

Abstract
Objective
Drugs that activate PPARγ improve glucose sensitivity and lower blood pressure, whereas dominant negative mutations in PPARγ cause severe insulin resistance and hypertension. We hypothesize that these PPARγ mutants regulate target genes opposite to that of ligand-mediated activation and tested this hypothesis on a genome-wide scale.
Methods and Results
We integrated gene expression data in aorta from mice treated with the PPARγ ligand rosiglitazone with data from mice containing a globally expressed knockin of the PPARγ P465L dominant negative mutation. We also integrated our data with publicly available datasets containing 1) gene expression profiles in many human tissues, 2) PPARγ target genes in 3T3-L1 adipocytes, and 3) experimentally validated PPARγ binding sites throughout the genome. Many classical PPARγ target genes were induced by rosiglitazone and repressed by dominant-negative PPARγ. A similar pattern was observed for about 90% of the gene sets regulated both by rosiglitazone and dominant-negative PPARγ. Genes exhibiting this pattern of contrasting regulation were significantly enriched for nearby PPARγ binding sites.
Conclusions
These results provide convincing evidence that the PPARγ P465L mutation causes transcriptional effects that are opposite to those mediated by PPARγ ligand thus validating mice carrying the mutation as a model of PPARγ interference.
Keywords: PPARγ, microarray, vasculature, transcription factor, bioinformatics
Introduction
PPARγ, a ligand activated nuclear hormone receptor, plays a key role in the regulation of cellular lipid metabolism.1 Thiazolidinedione medications that activate PPARγ have been prescribed to patients with documented success in improving glucose tolerance, improving insulin sensitivity and lowering blood pressure.2 In contrast, mutant forms of PPARγ that interfere with the PPARγ signaling pathway have been described in patients with high blood pressure and insulin resistance.3 As a transcription factor, the physiological actions of PPARγ are attributable to alterations in the cell's gene expression profile involving changes in transcription of many target genes. For example, PPARγ plays a key role in the coordinated regulatory changes in a large number of genes required for adipogenesis.4 Genome wide analysis suggests that over 5,000 binding sites for PPARγ in chromatin are induced during adipogenesis.5, 6 In addition, we have reported that ligand-mediated activation of PPARγ in blood vessel alters expression of hundreds of genes.7
At the present time it is not entirely clear how PPARγ mediates these large-scale changes in gene transcription. One model of PPARγ action involves binding of PPARγ to specific DNA sequences referred to as PPAR response elements (PPRE).8 In the absence of ligand, PPARγ, bound to the PPRE, recruits co-repressors and inhibits gene expression. Upon ligand binding, either endogenous or pharmacological, there is a conformational change in PPARγ, which leads to dismissal of the co-repressors, recruitment of co-activators, and activation of the target gene. Recent genome-wide chromatin immunoprecipitation studies support the PPRE-dependent mechanism of gene regulation in 3T3-L1 adipocytes.5, 6 In addition, it has been reported that PPARγ and other nuclear hormone receptors can decrease expression of target genes in a DNA-binding independent but ligand-dependent mechanism called trans-repression.9 This mechanism appears to be important in PPARγ mediated repression of pro-inflammatory genes.
Two rare but naturally occurring mutations in the ligand binding domain of PPARγ causing severe insulin resistance and hypertension in humans have been documented.3 Molecular evidence demonstrates that these mutations possess dominant negative (DN) activity and are able to compete against wild-type PPARγ for PPRE binding and to recruit co-repressors, but are resistant to the conformational changes needed to dismiss the co-repressors in the presence of ligand.10, 11 Thus, dominant negative PPARγ may regulate target genes in a direction opposite to ligand-induced activation.
To determine if PPARγ-mediated regulation of target genes fits the expected pattern, we examined aortic RNA, using microarray analysis, from C57BL/6J mice in which PPARγ was activated by treatment with a pharmacological ligand rosiglitazone (RZ) and from mice in which a globally expressed knockin of the PPARγ P465L DN mutation (G-DN) was used to interfere with the PPARγ signaling pathway.12
Materials and Methods
Experimental procedures
Care and use of mice met the standard set forth by the National Institutes of Health and all procedures were approved by the University Animal Care and Use Committee at the University of Iowa. Adult male mice (5-7 months of age) were used for all experiments. G-DN experimental mice were generated by breeding inbred 129/SvEv heterozygous P465L knockin mice with C57BL/6J mice, to produce control and heterozygous P465L mice on a F1 genetic background that is isogenic except for the mutation at the PPARγ locus.12 In these mice, the P465L mutation was knocked-in to the mouse PPARγ gene using standard gene-targeting methods. Importantly, these mice contain one copy each of the wild-type and mutant PPARγ genes, both under the control of the endogenous PPARγ promoter and expressed in the same tissues. PPARγ was activated in C57BL/6J mice (Jackson Laboratories) by administration of RZ for either 2 or 14 days at a dose of 3 or 10 mg/kg/day via a custom made diet (Teklad). Control littermates were fed standard mouse chow. Mice were killed by CO2 asphyxiation, and the thoracic aorta quickly removed and frozen. Tissues were homogenized in Tri-Reagent (Molecular Research Center, Cincinnati, OH) and the RNA was isolated as described by the manufacturer. The quality of the RNA was confirmed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California).
