

Abstract
Malaria is endemic in tropical and subtropical regions of Africa, Asia, and the Americas. The increasing prevalence of multi-drug-resistant Plasmodium falciparum drives the ongoing need for the development of new antimalarial drugs. In this light, novel scaffolds to which the parasite has not been exposed are of particular interest. Recently, workers at the Swiss Tropical Institute discovered two novel 4-oxo-3-carboxyl quinolones active against the intra-erythrocytic stages of P. falciparum while carrying out rationally directed low-throughput screening of potential antimalarial agents as part of an effort directed by the World Health Organization. Here we report the design, synthesis, and preliminary pharmacologic characterization of a series of analogues of 4-oxo-3-carboxyl quinolones. These studies indicate that the series has good potential for preclinical development.
Introduction
Malaria, caused by the protozoal species Plasmodium, is one of the most devastating infectious diseases in the world. More than 500 million cases are reported annually, with 1 to 3 million deaths.1, 2 The most lethal form of malaria is caused by Plasmodium falciparum and the rapid spread of chloroquine-resistant (CQ-R) and multi-drug-resistant (MDR) P. falciparum contributes to the deteriorating malaria situation.3–5 Thus, the development of new antimalarial drugs that are active against CQ-R and MDR strains is urgently needed.6–8
Among the potential classes of antimalarials, the acridone derivatives have been known since the 1940’s. Both 7-methoxy tetrahydroacridone (1) and endochin (2) have prophylactic and therapeutic activity in canaries infected with P. praecox or P. gallinaceum.9, 10 However, endochin is not effective against human malaria.11 Endochin-like 4(1H)-quinolones bearing extended alkenyl or alkyl side chains at the 3 position have potent activity against both chloroquine-sensitive (CQ-S) and MDR P. falciparum strains.11,12 The primary target of these compounds is thought to be the parasite cytochrome bc1 complex.12 Additionally, the 4(1H)-quinolone esters [WR197236 (3) and WR194905 (4)] have potent anti-relapse activity in rhesus monkeys infected with P. cynomolgi B.13 It has been reported that P. Berghei quickly developed resistance to WR197236 (3) when it was used alone.14 Acridinedione derivatives WR 243251 (5) and WR249685 (6) also have significant potency against CQ-R and CQ-S strains in vitro.15–17 WR 249685 (6) targets the quinol oxidation site of the bc1 complex.18
The World Health Organization’s Special Programme for Research and Training in Tropical Diseases (TDR) recently initiated a rationally directed screening campaign to explore the antimalarial potential of commercially available and proprietary compounds that resemble known antimalarials or compounds with known anti-parasitic effects. As part of this campaign, workers at the Swiss Tropical Institute recently identified two 4(1H)-quinolone derivatives, TDR42098 (7) and TDR17516 (8), with potent antimalarial activity against both the CQ-R K1 and CQ-S NF54 strains of P. falciparum. Although these compounds are structurally similar to the quinolones described above, their simpler structures, lacking the long fatty side chains, present some potential for improved physicochemical properties.19 We report here the development of synthetic routes that allow the rational perturbation of all potentially important functional groups on the 4(1H)-quinolone scaffold and the use of these routes to probe the fundamental structure activity relationships of the series. These studies led to the discovery of analogues with potency equal to or greater than that of mefloquine in drug-resistant strains.
Synthesis of targeted analogues of 7 and 8
There were several issues to be addressed in preliminary hit validation on this series. First, the prior work discussed above had indicated that absorption limited pharmacokinetics were a major potential worry with poor solubility and cellular permeability being the major physiochemical drivers. Second, this particular compound series shares significant pharmacophore overlap with the quinolone antibiotics, which may raise an issue of target mediated cyto- and geno-toxicity. Finally, the methylenedioxy and methoxy groups pose potential metabolic clearance challenges. The preliminary survey of structure-activity relationships was designed to assess liabilities related to all of these issues.
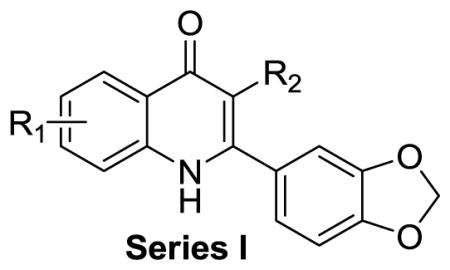
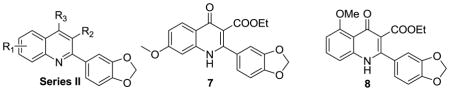
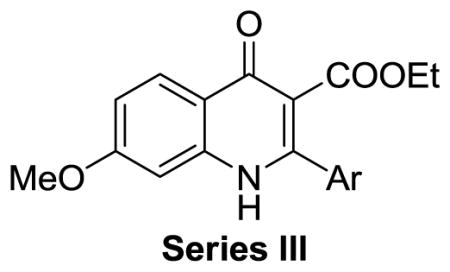
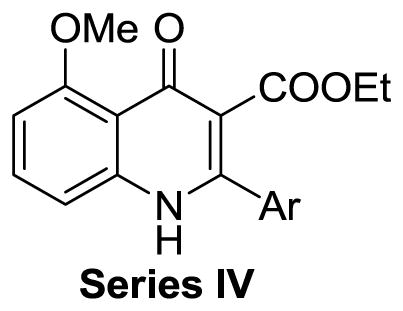
On the basis of their chemical structures, the 4-oxo-3-carboxyl quinolone analogues were divided into four groups. Quinolones containing 3-carboxylic acid constitute a class of antibacterial agents that target bacterial type II topoisomerase.20 Therefore, Series I consisted of derivatives with 3-carboxylic acid, 3-carboxylic amide, or a 3-acetyl group designed to explore whether this moiety was required and if any potential interactions with topoisomerases could be avoided while retaining activity. Series II included derivatives with reduced functionalities at the 3 and 4 positions, with the same overall goal.. Series III and IV focused on exploring substituted 2-aryl groups of 7-methoxy quinolones and 5-methoxy quinolones, respectively with the intention of identifying any alternate ring functionalities that would have lower metabolic liabilities with equivalent potency.
The synthetic route for the derivatives in Series I is illustrated in Schemes 1 and 2. The intermediate 4(1H)-quinolones (14) were prepared from anilines and diethyl (2-ethoxymethylene)malonate (11) or ethyl 2-(ethoxymethylene)acetoacetate (12), yielding enamines (13). Gould-Jacobs cyclization of 13, following the published procedure, gave the desired quinolones (14).21–24 The precursor 4(1H)-quinolone (15) was obtained by the reduction of 14b with H2 in the presence of a catalytic amount of 5% Pd/C.24 The 4(1H)-quinolones (14a, 14c, and 15) were converted to 4-chloro-quinolines (16) and then oxidized to yield N-oxide derivatives (17). Bromide 18 was produced from N-oxide 17 through bromination with POBr3. An m,p-methylenedioxyphenyl group was introduced at the 2 position by a Suzuki coupling reaction of 19 and the corresponding boronic acid at 75 °C. Control of the reaction temperature was important in this step to avoid cross-coupling at both the 2 and 4 positions. The desired 4(1H)-quinolones (7, 8, and 20) were prepared by the hydrolysis of 4-chloroquinolines (19) in AcOH/H2O.
Scheme 1. Synthesis of 4(1H)-quinolones.

a Reagents and conditions: a) EtOH, 130 °C, 3 h; b) Ph2O, reflux, 4–6 h; c) 5% Pd/C, H2, MeOH, rt, overnight; d) POCl3, 1,4-dioxane, 120 °C, 1 h; e) m-chloroperbenzoic acid, CHCl3, rt, 4 h; f) POBr3, CHCl3, rt, 1 h. g) 3,4-methylenedioxyphenyl boronic acid, Pd(PPh3)4, CsCO3, 1,4-dioxane/H2O, 75 °C, 3 h; h) AcOH/H2O (4:1), 120 °C, 1 h.
Scheme 2. Synthesis of 3-carboxylic acid and 3-carboxylic amide quinolones.

a Reagents and conditions: a) NaOH (1 N, aqueous), 130 °C, overnight; b) KOH (1 N, aqueous), 75 °C, overnight; c) N,N′-carbonyldiimidazole, DMF, 65 °C, 2.5 h, NH4OH (aqueous), rt, overnight.
Further derivatives in Series I were synthesized as depicted in Scheme 2. Decarboxylation of 7 and 8 yielded 21a and 21b. Hydrolysis of 7 gave 3-carboxylic acid quinolone (22a), which was easily converted to 3-carboxylic amide (23a).
