606-5. [DOI] [PubMed] [Google Scholar]
- 24.Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, Van Wagoner DR. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–2891. doi: 10.1161/hc4901.101760. [DOI] [PubMed] [Google Scholar]
- 25.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 26.Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel NaV1.8 is essential for pain at low temperatures. Nature. 2007;447:855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]
- 27.Rabert D, Koch BD, Ilnicka M, Obernolte RA, Naylor SL, Herman RC, Eglen RM, Hunter JC, Sangameswaran L. A tetrodotoxin-resistant voltage-gated sodium channel from human dorsal root ganglia, hPN3/SCN10A. Pain. 1998;78:107–114. doi: 10.1016/S0304-3959(98)00120-1. [DOI] [PubMed] [Google Scholar]
- 28.Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JC, Hofman A, Heckbert SR, O'Donnell CJ, Uitterlinden AG, Psaty BM, Lumley T, Larson MG, Stricker BH. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeufer A, Sanna S, Arking DE, Müller M, Gateva V, Fuchsberger C, Ehret GB, Orrú M, Pattaro C, Köttgen A, Perz S, Usala G, Barbalic M, Li M, Pütz B, Scuteri A, Prineas RJ, Sinner MF, Gieger C, Najjar SS, Kao WH, Mühleisen TW, Dei M, Happle C, Möhlenkamp S, Crisponi L, Erbel R, Jöckel KH, Naitza S, Steinbeck G, Marroni F, Hicks AA, Lakatta E, Müller-Myhsok B, Pramstaller PP, Wichmann HE, Schlessinger D, Boerwinkle E, Meitinger T, Uda M, Coresh J, Kääb S, Abecasis GR, Chakravarti A. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jay PY, Harris BS, Buerger A, Rozhitskaya O, Maguire CT, Barbosky LA, McCusty E, Berul CI, O'brien TX, Gourdie RG, Izumo S. Function follows form: cardiac conduction system defects in Nkx2-5 mutation. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:966–972. doi: 10.1002/ar.a.20102. [DOI] [PubMed] [Google Scholar]
- 32.Mesbah K, Harrelson Z, Théveniau-Ruissy M, Papaioannou VE, Kelly RG. Tbx3 Is Required for Outflow Tract Development. Circ Res. 2008;103:743–750. doi: 10.1161/CIRCRESAHA.108.172858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moskowitz IP, Kim JB, Moore ML, Wolf CM, Peterson MA, Shendure J, Nobrega MA, Yokota Y, Berul C, Izumo S, Seidman JG, Seidman CE. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell. 2007;129:1365–1376. doi: 10.1016/j.cell.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 34.Mori AD, Zhu Y, Vahora I, Nieman B, Koshiba-Takeuchi K, Davidson L, Pizard A, Seidman JG, Seidman CE, Chen XJ, Henkelman RM, Bruneau BG. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev Biol. 2006;297:566–586. doi: 10.1016/j.ydbio.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 35.Postma AV, van de Meerakker JB, Mathijssen IB, Barnett P, Christoffels VM, Ilgun A, Lam J, Wilde AA, Lekanne Deprez RH, Moorman AF. A gain-of-function TBX5 mutation is associated with atypical Holt-Oram syndrome and paroxysmal atrial fibrillation. Circ Res. 2008;102:1433–1442. doi: 10.1161/CIRCRESAHA.107.168294. [DOI] [PubMed] [Google Scholar]
- 36.Hoogaars WM, Engel A, Brons JF, Verkerk AO, de Lange FJ, Wong LY, Bakker ML, Clout DE, Wakker V, Barnett P, Ravesloot JH, Moorman AF, Verheijck EE, Christoffels VM. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007;21:1098–1112. doi: 10.1101/gad.416007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schinzel A. Ulnar-mammary syndrome. J Med Genet. 1987;24:778–781. doi: 10.1136/jmg.24.12.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004;94:1408–1417. doi: 10.1161/01.RES.0000129178.56294.17. [DOI] [PubMed] [Google Scholar]
- 39.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J, Jr, Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smits P, Li P, Mandel J, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B, Lefebvre V. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev Cell. 2001;1:277–290. doi: 10.1016/s1534-5807(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 41.Stankunas K, Shang C, Twu KY, Kao SC, Jenkins NA, Copeland NG, Sanyal M, Selleri L, Cleary ML, Chang CP. Pbx/Meis Deficiencies Demonstrate Multigenetic Origins of Congenital Heart Disease. Circ Res. 2008;103:702–709. doi: 10.1161/CIRCRESAHA.108.175489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandur P, Läsche M, Eisenberg LM, Kühl M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature. 2002;418:636–641. doi: 10.1038/nature00921. [DOI] [PubMed] [Google Scholar]
- 43.Su ZJ, Hahn CN, Goodall GJ, Reck NM, Leske AF, Davy A, Kremmidiotis G, Vadas MA, Gamble JR. A vascular cell-restricted RhoGAP, p73RhoGAP, is a key regulator of angiogenesis. Proc Natl Acad Sci U S A. 2004;101:12212–12217. doi: 10.1073/pnas.0404631101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pe'er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.