Gene Expression Profiling
For the microarray hybridizations, 3 separate biological replicates from each experimental group were used. Each biological replicate was separate RNA pools derived from 8-9 different mouse thoracic aortas. Thoracic aorta was chosen for study based on previous data showing minimal dysfunction in this vessel in the G-DN mice 13. Therefore the gene expression difference can be attributed to PPARγ mutation and not to vascular dysfunction. All the microarray procedures were conducted at the University of Iowa DNA Core facility using standard Affymetrix protocols. Approximately 3 μg of total RNA was used as input to a one-step amplification procedure to generate biotin-labeled RNA fragments for hybridization to the Affymetrix GeneChip Mouse Genome 430 2.0 array. Data from the microarray studies is publicly available at the Gene Expression Omnibus (GEO) at NCBI (array platform: GPL1261, series accession: GSE8949), as was described previously.13
Computational Analysis
Microarray data was analyzed using R statistical software and packages from the Bioconductor project.14 Raw microarray data (i.e., the *.CEL files) was imported into R and normalized using Robust Multi-array Average (RMA).15 The quality of the array hybridizations was confirmed by utilizing the array quality control (QC) functions in Bioconductor.16 One hybridization (RZ, 10 mg/kg/day, 14 days) failed quality control and was excluded from subsequent analysis. Differential expression of genes between groups and their corresponding genetically matched or vehicle treated controls was determined using the Linear Models for Microarray Analysis (limma) package.17 The adjusted p-value representing statistical significance was determined by analysis of the Affymetrix control probes as described by Smyth.18 Determination of whether expression of a set of genes, as a group, was statistically changed was accomplished using the JAVA-based command line version of the Gene Set Enrichment Analysis tool (GSEA, http://www.broad.mit.edu/gsea/).19 For GSEA, the number of gene set permutations was set at 1000, and a p-value less than 0.05 was considered statistically significant. Statistical tests of term enrichment (modified Fisher's exact test) were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) available at the NIH. Additional methods and the algorithms used in the computational search for PPREs can be found in the Supplemental Methods.
Real Time Q-PCR
Real-time quantitative PCR was performed using TaqMan Gene Expression Assays (Applied Biosystems) was performed as described in the Supplemental Methods.
Results
Genes activated by RZ are repressed by DN PPARγ
We first examined the expression of 37 PPARγ target genes (Table S1). These genes have been experimentally verified to be direct targets of PPARγ (see Supplemental Methods). Expression of the classic PPARγ target fatty acid binding protein 4 (FABP4), also known as aP2, was significantly increased 2-fold by RZ in aorta, and was decreased nearly 40% in mice containing the DN PPARγ (Table S2). The set of PPARγ targets, as a group, was significantly increased by RZ (GSEA, P<0.001 in all 4 groups) and repressed by DN PPARγ (GSEA, P=0.0031). For the 71 probe sets representing these PPARγ targets, an inverse relationship was evident between the gene expression changes induced by PPARγ activation and interference (Figures 1A and S1). There is a strong positive correlation (r>0.9) for comparisons between different RZ treatment groups, whereas there is a negative correlation (r<-0.6) between the RZ and DN PPARγ group (Figure S1).
Figure 1. PPARγ Target Genes and Co-expression of PPARγ, FABP4 and RBP7.
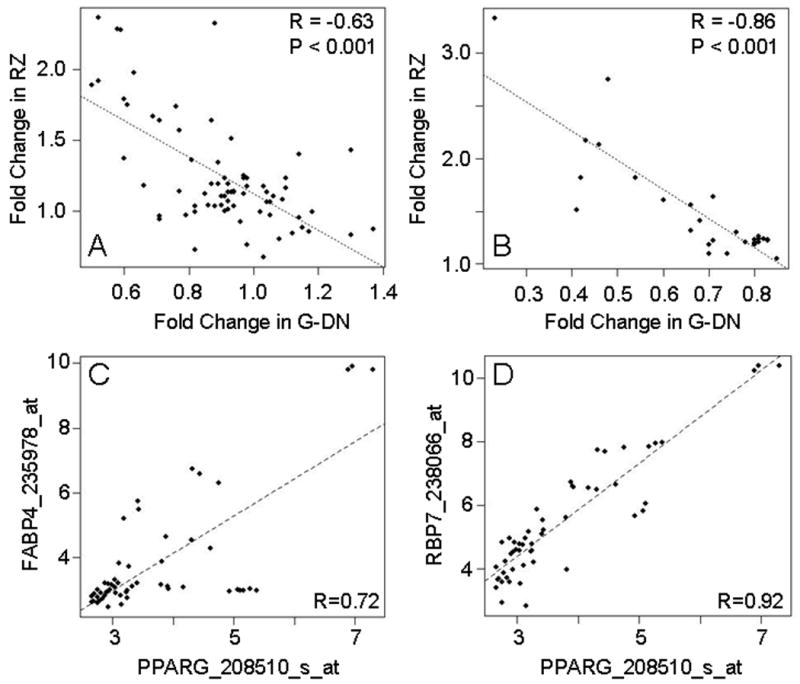
A. Scatterplot of fold-changes in expression of known PPARγ target genes (Table S1) relative to the appropriate control group for mice receiving RZ (3 mg/kg/day for 14 days) compared G-DN mice. B. Scatterplot of fold-changes in expression of PPARγ target genes displaying the most expected expression pattern (Table 1) relative to the appropriate control group for mice receiving RZ (3 mg/kg/day for 14 days) compared to G-DN mice. Similar results were seen for the other rosiglitazone treatments groups (data not shown). C-D. Scatterplots of expression values across a diverse range of human tissues for PPARγ, Rbp7, and FABP4. Values were obtained from a publicly available microarray dataset provided by Affymetrix (http://www.affymetrix.com/support/technical/sample_data/exon_array_data.affx) and Pearson's coefficient (r) was used as the metric for correlation. The identifier after the gene symbol is the Affymetrix probe set identifier.
We used GSEA to analyze the data (obtained from a publicly available repository, ArrayExpress ID: E-GEOD-1458, GEO Accession: GSE1458) from a control set of genes activated by RZ in 3T3-L1 differentiated adipocytes. The RZ-responsive genes identified in this study included known targets and genes without an association with PPARγ. Probesets considered “absent” (Affymetrix MAS 5.0) were removed and the data was sorted based on the level of induction by RZ. We then generated gene sets that were more stringent (only the 25 most induced genes) or less stringent (the top 150 induced genes). We also generated 4 additional gene sets of intermediate stringency (top 50, 75, 100, or 125 up-regulated genes) as well as a set of 50 randomly selected genes. The set of genes induced by RZ in 3T3-L1 cells were then examined in aorta where, as a set, increased (GSEA, P<0.05) after RZ treatment and were decreased (GSEA, P<0.05) by DN PPARγ (Table S3). On the contrary, there was no enrichment for the randomly selected genes in any of our experimental groups.