Compounds in Series II were synthesized as shown in Schemes 3–4. The 4-methoxy quinoline (24) and 4-methylamino quinoline derivatives (25) were prepared by the treatment of 19 with NaOMe and MeNH2, respectively (Scheme 3). Compounds 26a and 26b were prepared from the reduction of 19 with H2 in the presence of 5% Pd/C (Scheme 4). The treatment of 19a with diisobutylaluminum hydride (DIBAL) selectively reduced the carboxylate group of 19a and gave the 4-chloro-hydroxymethyl quinoline (27a). However, DIBAL reduced both 4-chloro and 3-carboxylate groups of 19b. The synthesis of 3-hydroxymethyl quinoline (29a and 29b) was performed by the reduction of 19a and 19b in the presence of LiAlH4 as the reducing agent. Alkylation of 27a and 29b was performed to give the corresponding ethyl ethers (28a and 30b).
Scheme 3. Synthesis of 4-methoxy and 4-methylamino derivatives.

a Reagents and conditions: a) NaOMe, MeOH/THF, 85 °C, 30 min; b) MeNH2 (2 N in MeOH), THF, 85 °C, 30 min.
Scheme 4. Reduction of 4-chloroquinolines 19.

a Reagents and conditions: a) 5% Pd/C, H2, Et3N, MeOH/THF, rt, overnight; b) DIBAL, THF, 0 °C to rt. 7 h; c) LiAlH4, THF, −42 °C, 1 h, rt, overnight; d) NaH (60% dispersion on mineral oil), EtI, THF, overnight.
The synthesis of Series III (31) and Series IV (32), with various substituted aromatic rings at the C-2 position, is shown in Scheme 5. 2-Aryl quinolone derivatives (31–32) were prepared from the cross-coupling of 2-bromo quinoline (19a and 19b) with appropriate boronic acids, followed by refluxing in AcOH/H2O.
Scheme 5. Exploring the 2-aryl group.

a Reagents and conditions: a) ArB(OH)2, Pd(PPh3)4, CsCO3, 1,4-dioxane/H2O, 75 °C, 3 h; b) AcOH/H2O (4:1), 120 °C, 1 h.
Extensive prior work in the antibacterial field has indicated that it is important to be aware of the quinolone/quinoline tautomerism, which determines where the hydrogen bond donor is located in the inhibitor and influences the interaction between inhibitors and both targets and membranes. The relative thermodynamic stabilities of the quinolone and quinoline forms are predicted to be similar.25, 26, 27 The more polar quinolone tautomer is predicted to be predominantly adopted in the solid state and in aqueous solution.25, 26, 28 The presence of a carboxylate group at the 3 position could act as a hydrogen bond acceptor for the 4-OH group and thus might stabilize the enol quinoline tautomer.25 To explore this tautomerism, we studied the Fourier transform infrared (FTIR) spectrum (KBr pellet) and nuclear Overhauser effects (NOE; in DMSO) of compound 7. We observed carbonyl vibrations at 1722.47 cm−1, 1632.53 cm−1, and 1627.52 cm−1 by FTIR, indicating the presence of the quinolone tautomer of compound 7 in the solid state. The quinolone tautomer was also supported by the NOE studies of compound 7, as shown in Figure 2. A characteristic NOE was observed between the N-proton and the quinolone proton at the 8 position, as well as between the N-proton and the ortho-protons of the m,p-methylenedioxyphenyl group.
Figure 2.

Determination of quinolone/quinoline tautomerism of compound 7 in DMSO by NOE.
Biological activities and physical properties of analogue series
All compounds described in this work were evaluated for antimalarial activity against the CQ-S P. falciparum strain 3D7 and the CQ-R strain K1 using a previously described assay.29 For all compounds, concentration response curves were defined using 10-point, 2-fold dilution schemes. Each experiment was performed in triplicate, and all experiments were independently replicated at least twice. Data are reported as average values with standard deviations based upon all replications of the experiments. Additionally, for the reasons discussed above, the aqueous solubility and passive membrane permeability of the compounds were measured in neutral physiologically isotonic buffers. Each experiment was performed in triplicate, and all experiments were independently replicated at least twice. Data are reported as average values with standard deviations based upon all replications of the experiments.
Series I: Quinolones with various substituents at the 3-position
The antimalarial activity of 7-methoxy quinolones varied within a large range of EC50 values, as shown in Table 1. The carboxyl ester 7 had the best antimalarial potency (EC50 ~0.25 μM against both the K1 and 3D7 strains). The replacement of the 3-carboxyl ester group by 3-carboxylic acid or 3-carboxylic amide abolished its activity against both strains (7 vs. 22a, and 7 vs. 23a, Table 1). The introduction of an acetyl group at the 3 position caused a nearly 90% drop in potency (7 vs. 20, Table 1). We also observed that the absence of a carboxylic ester group in compound 21a resulted in a lack of activity against both strains. The 5-methoxy quinolone was < 10% as active as 7-methoxy quinolone (7 vs. 8, Table 1). The solubility of these compounds was dependent on 3-substituents. Compounds with 3-carboxylate, 3-acid, and 3-amide groups, such as 7, 22a, 23a, and 8, were highly soluble (> 100 μM in phosphate-buffered saline [PBS] with DMSO at pH 7.4). Compounds 21a and 21b, which lacked 3-substituents, were less soluble (3 and 1 μM, respectively). In addition, we observed tolerable permeability (18–173×10−6 cm/s) for all compounds in Series I. Compounds with hydrophilic groups, such as 22a and 23a, did not penetrate the lipid layer as well as compound 7. Because of the limitations of 7-methoxy and 5-methoxy quinolones containing carboxylate at the 3-position, we focused our attention on compounds containing the 7-methoxy and 3-carboxyl ester substituents.
Table 1.
Summary of Series I compounds: antimalarial activity, solubility, and permeability across an artificial membrane (PAMPA)
 | ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | EC50 (μM)a K1 strain | EC50 (μM)a 3D7 strain | Sol. (μM)b | Perm (10−6 cm/s)c | |
| 7 | 7-OMe | COOEt | 0.25±0.01 | 0.24±0.02 | >100 | 100±13 |
| 22a | 7-OMe | COOH | >10 | >10 | >100 | 19±3 |
| 23a | 7-OMe | CONH2 | >10 | >10 | >100 | 18±2 |
| 20 | 7-OMe | COCH3 | 2.32±0.02 | 2.00±0.30 | 36±1 | 88±17 |
| 21a | 7-OMe | H | >10 | >10 | 3±0 | 146±15 |
| 8 | 5-OMe | COOEt | 3.01±0.08 | 2.81±0.11 | >100 | 47±7 |
| 21b | 5-OMe | H | >10 | >10 | 1±0 | 173±24 |
EC50 values are reported as the mean ± SD of at least two independent experiments. Positive controls were mefloquine (EC50 in K1: 0.04±0.02 μM; EC50 in 3D7: 0.16±0.02 μM) and chloroquine (EC50 in K1: 1.61±0.30 μM; EC50 in 3D7: 0.09±0.02 μM).
Solubility in PBS buffer, pH 7.4, containing 5% DMSO. The detection limit of the solubility assay was 100 μM. Two standards were tested in the same assay: albendazole (3±0.1 μM, poorly soluble) and carbamazepine (>100 μM, highly soluble).
Permeability at pH 7.4. Three standards were tested in the same assay: ranitidine HCl (1.2±0.7×10−6 cm/s, poorly permeable), carbamazepine (145±13×10−6 cm/s, moderately permeable), and verapamil HCl (1960±290×10−6 cm/s, highly permeable).
Series II: Quinoline analogues
The changes in properties arising from reduced functionalities at the 3 and 4 positions are shown in Table 2. Changing the functional keto at the 4 position to a 4-methoxy group led to a 2–3 fold decrease in potency (7 vs. 24a, and 8 vs. 24b, Table 2), suggesting that the 4-keto moieties in 7 and 8 makes them more active than 4-methoxy quinolines. This finding motivated us to evaluate the activity of N-alkyl derivatives. However, we found that N-ethylated 730 was inactive against both K1 and 3D7 strains (EC50 > 10 μM). In Series II, other reduced functionalities at the 4 position or at both the 3 and 4 positions all caused a complete loss of activity (25a–b, 26a–b, 28a, 29b, and 30b, Table 2). The solubility of this series ranged widely (from 0.4 to > 100 μM). Compounds containing 4-methoxy, 4-methylamino, or 3-hydroxymethyl (24a–b, 25a–b, 29a–b) were acceptably soluble (24–69 μM). All the tested quinolines were moderately or highly permeable (193–2796×10−6 cm/s). The results of these two sets of experiments prompted us to strongly focus our subsequent work on compounds with the 4(1H)-quinolone moiety.