GSEA was used to examine expression patterns from 2457 functional categories from Gene Ontology (GO) and the UniProt knowledgebase (KW). There were 56 gene sets consistently up-regulated and 100 gene sets down-regulated by RZ (i.e., different at a GSEA P<0.05 in all 4 RZ treatment groups). 49 of the up-regulated sets were either significantly repressed (GSEA, P<0.05) or showed a tendency to be repressed by DN PPARγ (Table S4). Among them, gene sets were involved in cellular metabolism and ion transport. Of the 100 gene sets showing decreased expression by RZ, 25 were significantly up-regulated (GSEA, P<0.05) in the G-DN mice, and included genes involved in the inflammatory response, consistent with the known antiinflammatory actions of PPARγ (Table S4).
Identification of PPARγ induced target genes
To generate a prioritized list of PPARγ targets (both direct and secondary targets), we integrated probeset level analyses from each of the experimental groups, combining the data from RZ-treated and G-DN datasets. The resulting set of genes is more likely to consist of genuine PPARγ targets and less likely to include genes whose expression changes were RZ-specific but unrelated to PPARγ. There were a similar number of genes up (322) and down (323) regulated in the mice containing DN PPARγ. In contrast, more genes were down-regulated (between 195 and 1020 depending on dose and duration) than up-regulated (186-315) after RZ-treatment, particularly in the mice receiving the higher dose (10 mg/kg/day). In total, 1679 unique genes were significantly regulated by RZ in at least 1 of the treatment groups. To increase specificity, we queried the datasets for genes that were regulated in at least 2 of the RZ groups and were oppositely regulated by DN. 28 RZ-induced genes involved in metabolism, lipid binding, and the peroxisome passed these criteria (Table 1). There is a strong positive correlation (r>0.9) between the RZ-treated groups (data not shown) and a negative correlation (r<-0.8) between the RZ and DN PPARγ groups (Figure 1B). We validated the pattern of RZ-induced and DN-repressed expression of 4 of the highest ranked genes by Q-PCR (Figure 2).
Table 1.
Gene expression patterns in mouse aorta: Prioritized list of potential PPARγ targets up-regulated by rosiglitazone.
| ProbeSet | rosiglitazone | gdn | Gene | Description | |||
|---|---|---|---|---|---|---|---|
| low2d | low14d | high2d | high14d | ||||
| 1424451_at | 3.14 | 3.33 | 4.41 | 7.64 | 0.23 | Acaa1b | Acetyl-Coenzyme A acyltransferase 1B |
| 1451679_at | 2.03 | 1.51 | 2.12 | 2.09 | 0.41 | 6530401D17Rik | RIKEN cDNA 6530401D17 gene |
| 1428333_at | 2.71 | 1.82 | 3.01 | 2.83 | 0.42 | 2900062L11Rik | RIKEN cDNA 2900062L11 gene |
| 1436233_at | 2.25 | 2.17 | 2.44 | 4.58 | 0.43 | AI117581 | Expressed sequence AI117581 |
| 1449218_at | 2.20 | 2.13 | 1.51 | 2.64 | 0.46 | Cox8b | Cytochrome c oxidase, subunit VIIIb |
| 1449461_at | 3.51 | 2.75 | 3.97 | 6.88 | 0.48 | Rbp7 | Retinol binding protein 7, cellular |
| 1416023_at | 1.47 | 1.82 | 1.21 | 2.50 | 0.54 | Fabp3 | Fatty acid binding protein 3, muscle and heart |
| 1448499_a_at | 1.69 | 1.61 | 1.71 | 2.34 | 0.60 | Ephx2 | Epoxide hydrolase 2, cytoplasmic |
| 1448382_at | 1.77 | 1.32 | 1.54 | 1.85 | 0.66 | Ehhadh | Enoyl-Coenzyme A, hydratase/3-hydroxyacyl Coenzyme |
| 1454067_a_at | 1.57 | 1.56 | 1.71 | 1.93 | 0.66 | 4931406C07Rik | RIKEN cDNA 4931406C07 gene |
| 1431789_s_at | 1.40 | 1.41 | 1.51 | 1.57 | 0.68 | Tmed5 | Transmembrane emp24 protein transport domain |
| 1447558_at | 1.40 | 1.10 | 1.56 | 0.97 | 0.70 | Atxn7l4 | Ataxin 7-like 4 (Atxn7l4), mRNA |
| 1428435_at | 1.16 | 1.18 | 1.40 | 1.38 | 0.70 | Muc2 | Mucin 2 |
| 1453592_at | 1.20 | 1.22 | 1.42 | 1.44 | 0.71 | Lrrc39 | Leucine rich repeat containing 39 |
| 1417023_a_at | 1.71 | 1.64 | 1.86 | 1.90 | 0.71 | Fabp4 | Fatty acid binding protein 4, adipocyte |
| 1444063_at | 1.24 | 1.10 | 1.32 | 1.32 | 0.74 | 5430435G22Rik | RIKEN cDNA 5430435G22 gene |
| 1445802_at | 1.22 | 1.30 | 1.35 | 1.26 | 0.76 | AU017455 | Expressed sequence AU017455 |
| 1441306_at | 0.98 | 1.21 | 1.21 | 1.07 | 0.78 | 6820408C15Rik | RIKEN cDNA 6820408C15 gene |
| 1420571_at | 1.06 | 1.20 | 1.24 | 1.10 | 0.80 | Prlpb | Prolactin-like protein B |
| 1442980_at | 1.18 | 1.23 | 1.25 | 1.00 | 0.80 | Srpk2 | Serine/arginine-rich protein specific kinase 2 |
| 1446574_at | 1.37 | 1.18 | 1.30 | 1.27 | 0.80 | Ncor1 | Nuclear receptor co-repressor 1 |
| 1423578_at | 1.23 | 1.26 | 1.25 | 1.37 | 0.81 | Col11a2 | Procollagen, type XI, alpha 2 |
| 1443670_at | 1.24 | 1.24 | 1.27 | 1.27 | 0.81 | 2010001J22Rik | RIKEN cDNA 2010001J22 gene |
| 1433238_at | 1.06 | 1.21 | 1.22 | 1.17 | 0.81 | 4930448E06Rik | RIKEN cDNA 4930448E06 gene |
| 1441211_at | 1.14 | 1.24 | 1.29 | 1.25 | 0.82 | Kcns2 | K+ voltage-gated channel, subfamily S, 2 |
| 1426391_at | 1.01 | 1.23 | 1.25 | 1.30 | 0.83 | LOC545267 | Similar to ADP-ribosylation factor 1 |
| 1437646_at | 1.18 | 1.22 | 1.25 | 1.19 | 0.83 | LOC631806 | Similar to Goliath homolog precursor (Ring finger |
| 1450736_a_at | 1.31 | 1.05 | 1.21 | 0.92 | 0.85 | Hbb-bh1 | Hemoglobin Z, beta-like embryonic chain |
Bold indicates statistical significance. Low and high correspond to rosiglitazone doses of 3 and 10 mg/kg/day, respectively. G-DN mice contain a globally expressed dominant negative isoform of PPARγ.
Figure 2. Validation of PPARγ Target Genes.
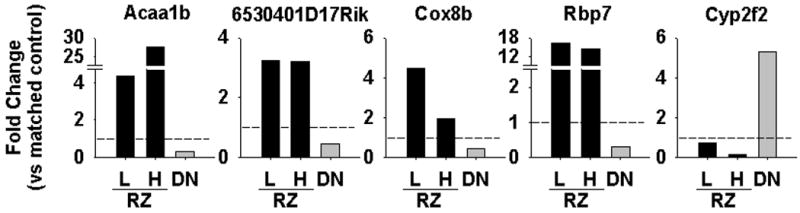
Quantitative real time PCR was performed on 5 genes identified in the microarray analysis. The fold change vs appropriate control is shown for RZ (3 mg/kg/day, L; and 10 mg/kg/day, H) vs G-DN (DN). Shown is the mean of 3 biological replicates.
Retinol binding protein 7 (Rbp7), which shares homology with FABP4 exhibited one of the most robust changes. To further examine the relationship between PPARγ and Rbp7, we compared their expression across a diverse range of human tissues using a publicly available microarray dataset (http://www.affymetrix.com/support/technical/sample_data/exon_array_data.affx). Interestingly, the levels of expression of both FABP4 (Figure 1C, r=0.72) and Rbp7 (Figure 1D, r=0.92) were highly correlated with the level of expression of PPARγ.
There was a smaller group of genes significantly induced by RZ that exhibited either no change or a paradoxical increase by DN PPARγ (Figure 3A, Table S5, Table S7). For example, heat shock proteins were enriched in this group (DAVID, P=7.7×10-5, Figure S2).
Figure 3. Expression Clustering and Functionally Validated PPRE.
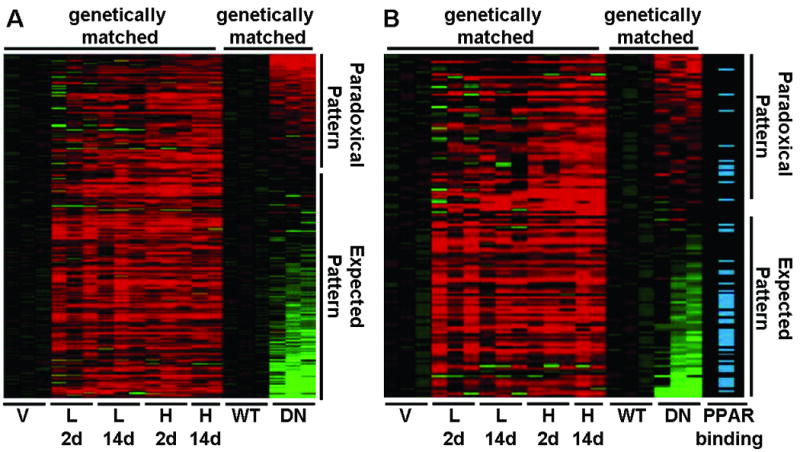
A. Expression clustering of genes up-regulated in at least one RZ-treatment group. All gene expression values have been log2 transformed and normalized to the appropriate control. Mice received rosiglitazone at either a low (3 mg/kg/day, L) or high (10 mg/kg/day, H) dose for either 2 (2d) or 14 (14d) days. Each column represents a sample, and each row a gene. Increasing intensity of red or green color indicates greater up- or down-regulation, respectively. Black indicates no change in expression. B. Association of PPARγ binding sites identified by Chip-ChIP in 3T3-L1 differentiated adipocytes with genes up-regulated in aorta in at least one RZ-treatment group. Only those genes expressed both in 3T3-L1 cells and aorta were included.
Identification of genes repressed by PPARγ
Unlike transcriptional induction by PPARγ, ligand-bound PPARγ has been shown to decrease expression of target genes by blocking the action of other transcription factors by transrepression. This mechanism plays a role in PPARγ-mediated repression of inflammatory cytokines; however, it is not known if the P465L mutation in PPARγ impairs its transrepression function and results in up-regulation of repressed genes. Among genes down-regulated by RZ, 22 were also significantly up-regulated in the G-DN mice (Table 2). As above, we validated the expression pattern of Cyp2f2 by Q-PCR (Figure 2). The mouseNET database (http://mousenet.princeton.edu/) combines experimental, genetic, and genomic data from a variety of public resources. We queried this database for genes from this set for evidence of linkage to inflammatory or NFκB pathways. Linkage was found between LIMA1, a cytoskeleton-associated protein, with the NFκB essential modulator protein (IKBKG or NEMO), and between four other genes (LIMD2, CCR1, IL2RG, and IKZF1) with inflammatory- or immune-related signaling. FAM120b, also known as CCPG, was not found in the mouseNET database, but was recently reported to be a co-activator for PPARγ and to promote adipogenesis in a PPARγ dependent manner.20
Table 2.