Table 2.
Summary of Series II compounds: antimalarial activity, solubility, and permeability across an artificial membrane (PAMPA)
 | |||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | EC50 (μM)a K1 strain | EC50 (μM)a 3D7 strain | Solubility (μM)b | Permeability (10−6 cm/s)c | |
| 7 | - | - | - | 0.25±0.01 | 0.24±0.02 | >100 | 100±13 |
| 8 | - | - | - | 3.01±0.08 | 2.81±0.11 | >100 | 47±7 |
| 24a | 7-OMe | COOEt | OMe | 0.71±0.09 | 0.61±0.01 | 24±3 | 783±139 |
| 25a | 7-OMe | COOEt | NHMe | >10 | >10 | 59±11 | 2013±179 |
| 26a | 7-OMe | COOEt | H | >10 | >10 | 11±2 | 649±44 |
| 19a | 7-OMe | COOEt | Cl | >10 | >10 | 0.4±0.1 | 735±0 |
| 29a | 7-OMe | CH2OH | H | >10 | >10 | 65±6 | 356±22 |
| 28a | 7-OMe | CH2OEt | Cl | >10 | >10 | 2±0 | 802±370 |
| 24b | 5-OMe | COOEt | OMe | 6.00±0.20 | 6.10±0.90 | 51±4 | 193±14 |
| 25b | 5-OMe | COOEt | NHMe | >10 | >10 | 67±0 | 1240±154 |
| 26b | 5-OMe | COOEt | H | >10 | >10 | 10±2 | 918±171 |
| 29b | 5-OMe | CH2OH | H | >10 | >10 | 69±0 | 644±20 |
| 30b | 5-OMe | CH2OEt | H | >10 | >10 | 16±3 | 2796±1746 |
EC50 values are reported as the mean ± SD of at least two independent experiments. Positive controls were mefloquine (EC50 in K1: 0.04±0.02 μM; EC50 in 3D7: 0.16±0.02 μM) and chloroquine (EC50 in K1: 1.61±0.30 μM; EC50 in 3D7: 0.09±0.02 μM)
Solubility in PBS buffer, pH 7.4, containing 5% DMSO. The detection limit of the solubility assay was 100 μM. Two standards were tested in the same assay: albendazole (3±0.1 μM, poorly soluble) and carbamazepine (>100 μM, highly soluble).
Permeability at pH 7.4. Three standards were tested in the same assay: ranitidine HCl (1.2±0.7×10−6 cm/s, poorly permeable), carbamazepine (145±13×10−6 cm/s, moderately permeable), and verapamil HCl (1960±290×10−6 cm/s, highly permeable).
Series III: 7-Methoxy quinolones with various 2-aryl groups
In the Series III compounds, the influence of the aryl group at the C-2 position of 7-methoxy quinolones was explored (Table 3). Compared with reference compound 7 (EC50: 0.25 μM), the unsubstituted phenyl group (31a) and the 2′-thienyl ring (31b) at the C-2 position led to 3-fold and 12-fold decreases in antimalarial potency, respectively. Meta substitutions generally gave increased potency, depending on the nature of the substituent. The meta-methoxy phenyl compound (31c) was the most potent analogue (EC50 in K1: 0.13 μM; EC50 in 3D7: 0.10). Meta-methyl phenyl (31d), meta-trifluoromethyl phenyl (31e), and meta-chloro phenyl (31f) analogues all had potencies approximately equivalent to that of the lead compound (7). However, the meta-fluoro phenyl (31g) and meta-nitro phenyl (31h) analogues were slightly less potent. The presence of para substituents on the phenyl ring, such para-methoxy (31i), para-t-butyl (31j), para-trifluoromethyl (31k), and para-chloro (31l), led to an overall decrease in antimalarial potency, with EC50 values between 1 μM and > 10 μM. On the other hand, ortho substitution on the phenyl at the C-2 position caused the complete loss of potency against both strains (31n and 31o). The solubility of Series III compounds varied between 2 and > 100 μM. Interestingly, para substituents on the 2-aryl group caused poor solubility, as shown in compounds 31i-l with 2–8 μM of solubility. All 7-methoxy quinolones in Series III had moderate to high permeability. These studies made it clear that meta substituted 2-phenyl rings gave the best overall average potency. They also confirmed that the potency of the original lead compounds could be retained while replacing the potentially metabolically labile and genotoxic methylenedioxy substitution pattern with more stable and less toxicologically troublesome substituents.
Table 3.
Summary of Series III compounds: antimalarial activity on CQ-resistant strain K1 and CQ-sensitive strain 3D7, solubility, and permeability across an artificial membrane (PAMPA)
 | |||||
|---|---|---|---|---|---|
| Aryl group | EC50 (μM)a K1 strain | EC50 (μM)a 3D7 strain | Solubility (μM)b | Permeability (10−6 cm/s)c | |
| 7 | m,p-methylenedioxy-phenyl | 0.25±0.01 | 0.24±0.02 | >100 | 100±13 |
| 31a | phenyl | 0.82±0.07 | 0.79±0.04 | >100 | 194±41 |
| 31b | 2-thienyl | 3.10±0.60 | 2.87±0.01 | >100 | 105±10 |
| 31c | m-MeO-phenyl | 0.13±0.01 | 0.10±0.01 | >100 | 172±19 |
| 31d | m-Me-phenyl | 0.30±0.01 | 0.29±0.01 | 48±8 | 227±7 |
| 31e | m-CF3-phenyl | 0.22±0.02 | 0.21±0.02 | 19±1 | 569±80 |
| 31f | m-Cl-phenyl | 0.18±0.02 | 0.20±0.01 | 58±8 | 948±39 |
| 31g | m-F-phenyl | 0.55±0.02 | 0.59±0.05 | >100 | 188±14 |
| 31h | m-NO2-phenyl | 0.87±0.01 | 0.84±0.02 | 13±1 | 397±19 |
| 31i | p-MeO-phenyl | 1.15±0.22 | 1.35±0.12 | 8±2 | 196±20 |
| 31j | p-tBu-phenyl | 1.02±0.03 | 1.93±1.69 | 2±0 | 876±54 |
| 31k | p-CF3-phenyl | >10 | 3.60±0.07 | 8±0 | 312±15 |
| 31l | p-Cl-phenyl | >10 | 2.09±0.27 | 3±0 | 501±71 |
| 31m | m,p-diCl-phenyl | 2.90±2.80 | 2.52±0.90 | 17±3 | 763±19 |
| 31n | o-Me-phenyl | >10 | >10 | >100 | 100±13 |
| 31o | o-Cl-phenyl | >10 | >10 | 28±1 | 674±81 |
EC50 values are reported as the mean ± SD of at least two independent experiments. Positive controls were mefloquine (EC50 in K1: 0.04±0.02 μM; EC50 in 3D7: 0.16±0.02 μM) and chloroquine (EC50 in K1: 1.61±0.30 μM; EC50 in 3D7: 0.09±0.02 μM)
Solubility in PBS buffer, pH 7.4, containing 5% DMSO. The detection limit of the solubility assay was 100 μM. Two standards were tested in the same assay: albendazole (3±0.1 μM, poorly soluble) and carbamazepine (>100 μM, highly soluble).
Permeability at pH 7.4. Three standards were tested in the same assay: ranitidine HCl (1.2±0.7×10−6 cm/s, poorly permeable), carbamazepine (145±13×10−6 cm/s, moderately permeable), and verapamil HCl (1960±290×10−6 cm/s, highly permeable).
Series IV: 5-Methoxy quinolones with various 2-aryl groups
In this series, 12 compounds with various aryl groups at the 2-position had no measurable antimalarial activity against either the K1 or 3D7 strains. In general, the compounds in Series IV that did have activity were significantly less potent than those in Series III. We observed moderate (22 μM) to high solubility (>100 μM) for compounds in Series IV, except for 32j (1 μM), which included a hydrophobic t-butyl group. The permeability of Series IV compounds varied from 37×10−6 cm/s to 1143×10−6 cm/s. The results of this study focused our work on 7-methoxy substituted compounds.