Gene expression patterns in mouse aorta: Prioritized list of potential PPARγ targets down-regulated by rosiglitazone.
| ProbeSet | rosiglitazone | gdn | Gene | Description | |||
|---|---|---|---|---|---|---|---|
| low2d | low14d | high2d | high14d | ||||
| 1448792_a_at | 0.62 | 0.38 | 0.32 | 0.39 | 3.97 | Cyp2f2 | Cytochrome P450, family 2, subfamily f |
| 1418872_at | 0.71 | 0.64 | 0.70 | 0.89 | 1.45 | Abcb1b | ATP-binding cassette, sub-family B (MDR/TAP) |
| 1419609_at | 0.80 | 0.82 | 0.74 | 0.72 | 1.45 | Ccr1 | Chemokine (C-C motif) receptor 1 |
| 1456377_x_at | 0.69 | 0.68 | 0.58 | 0.75 | 1.44 | Limd2 | LIM domain containing 2 |
| 1417045_at | 0.83 | 0.77 | 0.63 | 0.61 | 1.40 | Bid | BH3 interacting domain death agonist |
| 1438427_at | 0.82 | 0.72 | 0.77 | 0.73 | 1.39 | Fam120b | family with sequence similarity 120, member B |
| 1451335_at | 0.68 | 0.68 | 0.61 | 0.75 | 1.38 | Plac8 | Placenta-specific 8 |
| 1427183_at | 0.72 | 0.65 | 0.71 | 0.89 | 1.33 | Efemp1 | Epidermal growth factor-containing fibulin-like |
| 1416296_at | 0.89 | 0.79 | 0.73 | 0.91 | 1.32 | Il2rg | Interleukin 2 receptor, gamma chain |
| 1451716_at | 0.87 | 0.80 | 0.80 | 0.84 | 1.32 | Mafb | V-maf musculoaponeurotic fibrosarcoma oncogene |
| 1460465_at | 0.74 | 0.73 | 0.80 | 0.95 | 1.32 | A930038C07Rik | RIKEN cDNA A930038C07 gene |
| 1430038_at | 0.90 | 0.79 | 0.80 | 0.77 | 1.32 | Gphn | Gephyrin |
| 1422744_at | 0.77 | 0.74 | 0.87 | 0.78 | 1.31 | Phka1 | Phosphorylase kinase alpha 1 |
| 1437414_at | 0.76 | 0.71 | 0.84 | 0.79 | 1.31 | Zfp217 | Zinc finger protein 217 |
| 1437410_at | 0.68 | 0.75 | 0.82 | 0.89 | 1.30 | Aldh2 | Aldehyde dehydrogenase 2, mitochondrial |
| 1424965_at | 0.68 | 0.71 | 0.61 | 0.53 | 1.29 | Lpxn | Leupaxin |
| 1455224_at | 0.70 | 0.59 | 0.72 | 0.68 | 1.28 | Angptl1 | Angiopoietin-like 1 |
| 1434401_at | 0.79 | 0.80 | 0.85 | 0.91 | 1.25 | Zcchc2 | Zinc finger, CCHC domain containing 2 |
| 1446834_at | 0.88 | 0.98 | 0.83 | 0.74 | 1.24 | Ctsc | Cathepsin C |
| 1436312_at | 0.95 | 0.84 | 0.79 | 0.79 | 1.24 | Ikzf1 | IKAROS family zinc finger 1 |
| 1460391_at | 0.95 | 0.83 | 0.86 | 0.78 | 1.24 | Gtpbp9 | GTP-binding protein 9 (putative) |
| 1450629_at | 0.82 | 0.84 | 0.78 | 0.81 | 1.19 | Lima1 | LIM domain and actin binding 1 |
Bold indicates statistical significance. Low and high correspond to rosiglitazone doses of 3 and 10 mg/kg/day, respectively. G-DN mice contain a globally expressed dominant negative isoform of PPARγ.
Interestingly, a higher percentage of genes that were down-regulated by RZ exhibited a paradoxical pattern of down-regulation in G-DN mice suggesting that PPARγ carrying the DN mutation may retain an ability to repress transcription (Figure S3, Table S6-S7).
List of genes altered specifically by DN PPARγ
We next determined if there were clusters of genes whose expression was altered by DN but not by RZ. 150 and 92 probesets displayed DN-specific, up- and down- regulation, respectively. In this set of RZ-unresponsive genes, there was an enrichment for genes involved in regulation of transcription (P=0.027) and oxidoreductase activity (P=0.003) (Table S8). Of note, the nuclear receptor NR2F2, also known as COUP-TFII, was one of the transcriptional regulators showing DN-specific activation. COUP-TFII has been reported to be able to bind to the PPRE upstream of the PEPCK (PCK1) gene, a known PPARγ target gene, and to negatively regulate its expression.21 Therefore, some of the effects of DN PPARγ may be mediated by its effects on other transcriptional regulators.
Search for PPARγ response elements
The conventional model of PPARγ-mediated gene activation requires binding to a PPRE, and thus RZ-induced genes should be enriched for nearby PPREs. Approximately 60% of the differentially expressed genes were associated with at least one computationally identified upstream PPRE. However, this did not represent an enrichment compared to a random set of genes, and the patterns of gene expression were similar regardless of the presence or absence of an upstream PPRE-like sequence (Figure S4). We therefore used a publicly available dataset mapping the location of PPREs in 3T3-L1 adipocytes on the basis of function (chromatin immunoprecipitation and microarray hybridization, ChIP-chip) not computational prediction.5 Independent ChIP assays using quantitative PCR and custom microarrays demonstrated that the false discovery rate for this ChIP-chip experiment was low (3-4%). To merge datasets, we limited our analysis to those genes that were consistently expressed in both aorta and 3T3-L1 adipocytes (3T3-L1 expression data, NCBI GEO Accession GSE14004 and GSE8682). The set of genes activated by RZ was significantly enriched (P<0.01 by Fisher's exact test) for PPREs compared either to the set of genes down-regulated by RZ or to the set of all genes expressed both in aorta and 3T3-L1 adipocytes (Table 3). The enrichment was particularly robust for those genes activated at multiple times or doses of RZ. There was also an enrichment in PPREs in genes which exhibited the pattern of opposite regulation (increased to RZ; decreased to DN) than those which exhibit a similar pattern of increased expression in response to RZ and DN (Figure 3B). Finally, we examined our prioritized sets of candidate PPARγ target genes. All 7 genes induced by RZ and repressed by DN and co-expressed in 3T3-L1 adipocytes were associated with a functionally validated PPRE (Table 3). Alternatively, only one gene repressed by RZ and induced by DN was associated with a PPRE (Table 3).
Table 3.
Association of differentially expressed genes with experimentally determined PPARγ binding sites.
| Group | Total | PPRE+ | PPRE- | PPRE+ % | p-value |
|---|---|---|---|---|---|
| RZ up # changes >= 1 | 117 | 34 | 83 | 29.06 | 2.50E-06 |
| RZ up # changes >= 2 | 65 | 25 | 40 | 38.46 | 1.57E-07 |
| RZ up # changes >= 3 | 26 | 15 | 11 | 57.69 | 7.14E-08 |
| RZ up # changes >= 4 | 12 | 11 | 1 | 91.67 | 1.49E-09 |
| RZ down # changes >= 1 | 706 | 89 | 617 | 12.61 | 5.33E-01 |
| RZ down # changes >= 2 | 369 | 52 | 317 | 14.09 | 2.30E-01 |
| RZ down # changes >= 3 | 143 | 24 | 119 | 16.78 | 9.29E-02 |
| RZ down # changes >= 4 | 12 | 0 | 12 | 0.00 | 1.00E+00 |
| RZ up and G-DN down (Table 1) | 7 | 7 | 0 | 100.00 | 5.31E-07 |
| RZ down and G-DN up (Table 2) | 10 | 1 | 9 | 10.00 | 7.42E-01 |
| Genes expressed in aorta and 3T3-L1 adipocytes | 8661 | 1096 | 7565 | 12.65 | NA |
RZ = rosiglitazone, G-DN = mice with dominant negative PPARγ, up = up-regulated, down = down-regulated.
Discussion
We examined global gene expression changes in aorta in response to RZ and in gene-targeted mice containing a DN isoform of PPARγ. Integrated analyses of results from these models suggest that the pathways regulated by PPARγ in the aorta are diverse and are most often regulated in the contrasting direction by the DN PPARγ. The main findings of the study are: 1) known validated PPARγ target genes are regulated in aorta as they are in adipocytes, 2) known PPARγ target genes exhibit the expected opposite pattern of expression in response to PPARγ ligand and DN PPARγ, 3) the changes in expression caused by PPARγ ligand- and DN-PPARγ can be used to generate a prioritized list of target genes, in particular, genes induced by PPARγ, 4) the expression of some PPARγ targets closely parallels the expression of PPARγ in tissues, 5) genes induced by RZ and repressed by DN-PPARγ are often associated with functionally validated PPARγ binding sites, and 6) the identification of PPARγ binding sites from one tissue can be used to potentially predict PPARγ target genes in another, as long as the genes are expressed in both tissues. Because of the commonality of PPARγ targets and associated PPARγ binding sites among cell types, PPARγ likely serves similarly conserved functions across diverse cells. In addition, because the pattern of gene expression in response to DN-PPARγ was generally opposite to the response to PPARγ ligand suggests that models employing DN-PPARγ accurately reflect interference with PPARγ signaling.
Transcriptional Induction and Repression by PPARγ
Based on our analysis of a publicly available dataset, we determined that many of the genes regulated by PPARγ in mouse aorta, particularly metabolism-related genes, are regulated by PPARγ in a similar manner as in a non-vascular cell type (i.e., adipocytes). Therefore, it is tempting to speculate that at least part of the physiological actions of PPARγ in the vasculature may be secondary to a PPARγ-mediated change in the cellular metabolic phenotype. For example, activation of PPARγ in monocyte-derived dendritic cells was reported to be associated with up-regulation of genes involved in lipid metabolism and a reduction in lipid content, changes that might influence the immune response in these cells.22 Whether PPARγ plays additional roles such as providing antioxidant and antiinflammatory defenses that are unique to the vasculature remain to be determined.
While the most studied mechanism of ligand-mediated transactivation of PPARγ involves the PPRE, other indirect means may account for a significant fraction of the response to PPARγ (i.e. the large number of genes down-regulated by RZ). In fact, a much smaller percentage of the genes repressed by RZ had functionally validated PPRE sequences than those induced by RZ. This suggests that the decrease in expression of non-PPRE-containing genes in response to ligand might be due to trans-repression, a process requiring PPARγ ligand but not PPRE binding. Transrepression prevents the transcriptional induction caused by another transcription factor. For example LPS-induced transcription of iNOS by NFκB in macrophages is prevented by ligand-activated PPARγ via a PPRE-independent mechanism.9 The prevalence of PPARγ-mediated trans-repression in the vasculature and the impact of DN PPARγ on this response is not known. If PPARγ trans-repression is impaired by DN, it might explain the cluster of genes that are down-regulated by RZ and up-regulated by DN PPARγ. More difficult to explain are the gene clusters whose expression changes in the same direction during both PPARγ activation and interference. This could potentially be interpreted as retention of the transrepression activity of DN PPARγ. To our knowledge, there have been no studies examining the transrepression potential of DN mutants of PPARγ.