Toxicity studies
To provide preliminary assessment of inherent toxicity, all compounds were screened using a panel of mammalian cell lines, including an embryonic kidney cell line (HEK 293), a lymphoblastoid cell line (Raji), a hepatocellular carcinoma cell line (HepG2), and a fibroblast cell line (BJ). The cytotoxicity of each compound was first evaluated by screening for effects on cell viability at a fixed concentration of 27.5 μM. No compound was more than 60% cytotoxic at this concentration in any cell line. Compounds that were 30–60% cytotoxic at 27.5 μM were further tested to determine their potencies. Each experiment was performed in triplicate, and all experiments were independently replicated at least twice. Data are reported as average values with standard deviations based upon all replications of the experiments.
Since HEK 293 cells were the most sensitive of the four cell lines, a therapeutic index (TI) was defined as the ratio between the EC50 in the cytotoxicity assay on HEK 293 cells and the EC50 in the antimalarial assay on the K1 strain. For compounds < 30% cytotoxic at 27.5 μM, the potency of these compounds as a cytotoxin was conservatively estimated at 27.5 μM. Eight compounds had a TI > 10, with three having a TI > 100 and two of those having a TI > 200. This reflected the ability of compounds to inhibit parasite growth at concentrations significantly lower than those producing mammalian cytotoxicity.
Quinolones are a well known class of bactericidal drugs that inhibit bacterial topoisomerase II or topoisomerase IV, thereby blocking bacterial DNA replication.20,31–33 Quinolone antibacterial drugs have been reported to have weak antimalarial activity against P. falciparum parasites, suggesting that the malarial topoisomerase shares some overlapping sensitivity with the bacterial.34, 35 Because of the similarity of analogues of 7 to antibacterial quinolones, we evaluated the antibacterial activity of five compounds, 7, 21a, 24a, 31a, and N-ethylated 7 on the Escherichia coli BL21 strain. None of the tested compounds inhibited bacterial growth at concentrations up to 200 μM, whereas the control fluoroquinolone was highly active. Moreover, our antimalarial testing showed that the introduction of the 3-carboxylic acid or an N-ethyl group on the quinolone nitrogen, functional features of quinolone antibacterial agents, abolished antimalarial potency. These limited findings suggest that topoisomerase II of P. falciparum is not a target for the quinolone ester derivatives reported herein.
Because the MMV/WHO product profile specifies orally available drugs, drug-like antimalarial candidates should possess a balance between antimalarial potency, solubility, permeability, and toxicity. Among the subseries studied, only analogues (31c–h) in Series III containing a meta-substituted phenyl at the 2-position had such properties. Compound 31c, equipotent with mefloquine against the 3D7 strain, was identified as the most active analogue with high solubility (>100 μM) and reasonable permeability (172×10−6 cm/s). Exploring meta-substituents of the 2 aromatic ring should provide the possibility to improve both potency and physicochemical properties. Moreover, isosteres of 3-carboxylate would likely be valuable in future investigations.
Conclusion
Our structure-activity relationship studies revealed that several factors influence the antimalarial potency of quinolone analogues: 1) an ethyl carboxylate at the 3 position was superior to a carboxylic acid, carboxylic amide, or other replacements; 2) removal of the 4-oxo moiety led to a strong decrease in antimalarial potency; 3) a 7-methoxy group was favored over a 5-methoxy group; and 4) an aryl ring at the C-2 position caused large variations in antimalarial potency. With respect to the C-2 aryl ring, meta-substituents were superior to para-substituents and ortho-substituents. Compounds 31c–h had reasonably drug-like properties. In particular, compound 31c was the most promising analogue.
With respect to structure-property relationships, there were no clear structural associations that governed either solubility or hydrophobicity other than a loose correlation between hydrophobicity of introduced groups and decreased solubility. There were no clear associations between potency and either solubility or permeability.
Overall, this series shows promise for further development with a particular focus on understanding absorption, distribution, metabolism, excretion, and toxicology (ADMET). Clearly two early goals are improving potency and aqueous solubility. Additionally, isosteres of the carboxyl ester, isosteres of the 4(1H)-quinolone, and exploration of solubility-enhancing substituents at the meta position of the 2 aromatic ring are all appropriate venues for future work.
Experimental Section
General methods
3,4-Methylenedioxphenyl boronic acid was purchased from Boron Molecular, and all other chemical reagents were from Acros, Aldrich, or Combi-Blocks. All materials were obtained from commercial suppliers and used without further purification. Thin layer chromatography was performed using a silica gel 60 F254 plate from EMD. Purification of compounds was done by normal phase column chromatography (Biotage SP1). 1H NMR, 13C NMR, and 2D NOE spectra were recorded on a Bruker 400 MHz. Chemical shifts were expressed in ppm relative to tetramethylsilane, which was used as an internal standard. Purity was estimated using high-performance liquid chromatography/mass spectrometry (Alliance HT, Micromass ZQ 4000, and RP-C18 Xterra column, 5 μm, 6 × 50 mm [Waters]) or ultra-performance liquid chromatography/mass spectrometry (Acquity PDA detector, Acquity SQ detector, and Acquity UPLC BEH-C18 column, 1.7 μm, 2.1 × 50 mm [Waters]). FTIR spectrums were recorded on a Thermo Nicolet IR 100 FTIR spectrometer. Compounds prepared in our laboratory were generally 90–98% pure. Efficacy data were obtained only on compounds that were at least 95% pure.
Ethyl 7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate (14a) and 3-acetyl-7-methoxyquinolin-4(1H)-one (14c)
Ethyl 7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate (14a) was prepared from m-aminoanisole (9) (15 mmol) and diethyl 2-(ethoxymethylene)malonate (11) (3.309 mL, 16.5 mmol), followed by Gould-Jacobs cyclization according to the published procedure.22 3-Acetyl-7-methoxyquinolin-4(1H)-one (14c) was prepared in a similar manner from m-aminoanisole and ethyl 2-(ethoxymethylene)acetoacetate.23
Ethyl 5-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate (15)
The intermediate compound ethyl 8-chloro-5-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate (14b) was prepared in a manner similar to that of 14a from 2-chloro-5-methoxyanilin (10) and diethyl 2-(ethoxymethylene)malonate (11). Following the treatment of 14b with H2 in the presence of a catalytic amount of 5 wt% Pd/C at room temperature (rt) overnight, the catalyst was removed by filtration through Celite, and the filtrate was concentrated under vacuum to produce compound 15 at a 97% yield.
Ethyl 2-bromo-4-chloro-7-methoxyquinoline-3-carboxylate (18a)
Phosphorus oxychloride (0.72 mL, 7.8 mmol) was added to a suspended solution of 3-carboxylate quinolone (14a) (1.6 g, 6.5 mmol) in 6 mL of 1,4-dioxane. The mixture was sealed in a reaction tube and stirred at 120 °C for 1 h. After being cooled to rt, the reaction mixture was poured into ice water and then neutralized by adding K2CO3 until the pH was approximately 7. 4-Chloro quinoline intermediate 16a was extracted by CH2Cl2 (30 mL × 2). Organic layers were combined, dried over Na2SO4, filtered, and concentrated to produce 16a at an 86% yield. No purification was performed on 16a.
4-Chloro quinoline intermediate 16a was treated with m-chloroperbenzoic acid (1.74 g, 7.8 mmol) in 30 mL of CHCl3 at rt for 4 h. The reaction was quenched by adding NaHCO3 aqueous solution. The aqueous layer was extracted by CH2Cl2 (30 mL × 2). The combined organic layers were dried over Na2SO4, filtered, and concentrated to produce N-oxide intermediate 17a at a 92% yield, which was used for the next step without the further purification.
Phosphorus oxybromide (2.06 g, 7.2 mmol) was added to the solution of N-oxide intermediate 17a in 30 mL of CHCl3. After being stirred at rt for 1 h, the reaction was quenched by adding ice water and then neutralized by adding K2CO3 until the pH was approximately 7. The aqueous layer was extracted by CH2Cl2 (30 mL × 2). The combined organic layers were dried over Na2SO4, filtered, and concentrated. Purification was done by flash column chromatography to give the corresponding compounds 18a at a 71% yield over three steps. (SP1, 40S, SiO2, flow rate: 40 mL/min, gradient: 1–20% ethyl acetate in hexanes over 7 CV; 20% ethyl acetate in hexanes over 10 CV). 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 9.2, 1H), 7.25 (dd, J = 6.3, 15.4, 2H), 4.45 (q, J = 7.2, 2H), 3.88 (s, 3H), 1.39 (t, J = 7.2, 3H). 13C NMR (401 MHz, CDCl3) δ 164.63, 162.83, 150.13, 140.35, 136.96, 127.43, 125.79, 121.86, 119.65, 107.16, 62.82, 55.89, 14.05. Electrospray ionization mass spectrometry (MS [ESI]) calculated for C13H11BrClNO3 [M+H]+, 343.97; found, 344.65.