PPARγ and the Vasculature
An association between PPARγ, lipid metabolism, and vascular function is suggested from mice in which the PPARγ gene was deleted specifically from endothelial cells.23 These mice have normal blood pressure at baseline but become hypertensive after a high fat diet. Control mice with intact PPARγ were resistant to the blood pressure elevating effects of the high fat diet suggesting that PPARγ is protective. Similarly, we showed that mice specifically expressing DN PPARγ in endothelial cells exhibit high-fat diet induced vascular dysfunction.24 Because various lipid molecules can act as a ligand to activate PPARγ, it has been suggested that PPARγ acts as a “fatty acid sensor”.25 Activated PPARγ then, via its actions on gene transcription, could reprogram the cellular gene expression profile in order to adapt to the new environmental inputs. It remains unclear which endogenous lipids act as true ligands to PPARγ in vivo.
In addition to the endothelium, we showed that vascular muscle PPARγ is crucial to the regulation of arterial pressure and vascular function. Targeting the DN P467L mutation in PPARγ to vascular muscle in transgenic mice resulted in severely impaired vasodilatation, augmented vasoconstriction, and moderate hypertension.26 Based on our data, we were surprised by recent reports presenting contradictory data on blood pressure in mice lacking PPARγ in vascular muscle cells.27, 28. Wang et al 27 reported increased arterial pressure whereas Chang et al 28 reported hypotension in similar models of smooth muscle PPARγ-deficiency using the Cre-loxP system. The contradictory data may be attributable to differences in the SM22α-cre models employed by both groups, one being a transgenic 27 and the other a knockin 28. Despite these differences, the data beg the question of how vascular muscle-specific DN interference causes hypertension whereas vascular muscle-specific ablation causes hypotension?
Does this mean that interference and deficiency are not equivalent? Perhaps this may be explained by the mechanism of gene induction by PPARγ. In the absence of ligand, PPARγ/RXR heterodimers can interact with a PPRE and recruit co-repressors, thus repressing transcription. The co-repressors are dismissed and replaced by co-activators when PPARγ ligand is present (i.e. ligand-mediated transactivation). Evidence suggests that DN PPARγ can out-compete wild-type PPARγ for the PPRE binding due to a reduction in receptor recycling rate thus preserving or extending the state of transcriptional repression.10 Alternatively, PPARγ deficiency may reduce or remove the repressive state by eliminating the recruitment of co-repressors, thus causing some level of transcriptional activation through other elements of the transcriptional machinery. Indeed Mortensen and colleagues suggested the concept that the phenotype of gene-deficiency may mimic agonist-mediated induction due to gene suppression that both agonist and genetic deficiency are capable of relieving.29 Support for this differential model of DN interference vs PPARγ-deficiency comes from 1) our data showing reproducible repression of genes induced by RZ, in particular those with functionally validated PPREs, and 2) data reporting activation of the PPARγ target gene β2-adrenergic receptor (β2AR) expression by RZ or genetic-deficiency or shRNA-mediated ablation of PPARγ.28 Consistent with this, PPARγ-mediated repression of β2AR required DNA binding as it was abolished by mutations in the DNA binding domain of PPARγ. Consequently, whereas DN PPARγ appears to act as a bonafide inhibitor of PPARγ-mediated induction, PPARγ-deficiency may actually provide a gain-of-function thus emulating some of the phenotypes associated with TZD treatment (i.e. lowered arterial pressure).
Perspectives
The large number of differentially expressed genes in most microarray experiments has made prioritizing the gene list, so that experimental efforts are directed toward the most attractive genes or pathways, a substantial challenge. For studies involving pharmacological agents, off-target and dose- or time- dependent effects can result in changes in gene expression that hinder identification of the primary target genes. By integrating results from pharmacological studies and gene-targeted mouse models in the present study, we have generated a list of approximately 30 target genes that demonstrate significant responses consistent with the conventional model of PPARγ action. Further bioinformatic analysis of freely available datasets revealed some of these to be co-expressed in 3T3-L1 cells and to contain PPREs. One of these, RBP7 for example, must be promoted on the list because it was one of the most robustly induced by RZ and repressed by DN, and its expression correlated well with PPARγ in many tissues. RBP7 expression is induced several days after the start of differentiation of 3T3-L1 cells, and its promoter contains several PPRE sequences that were shown to bind PPARγ by EMSA and ChIP, and were functional in transfection assays.5, 30 Studies of RBP7-deficient mice revealed a role in lipid and whole body energy metabolism.30 RBP7 becomes an even more attractive candidate when one considers that Caprioli et al. showed that it is expressed in microvascular endothelial cells.31 These two observations, an involvement in lipid and energy metabolism, along with its expression in endothelium are particularly interesting in light of our data showing high fat diet induced vascular dysfunction in mice specifically expressing DN PPARγ in the endothelium.24 Consequently, examining the role of RBP7 in the blood vessel wall is a necessary next step.
Supplementary Material
Acknowledgments
a) Sources of Funding: This work was supported by grants from the National Institutes of Health (HL062984, HL084207 to CDS, and NS024621 to FMF) and American Heart Association (AMB). We gratefully acknowledge the generous research support of the Roy J. Carver Trust.