Ethyl 2-bromo-4-chloro-5-methoxyquinoline-3-carboxylate (18b)
The compound was prepared in a manner similar to that of 18a from 15 for a 48% yield over three steps. 1H NMR (400 MHz, CDCl3) δ 7.59–7.68 (m, 3H), 6.96 (d, J = 7.5, 1H), 4.49 (q, J = 7.1, 2H), 3.95 (s, 3H), 1.43 (t, 3H), 13C NMR (400 MHz, CDCl3) δ 164.82, 156.82, 150.09, 139.31, 136.71, 132.02, 130.79, 121.60, 116.74, 108.10, 62.73, 56.17, 13.99. MS (ESI) calculated for C13H11BrClNO3 [M+H]+, 343.97; found, 344.65.
1-(2-Bromo-4-chloro-7-methoxyquinolin-3-yl)ethanone (18c)
The compound was prepared in a manner similar to that of 18a from 14c for a 23% yield over three steps. 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 9.2, 1H), 7.14-7.02 (m, 2H), 3.72 (s, 3H), 2.45 (s, 3H). 13C NMR (401 MHz, CDCl3) δ 199.08, 162.73, 150.11, 138.54, 135.50, 133.07, 125.52, 121.94, 119.88, 107.12, 55.91, 31.28. MS (ESI) calculated for C12H9BrClNO2 [M+H]+, 313.96; found, 314.09.
General Procedure for 7, 8, 20, 31a–o, 32a–o
The flask was charged with 2-bromo-4-chloro quinoline intermediate 18 (0.3 mmol), proper boronic acid (0.31 mmol), Pd(PPh3)4 (8.6 mg, 0.0075 mmol) in 2 mL of 1,4-dioxane. The flask was degassed three times. To the mixture was added the solution of CsCO3 (0.195 g, 0.6 mmol) in 0.6 mL of H2O. The flask was degassed again three times. The reaction mixture was stirred at 75 °C for 3 h. After being cooled to rt, the bottom aqueous layer was removed, and the organic layer was concentrated and purified by flash column chromatography to produce the intermediate compound ethyl 2-aryl-4-chloroquinoline-3-carboxylate. (SP1, 12S, SiO2, flow rate: 6 mL/min, gradient: 1–20% ethyl acetate in hexanes over 7 CV; 20% ethyl acetate in hexanes over 10 CV).
The solution of ethyl 2-aryl-4-chloroquinoline-3-carboxylate intermediate (0.2 mmol) in 1 mL of AcOH/H2O (4:1) was refluxed at 120 °C for 1 h. The reaction mixture was concentrated and neutralized by adding 5 drops of NH4OH. Purification was performed by flash column chromatography to produce the desired compounds. (SP1, 12S, SiO2, flow rate: 12 mL/min, gradient: 40% ethyl acetate in hexanes over 5 CV; 100% ethyl acetate over 10 CV). 7 is given as an example. For characterization of 8, 20, 31a–o, and 32a–o, see Supporting Information.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylate (7)
A 59% yield was produced over two steps. 1H NMR (400 MHz, DMSO) δ 11.77 (s, 1H), 8.01 (d, J = 9.0, 1H), 7.07-6.98 (m, 6H), 6.14 (d, J = 4.3, 2H), 4.03 (d, J = 7.1, 3H), 3.86 (s, 3H), 1.03 (t, J = 7.1, 3H). 13C NMR (400 MHz, DMSO) δ 173.12, 166.46, 162.29, 148.82, 148.32, 147.36, 141.29, 127.29, 126.74, 122.55, 118.70, 115.22, 113.83, 108.45, 101.76, 99.74, 60.17, 55.48, 13.80. MS (ESI) calculated for C20H17NO6 [M+H]+, 368.11; found, 368.02.
2-(Benzo[d][1,3]dioxol-5-yl)-7-methoxyquinolin-4(1H)-one (21a)
The suspended solution of quinolone 7 (0.73 g, 2 mmol) in 10 mL of 1 N NaOH was stirred at 130 °C overnight. The cooled solution was neutralized by adding 1 N HCl solution until the pH was approximately 7. The white precipitate was formed, collected by filtration, and dried under a vacuum for a 75% yield. 1H NMR (400 MHz, DMSO) δ 11.40 (s, 1H), 7.96 (d, J = 8.9, 1H), 7.42-7.35 (m, 2H), 7.13 (dd, J = 5.3, 27.9, 2H), 6.89 (dd, J = 2.4, 8.9, 1H), 6.23 (s, 1H), 6.13 (s, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, DMSO) δ 161.50, 148.77, 148.58, 147.79, 126.28, 121.36, 119.42, 112.82, 108.52, 107.32, 101.63, 55.30. MS (ESI) calculated for C17H13NO4 [M+H]+, 296.09; found, 296.07.
2-(Benzo[d][1,3]dioxol-5-yl)-5-methoxyquinolin-4(1H)-one (21b)
The compound was prepared in a manner similar to that of 21a from 8 for an 80% yield. 1H NMR (400 MHz, DMSO) δ 11.17 (br, 1H), 7.43 (dd, J = 25.9, 54.7, 4H), 7.09 (d, J = 8.2, 1H), 6.77 (s, 1H), 6.13 (s, 3H), 3.33 (s, 9H). 13C NMR (101 MHz, DMSO) δ 177.95, 148.91, 147.81, 131.74, 121.50, 114.93, 108.54, 107.35, 104.28, 101.66, 55.73. MS (ESI) calculated for C17H13NO4 [M+H]+, 296.09; found, 295.97.
2-(Benzo[d][1,3]dioxol-5-yl)-7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (22a)
Ethyl ester 7 (0.110 g, 0.3 mmol) underwent hydrolysis in 5 mL of 1 N KOH at 75 °C overnight. The mixture was neutralized by adding 1 N HCl (aqueous), followed by filtration to produce the compound at a 55% yield. 1H NMR (400 MHz, DMSO) δ 12.75 (s, 1H), 8.20 (d, J = 9.0, 1H), 7.24-6.99 (m, 5H), 6.13 (s, 2H), 3.89 (s, 3H). 13C NMR (101 MHz, DMSO) δ 178.08, 165.30, 163.43, 156.61, 148.32, 146.59, 140.52, 128.70, 126.98, 122.51, 117.52, 116.15, 109.44, 107.79, 106.68, 101.44, 100.12, 55.78. MS (ESI) calculated for C18H13NO6 [M+H]+, 340.08; found, 339.86.
2-(Benzo[d][1,3]dioxol-5-yl)-7-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxamide (23a)
N,N′-carbonyldiimidazole (49 mg, 0.3 mmol) was added to the corresponding carboxylic acid 22a (50 mg, 0.15 mmol) in 1 mL of DMF. After being stirred at 65 °C for 2.5 h, the mixture was cooled to rt and then poured into 5 mL of iced NH4OH. The solvent was evaporated under a vacuum. To the residue was added 5 mL of iced water, and precipitation was observed. The desired product was collected by filtration at a 56% yield. 1H NMR (400 MHz, DMSO) δ 11.63 (s, 1H), 8.06 (d, J = 8.9, 1H), 7.12-6.89 (m, 6H), 6.12 (s, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, DMSO) δ 174.58, 167.18, 162.15, 149.85, 148.19, 146.80, 140.86, 128.99, 126.99, 122.45, 119.07, 116.10, 113.77, 108.92, 108.01, 101.46, 99.50, 55.44. MS (ESI) calculated for C18H14N2O5 [M+H]+, 339.10; found, 338.94.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-4,7-dimethoxyquinoline-3-carboxylate (24a)
To fresh sodium methoxide solution, which was prepared from Na (9 mg, 0.4 mmol) in 1 mL of MeOH, was added the corresponding 4-chloro quinoline 19a (77 mg, 0.2 mmol) in 0.5 mL of dried tetrahydrofuran (THF). After the mixture was refluxed at 85 °C for 30 min, the solvent was evaporated under a vacuum. The obtained residue was purified by flash column chromatography to produce the compound at a 44% yield (SP1, 12S, SiO2, flow rate: 6 mL/min, gradient: 1–20% ethyl acetate in hexanes over 7 CV; 20% ethyl acetate in hexanes over 10 CV). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 9.2, 1H), 7.49 (s, 1H), 7.20 (ddd, J = 1.7, 8.2, 23.1, 3H), 6.91 (d, J = 8.0, 1H), 6.04 (s, 2H), 4.27 (q, J = 7.2, 2H), 4.16 (s, 3H), 3.98 (s, 3H), 1.20 (t, J = 7.2, 3H). 13C NMR (101 MHz, CDCl3) δ 167.60, 162.08, 161.98, 157.82, 151.17, 148.32, 147.78, 134.11, 123.65, 122.56, 119.81, 116.79, 116.28, 109.14, 108.19, 107.51, 101.28, 62.83, 61.77, 55.67, 13.88. MS (ESI) calculated for C21H19NO6 [M+H]+, 382.13; found, 382.03.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-4,5-dimethoxyquinoline-3-carboxylate (24b)
The compound was prepared in a similar manner from 19b for a 77% yield. 1H NMR (400 MHz, CDCl3) δ 7.70-7.57 (m, 2H), 7.18 (d, J = 8.0, 1H), 6.84 (dd, J = 7.9, 16.0, 2H), 5.96 (s, 2H), 4.24 (q, J = 7.0, 2H), 3.98 (s, 3H), 3.96 (s, 3H), 1.17 (t, J = 7.1, 3H). 13C NMR (101 MHz, CDCl3) δ 167.27, 162.79, 156.76, 155.77, 151.62, 148.56, 147.89, 133.46, 130.78, 122.76, 122.36, 121.82, 114.04, 109.17, 108.24, 106.17, 101.31, 64.15, 61.68, 56.33, 14.02. MS (ESI) calculated for C21H19NO6 [M+H]+, 382.13; found, 382.03.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-7-methoxy-4-(methylamino)quinoline-3-carboxylate (25a)
The 4-chloro quinoline 19a (77 mg, 0.2 mmol) and 2 mL of MeNH2 (2 M in THF) were refluxed at 85 °C for 30 min. After the solution was cooled to rt, the solvent was evaporated. Then, to the residue was added 2 mL of H2O. The precipitate was collected by filtration. The compound was produced at a 63% yield. 1H NMR (400 MHz, MeOD) δ 8.09 (d, J = 9.2, 1H), 7.22 (s, 1H), 7.10 (d, J = 9.2, 1H), 6.91 (dd, J = 10.2, 20.3, 2H), 6.01 (s, 2H), 4.02 (q, J = 7.1, 2H), 3.92 (s, 3H), 3.07 (s, 3H), 1.01 (t, J = 7.1, 3H). 13C NMR (101 MHz, MeOD) δ 170.17, 161.59, 158.46, 150.74, 148.81, 147.82, 147.30, 134.96, 123.06, 121.76, 116.64, 112.40, 108.57, 107.27, 106.28, 105.58, 101.18, 60.97, 54.48, 30.94, 12.45. MS (ESI) calculated for C21H20N2O5 [M+H]+, 381.15; found, 381.32.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-5-methoxy-4-(methylamino)quinoline-3-carboxylate (25b)
The compound was prepared in a manner similar to that of 25a from 19b for a 56% yield. 1H NMR (400 MHz, MeOD) δ 7.55 (t, J = 8.2, 1H), 7.42 (d, J = 8.5, 1H), 6.94 (dd, J = 10.9, 28.2, 4H), 6.00 (d, J = 3.0, 2H), 4.07 (s, 3H), 4.01 (q, J = 7.2, 2H), 2.96 (s, 3H), 1.03 (t, J = 7.2, 2H). 13C NMR (101 MHz, MeOD) δ 170.13, 158.15, 157.54, 152.34, 148.75, 147.78, 147.25, 134.45, 129.94, 121.61, 120.37, 109.65, 108.51, 107.24, 106.57, 104.97, 101.17, 61.12, 55.53, 30.48, 12.44. MS (ESI) calculated for C21H20N2O5 [M+H]+, 381.15; found, 380.98.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-7-methoxyquinoline-3-carboxylate (26a)
The corresponding 4-chloro quinoline 19a (93 mg, 0.24 mmol) in 5 mL of MeOH and 2 mL of THF was treated with H2 in the presence of Et3N (34 μL, 0.24 mmol) and 5 mol% of 5 wt% Pd/C. After the mixture was stirred at rt overnight, the Pd/C was filtered out though Celite. The filtrate was concentrated and then purified by flash column chromatography to produce the compound at a 65% yield. (SP1, 12S, SiO2, flow rate: 6 mL/min, gradient: 1–20% ethyl acetate in hexanes over 7 CV; 20% ethyl acetate in hexanes over 10 CV). 1H NMR (400 MHz, CDCl3) δ 8.55 (s, 1H), 7.76 (d, J = 9.0, 1H), 7.23-7.20 (m, 1H), 7.14 (d, J = 1.8, 1H), 7.06 (d, J = 8.0, 1H), 6.88 (d, J = 8.0, 1H), 6.00 (s, 2H), 4.23 (q, J = 7.2, 2H), 3.95 (s, 3H), 1.17 (t, J = 7.1, 3H). 13C NMR (101 MHz, CDCl3) δ 167.85, 162.77, 157.87, 148.25, 147.61, 139.21, 129.31, 123.01, 121.08, 120.81, 109.37, 108.15, 107.00, 106.71, 101.27, 61.48, 55.80, 13.96. MS (ESI) calculated for C20H17NO5 [M+H]+, 352.12; found, 352.02.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-5-methoxyquinoline-3-carboxylate (26b)
The compound was prepared in a manner similar to that of 26b from 19b for a 41% yield. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.68-6.85 (dd, J = 6.7, 13.9, 2H), 7.17 (d, J = 1.8, 1H), 7.07 (dd, J = 1.8, 8.0, 1H), 6.87 (m, 2H), 5.99 (s, 2H), 4.24 (q, J = 7.2, 2H), 4.02 (s, 3H), 1.19 (t, J = 7.1, 3H). 13C NMR (101 MHz, CDCl3) δ 167.12, 156.62, 154.66, 147.90, 147.22, 146.63, 133.28, 130.84, 123.43, 121.98, 120.24, 117.34, 108.35, 107.11, 103.78, 100.23, 60.51, 54.89, 12.96, 0.02. MS (ESI) calculated for C20H17NO5 [M+H]+, 352.12; found, 352.02.
(2-(Benzo[d][1,3]dioxol-5-yl)-4-chloro-7-methoxyquinolin-3-yl)methanol (27a)
To the 4-chloro quinoline 19a (108 mg, 0.28 mmol) in 3 mL of THF was added DIBAL (239 mg, 1.68 mmol) at 0 °C. The mixture was stirred at 0 °C for 10 min, and then at rt overnight. The reaction was quenched by adding saturated Na2SO4·10H2O solution at 0 °C. The mixture was stirred at 0 °C for 20 min, and then at rt for 30 min. The suspended solid was filtered out though Celite. The filtrate was concentrated and purified by flash column chromatography (SP1, 25S, SiO2, flow rate: 12 mL/min, gradient: 1–20% ethyl acetate in hexanes over 7 CV; 20% ethyl acetate in hexanes over 10 CV; 20–80% ethyl acetate in hexanes over 2 CV; 80% ethyl acetate in hexanes over 3 CV). The compound was obtained at an 82% yield. 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 9.3, 1H), 7.49 (br, 1H), 7.29-7.25 (m, 3H), 6.93 (d, J = 8.4, 1H), 6.02 (s, 2H), 4.89 (d, J = 6.4, 2H), 3.94 (s, 3H).
(2-(Benzo[d][1,3]dioxol-5-yl)-7-methoxyquinolin-3-yl)methanol (29a)
To LiAlH4 (15 mg, 0.4 mmol) in 1 mL of dry THF was added the solution of the corresponding 4-chloro quinoline 19a (50 mg, 0.13 mmol) in 1 mL of THF at −42 °C. The reaction mixture was stirred at −42 °C for 1 h, then at rt overnight. The reaction was quenched by adding 1 N NaOH aqueous solution. The suspended solid was filtered out though Celite. The filtrate was concentrated and purified by flash column chromatography (SP1, 12S, SiO2, flow rate: 6 mL/min, gradient: 1–30% ethyl acetate in hexanes over 7 CV; 30% ethyl acetate in hexanes over 10 CV). The compound was obtained at a 36% yield. 1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 7.72 (d, J = 9.0, 1H), 7.50 (s, 1H), 7.19 (dd, J = 2.5, 8.9, 1H), 7.14-7.09 (m, 2H), 6.90 (d, J = 8.0, 1H), 6.01 (s, 2H), 4.78 (s, 2H), 3.93 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.18, 161.21, 148.17, 147.76, 129.90, 128.48, 123.00, 122.52, 120.15, 109.59, 108.32, 106.63, 101.31, 60.41, 55.64. MS (ESI) calculated for C18H15NO4 [M+H]+, 310.11; found, 309.98.