Footnotes
c) Disclosure:
Henry L. Keen: none
Carmen M. Halabi: NIH NRSA Funding
Andreas M. Beyer: AHA Predoctoral Fellowship
Willem J. de Lange: none
Xuebo Liu: none
Nobuyo Maeda: NIH Grants
Frank M. Faraci: NIH Grants
Thomas L. Casavant: NIH Grants
Curt D. Sigmund: NIH Grants, Roy J. Carver Trust
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Sharma AM, Staels B. Review: Peroxisome proliferator-activated receptor gamma and adipose tissue--understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab. 2007;92:386–395. doi: 10.1210/jc.2006-1268. [DOI] [PubMed] [Google Scholar]
- 2.Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, Neves MF, Schiffrin EL. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation. 2002;105:2296–2302. doi: 10.1161/01.cir.0000016049.86468.23. [DOI] [PubMed] [Google Scholar]
- 3.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O'Rahilly S. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 4.Perera RJ, Marcusson EG, Koo S, Kang X, Kim Y, White N, Dean NM. Identification of novel PPARgamma target genes in primary human adipocytes. Gene. 2006;369:90–99. doi: 10.1016/j.gene.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 5.Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, Feng D, Zhuo D, Stoeckert CJ, Jr, Liu XS, Lazar MA. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008;22:2941–2952. doi: 10.1101/gad.1709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S, Stunnenberg HG. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keen HL, Ryan MJ, Beyer A, Mathur S, Scheetz TE, Gackle BD, Faraci FM, Casavant TL, Sigmund CD. Gene expression profiling of potential PPAR{gamma} target genes in mouse aorta. Physiological Genomics. 2004;18:33–42. doi: 10.1152/physiolgenomics.00027.2004. [DOI] [PubMed] [Google Scholar]
- 8.Glass CK. Going nuclear in metabolic and cardiovascular disease. J Clin Invest. 2006;116:556–560. doi: 10.1172/JCI27913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li G, Leff T. Altered promoter recycling rates contribute to dominant-negative activity of human peroxisome proliferator-activated receptor-gamma mutations associated with diabetes. Mol Endocrinol. 2007;21:857–864. doi: 10.1210/me.2006-0401. [DOI] [PubMed] [Google Scholar]
- 11.Agostini M, Gurnell M, Savage DB, Wood EM, Smith AG, Rajanayagam O, Garnes KT, Levinson SH, Xu HE, Schwabe JW, Willson TM, O'Rahilly S, Chatterjee VK. Tyrosine agonists reverse the molecular defects associated with dominant-negative mutations in human peroxisome proliferator-activated receptor gamma. Endocrinology. 2004;145:1527–1538. doi: 10.1210/en.2003-1271. [DOI] [PubMed] [Google Scholar]
- 12.Tsai YS, Kim HJ, Takahashi N, Kim HS, Hagaman JR, Kim JK, Maeda N. Hypertension and abnormal fat distribution but not insulin resistance in mice with P465L PPARgamma. J Clin Invest. 2004;114:240–249. doi: 10.1172/JCI20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beyer AM, Baumbach GL, Halabi CM, Modrick ML, Lynch CM, Gerhold TD, Ghoneim SM, deLange WJ, Keen HL, Tsai YS, Maeda N, Sigmund CD, Faraci FM. Interference with PPARγ Signaling Causes Cerebral Vascular Dysfunction, Hypertrophy, and Remodeling. Hypertension. 2008;51:867–871. doi: 10.1161/HYPERTENSIONAHA.107.103648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 16.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 17.Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 18.Smyth GK. Limma: Linear models for microarray data. In: Gentleman RC, Carey VJ, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Speth RC, Karamyan VT. The significance of brain aminopeptidases in the regulation of the actions of angiotensin peptides in the brain. Heart Fail Rev. 2008 doi: 10.1007/s10741-007-9078-2. [DOI] [PubMed] [Google Scholar]
- 21.Eubank DW, Duplus E, Williams SC, Forest C, Beale EG. Peroxisome proliferator-activated receptor gamma and chicken ovalbumin upstream promoter transcription factor II negatively regulate the phosphoenolpyruvate carboxykinase promoter via a common element. J Biol Chem. 2001;276:30561–30569. doi: 10.1074/jbc.M103019200. [DOI] [PubMed] [Google Scholar]
- 22.Szatmari I, Torocsik D, Agostini M, Nagy T, Gurnell M, Barta E, Chatterjee K, Nagy L. PPARgamma regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood. 2007;110:3271–3280. doi: 10.1182/blood-2007-06-096222. [DOI] [PubMed] [Google Scholar]
- 23.Nicol CJ, Adachi M, Akiyama TE, Gonzalez FJ. PPARgamma in endothelial cells influences high fat diet-induced hypertension. Am J Hypertens. 2005;18:549–556. doi: 10.1016/j.amjhyper.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 24.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res. 2008;103:654–661. doi: 10.1161/CIRCRESAHA.108.176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 26.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, Sigmund CD. Interference with PPARγ Function in Smooth Muscle Causes Vascular Dysfunction and Hypertension. Cell Metabolism. 2008;7:215–226. doi: 10.1016/j.cmet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang N, Yang G, Jia Z, Zhang H, Aoyagi T, Soodvilai S, Symons JD, Schnermann JB, Gonzalez FJ, Litwin SE, Yang T. Vascular PPARgamma controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab. 2008;8:482–491. doi: 10.1016/j.cmet.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang L, Villacorta L, Zhang J, Garcia-Barrio MT, Yang K, Hamblin M, Whitesall SE, D'Alecy LG, Chen YE. Vascular smooth muscle cell-selective peroxisome proliferator-activated receptor-gamma deletion leads to hypotension. Circulation. 2009;119:2161–2169. doi: 10.1161/CIRCULATIONAHA.108.815803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duan SZ, Ivashchenko CY, Whitesall SE, D'Alecy LG, Duquaine DC, Brosius FC, Gonzalez FJ, Vinson C, Pierre MA, Milstone DS, Mortensen RM. Hypotension, lipodystrophy, and insulin resistance in generalized PPARgamma-deficient mice rescued from embryonic lethality. J Clin Invest. 2007;117:812–822. doi: 10.1172/JCI28859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zizola CF, Schwartz GJ, Vogel S. Cellular retinol-binding protein type III is a PPARgamma target gene and plays a role in lipid metabolism. Am J Physiol Endocrinol Metab. 2008;295:E1358–E1368. doi: 10.1152/ajpendo.90464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caprioli A, Zhu H, Sato TN. CRBP-III:lacZ expression pattern reveals a novel heterogeneity of vascular endothelial cells. Genesis. 2004;40:139–145. doi: 10.1002/gene.20075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.