(2-(Benzo[d][1,3]dioxol-5-yl)-5-methoxyquinolin-3-yl)methanol (29b)
The compound was prepared in a manner similar to that of 29a from 19b for a 42% yield. 1H NMR (400 MHz, MeOD) δ 8.83 (s, 1H), 7.68-7.59 (m, 2H), 7.12-6.96 (m, 5H), 6.05 (s, 2H), 4.70 (s, 2H), 4.06 (s, 3H). 13C NMR (101 MHz, MeOD) δ 160.28, 156.62, 149.17, 148.55, 134.56, 133.53, 132.22, 131.30, 124.13, 121.20, 120.81, 110.47, 109.11, 105.76, 102.84, 62.55, 56.47. MS (ESI) calculated for C18H15NO4 [M+H]+, 310.11; found, 310.24.
2-(Benzo[d][1,3]dioxol-5-yl)-4-chloro-3-(ethoxymethyl)-7-methoxyquinoline (28a)
The corresponding 3-hydroxymethyl quinoline 27a (34 mg, 0.1 mmol) and NaH (60% dispersion in mineral oil, 8 mg, 0.2 mmol) in 1 mL of dry THF were stirred at rt for 1 h. To the reaction mixture was added Et3N (42 μL, 3 mmol). The mixture was stirred at 75 °C for 1 h, followed by quenching with H2O. The aqueous layer was extracted by dichloromethane (3 × 3 mL). Organic layers were combined, dried over Na2SO4, filtered, and concentrated. The crude product was purified by flash column chromatography (SP1, 12S, SiO2, flow rate: 6 mL/min, gradient: 1–20% ethyl acetate in hexanes over 7 CV; 20% ethyl acetate in hexanes over 10 CV). The compound was obtained at an 80% yield. 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 9.3, 1H), 7.43 (s, 1H), 7.26 (dd, J = 7.2, 9.3, 3H), 6.91 (d, J = 7.8, 1H), 6.01 (s, 2H), 4.60 (s, 2H), 3.93 (s, 3H), 3.60 (q, J = 7.0, 2H), 1.27 (t, J = 7.0, 3H). 13C NMR (101 MHz, CDCl3) δ 176.98, 168.05, 155.07, 147.59, 125.84, 124.88, 123.57, 120.90, 120.72, 109.95, 108.15, 101.29, 67.61, 66.39, 55.78, 15.27. MS (ESI) calculated for C20H18ClNO4 [M+H]+, 372.10; found, 371.99.
2-(Benzo[d][1,3]dioxol-5-yl)-3-(ethoxymethyl)-5-methoxyquinoline (30b)
The compound was prepared in a manner similar to that of 28a from 29b for a 45% yield. 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 7.72 (s, 1H), 7.59 (t, J = 8.2, 1H), 7.21-7.16 (m, 2H), 6.91 (d, J = 8.0, 1H), 6.84 (d, J = 7.8, 1H), 6.01 (s, 2H), 4.55 (s, 2H), 4.01 (s, 3H), 3.56 (q, J = 7.0, 2H), 1.25 (t, J = 7.0, 3H). 13C NMR (101 MHz, CDCl3) δ 155.13, 147.65, 128.76, 123.33, 119.71, 109.85, 108.20, 104.29, 101.24, 70.30, 66.15, 55.83, 15.27. MS (ESI) calculated for C20H19NO4 [M+H]+, 338.14; found, 338.39.
Biological Assay
Two P. falciparum strains, CQ-S 3D7 and CQ-R K1, were used in this study and were provided by the MR4 Unit of the American Type Culture Collection (Manassas, VA). Asynchronous parasites were maintained in culture based on the method of Trager.36 Parasites were grown in the presence of fresh group O-positive erythrocytes (Lifeblood, Memphis, TN) in Petri dishes at a hematocrit of 4–6% in RPMI-based medium. It consisted of RPMI 1640 supplemented with 0.5% AlbuMAX II, 25 mM HEPES, 25 mM NaHCO3 (pH 7.3), 100 μg/mL hypoxanthine, and 5 μg/mL gentamicin. Cultures were incubated at 37 °C in a gas mixture of 90% N2, 5% O2, and 5% CO2. For EC50 determinations, 20 μl of RPMI 1640 with 5 μg/mL gentamicin were dispensed per well in an assay plate (384-well microplate, clear-bottom, tissue-treated). Next, 40 nL of compound, previously serial diluted in a separate 384-well white polypropylene plate, were dispensed in the assay plate, and then 20 μL of a synchronized culture suspension (1% rings, 10% hematocrit) were added per well to make a final hematocrit and parasitemia of 5% and 1%, respectively. Assay plates were incubated for 72 h, and the parasitemia was determined by a method previously described.37 Briefly, 10 μL of 10X Sybr Green I, 0.5% v/v Triton, and 0.5 mg/mL saponin solution in RPMI were added per well. Assay plates were shaken for 30 s, incubated in the dark for 4 h, and then read with the Envision spectrofluorometer at Ex/Em 485 nm/535 nm. EC50s were calculated with Graphpad PRISM software.
Solubility
Solubility assays were carried out on a Biomek FX lab automation workstation (Beckman Coulter, Inc., Fullerton, CA) using μSOL Evolution software (pION Inc., Woburn, MA) as follows: 10 μL of compound stock was added to 190 μL of 1-propanol to make a reference stock plate. Next, 5 μL of this reference stock plate was mixed with 70 μL of 1-propanol and 75 μL of PBS (pH 7.4 and 4, respectively) to make the reference plate, and the UV spectrum (250–500 nm) of the reference plate was read. Then, 6 μL of 10 mM test compound stock was added to 600 μL of PBS in a 96-well storage plate and mixed. The storage plate was sealed and incubated at rt for 18 h. The suspension was then filtered through a 96-well filter plate (pION Inc., Woburn, MA). Next, 75 μL of filtrate was mixed with 75 μL of 1-propanol to make the sample plate, and the UV spectrum of the sample plate was read. Calculations were done using μSOL Evolution software based on the area under the curve (AUC) of the UV spectrum of the sample plate and the reference plate. All compounds were tested in triplicate.
Permeability
A parallel artificial membrane permeability assay (PAMPA) was conducted on a Biomek FX lab automation workstation (Beckman Coulter, Inc., Fullerton, CA) with PAMPA evolution 96 command software (pION Inc., Woburn, MA) as follows: 3 μL of 10 μM test compound stock was mixed with 600 μL of system solution buffer, pH 7.4 or 4 (pION Inc., Woburn, MA) to make diluted test compound. Then 150 μL of diluted test compound was transferred to a UV plate (pION Inc., Woburn, MA), and the UV spectrum was read as the reference plate. The membrane on a preloaded PAMPA sandwich (pION Inc., Woburn, MA) was painted with 4 μL of GIT lipid (pION Inc., Woburn, MA). The acceptor chamber was then filled with 200 μL of acceptor solution buffer (pION Inc., Woburn, MA), and the donor chamber was filled with 180 μL of diluted test compound. The PAMPA sandwich was assembled, placed on the Gut-Box controlled environment chamber and stirred for 30 min. The aqueous boundary layer was set to 40 μm for stirring. The UV spectrum (250–500 nm) of the donor and the acceptor were read. The permeability coefficient was calculated using PAMPA Evolution 96 Command software (pION Inc., Woburn, MA) based on the AUC of the reference plate, the donor plate, and the acceptor plate. All compounds were tested in triplicate.
Cytotoxicity screens
Cytotoxicity was tested on BJ, Raji, HEK 293, and HepG2 cell lines using the Alamar blue assay as described previously.38 The final concentration of the tested compounds was 27.5 μM in 0.5% DMSO. The cell viability was normalized to a DMSO control.
Antibacterial screens
E. coli BL21(DE3) competent cells and 1 mL of pre-warmed SOB medium were shaken at 300 rpm at 37 °C for 1 h. Cells were plated on prewarmed LB-agar selective plates and cultured at 37 °C overnight. Isolated colonies were diluted in LB broth and grown in broth for 4 h at 37 °C to an absorbance of ~0.6 at 600 OD. Cultures were further diluted by a 10-fold addition of broth. Then 196 μL of cultures and 4 μL of the test compound in DMSO were added into each well of a 96-well plate. Assay plates were incubated at 37 °C for 18 h. The positive control drug was ciprofloxacin.
Supplementary Material
Figure 1.

Reference compounds
Figure 3.

Activity and physicochemical property summary sorted by therapeutic index. Antimalarial activity is represented as the EC50 value in green, with darker squares indicating higher potency. Cytotoxicity data is represented as the percentage cell death relative to untreated control at a fixed 27.5 μM screening concentration in orange, with lighter squares indicating more cell death. The therapeutic index is represented as a ratio of the cytotoxicity value in HEK 293 cells to the EC50 in the K1 strain in blue, with darker squares indicating higher potency (LD50 in HEK 293 cells applied to estimate the therapeutic index.). Permeability (PAMPA) at pH 7.4 is represented in purple, with darker squares indicating higher permeability. Solubility is represented in yellow, with darker squares indicating higher solubility.
Table 4.
Summary of Series IV compounds: antimalarial activity on CQ-resistant strain K1 and CQ-sensitive strain 3D7, solubility, and permeability across an artificial membrane (PAMPA)
 | |||||
|---|---|---|---|---|---|
| Compound | Aryl group | EC50 (μM)a K1 strain | EC50 (μM)a 3D7 strain | Solubility (μM)b | Permeability (10−6 cm/s)c |
| 8 | m,p-methylenedioxy-phenyl | 3.01±0.08 | 2.81±0.11 | >100 | 47±7 |
| 32a | phenyl | >10 | >10 | >100 | 37±1 |
| 32c | m-MeO-phenyl | 7.98±3.09 | 9.94±0.13 | >100 | 64±13 |
| 32d | m-Me-phenyl | 9.60±2.00 | >10 | >100 | 126±14 |
| 32e | m-CF3-phenyl | >10 | >10 | >100 | 377±33 |
| 32f | m-Cl-phenyl | >10 | >10 | >100 | 196±38 |
| 32g | m-F-phenyl | >10 | >10 | >100 | 79±12 |
| 32h | m-NO2-phenyl | >10 | >10 | 49±2 | 110±6d |
| 32i | p-MeO-phenyl | >10 | 8.80±2.00 | >100 | 78±4 |
| 32j | p-tBu-phenyl | >10 | >10 | 1±0 | 1143±56 |
| 32k | p-CF3-phenyl | >10 | >10 | 22±3 | 457±72 |
| 32l | p-Cl-phenyl | >10 | >10 | >100 | 211±15 |
| 32m | m,p-diCl-phenyl | >10 | >10 | 39±5 | 781±77 |
| 32n | o-Me-phenyl | >10 | >10 | >100 | 92±13 |
| 32o | o-Cl-phenyl | >10 | >10 | >100 | 112±28 |
EC50 values are reported as the mean ± SD of at least two independent experiments. Positive controls were Mefloquine (EC50 in K1: 0.04±0.02 μM; EC50 in 3D7: 0.16±0.02 μM) and Chloroquine (EC50 in K1: 1.61±0.30 μM; EC50 in 3D7: 0.09±0.02 μM).
Solubility in PBS buffer, pH 7.4, containing 5% DMSO. The detection limit of the solubility assay was 100 μM. Two standards were tested in the same assay: albendazole (3±0.1 μM, poorly soluble) and carbamazepine (>100 μM, highly soluble).
Permeability at pH 7.4. Three standards were tested in the same assay: ranitidine HCl (1.2±0.7×10−6 cm/s, poorly permeable), carbamazepine (145±13×10−6 cm/s, moderately permeable), and verapamil HCl (1960±290×10−6 cm/s, highly permeable).
Permeability was measured at pH 4.0. Two standards were tested in the same assay: ranitidine HCl (0, poorly permeable) and carbamazepine (143±7×10−6 cm/s, moderately permeable).
Acknowledgments
We thank Jean-Marc Paris and Allan Reitz for helpful discussions during the course of these studies. This work was funded by World Health Organization grant 180738010, the American Lebanese Syrian Associated Charities (ALSAC), and St. Jude Children’s Research Hospital.
Abbreviations
- TDR
Special Programme for Research and Training in Tropical Diseases
- CQ-R
chloroquine-resistant
- CQ-S
chloroquine-sensitive
- MDR
multi-drug-resistant
- PAMPA
parallel artificial membrane permeability assay
- TI
therapeutic index
- NOE
nuclear Overhauser effect
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sachs J, Malaney P. Nature. 2002;415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 2.Chiba P, Burghofer S, Richter E, Tell B, Moser A, Ecker G. J Med Chem. 1995;38(14):2789–93. doi: 10.1021/jm00014a031. [DOI] [PubMed] [Google Scholar]
- 3.Wellems TE, Plowe CV. J Infect Dis. 2001;184(6):770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]
- 4.Sidhu ABS, Verdier-Pinard D, Fidock DA. Science. 2002;298:5591. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyde JE. Trends Parasitol. 2005;21(11):494–498. doi: 10.1016/j.pt.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridley RG. Nature. 2002;415(6872):686–693. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- 7.Wiesner J, Ortmann R, Jomaa H, Schlitzer M. Angew Chem Int Ed. 2003;42(43):5274–5293. doi: 10.1002/anie.200200569. [DOI] [PubMed] [Google Scholar]
- 8.Schlitzer M. ChemMedChem. 2007;2(7):944–986. doi: 10.1002/cmdc.200600240. [DOI] [PubMed] [Google Scholar]
- 9.Stephen JML, Tonkin IM, Walker J. J Chem Soc. 1947:1034–1039. doi: 10.1039/jr9470001034. [DOI] [PubMed] [Google Scholar]
- 10.Coatey GR, Cooper WC. J Parasitol. 1948;34(4):275–289. [PubMed] [Google Scholar]
- 11.Casey AC. J Med Chem. 1974;17(2):255–256. doi: 10.1021/jm00248a030. [DOI] [PubMed] [Google Scholar]
- 12.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK. Exp Parasitol. 2008;118(4):487–497. doi: 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puri SK, Dutta GP. Trans R Soc Trop Med Hyg. 1990;84(6):759–760. doi: 10.1016/0035-9203(90)90066-n. [DOI] [PubMed] [Google Scholar]
- 14.Ryley JF, Peters W. Ann Trop Med Parasitol. 1970;64(2):209–222. doi: 10.1080/00034983.1970.11686683. [DOI] [PubMed] [Google Scholar]
- 15.Kesten SJ, Degnan MJ, Hung J, McNamara DJ, Ortwine DF, Uhlendorf SE, Werbel LM. J Med Chem. 1992;35(19):3429–3447. doi: 10.1021/jm00097a001. [DOI] [PubMed] [Google Scholar]
- 16.Berman J, Brown L, Miller R, Andersen SL, McGreevy P, Schuster BG, Ellis W, Ager A, Rossan R. Antimicrob Agents Chemother. 1994;38(8):1753–1756. doi: 10.1128/aac.38.8.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorn A, Scovill JP, Ellis WY, Matile H, Ridley RG, Vennerstrom JL. Am J Trop Med Hyg. 2001;65(1):19–20. doi: 10.4269/ajtmh.2001.65.19. [DOI] [PubMed] [Google Scholar]
- 18.Biagini GA, Fisher N, Berry N, Stocks PA, Brigitte Meunier, Williams DP, Bonar-Law R, Bray PG, Owen A, O’Neill PM, Ward SA. Mol Pharmacol. 2008;73(5) doi: 10.1124/mol.108.045120. [DOI] [PubMed] [Google Scholar]
- 19.Kerns EH, Di L. Drug-like properties: concepts, ctructure design and methods: from ADME to toxicity optimization. Elsevier; Oxford: 2008. [Google Scholar]
- 20.Mitscher LA. Chem Rev. 2005;105(2):559–592. doi: 10.1021/cr030101q. [DOI] [PubMed] [Google Scholar]
- 21.Gould RG, Jacobs WA. J Am Chem Soc. 1939;61(10):2890–2895. [Google Scholar]
- 22.Santo RD, Costi R, Roux A, Miele G, Crucitti GC, Iacovo A, Rosi F, Lavecchia A, Marinelli L, Giovanni CD, Novellino E, Palmisano L, Andreotti M, Amici R, Galluzzo CM, Nencioni L, Palamara AT, Pommier Y, Marchand C. J Med Chem. 2008;51(15):4744–4750. doi: 10.1021/jm8001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucero BdA, Gomes CRB, Frugulhetti ICdPP, Faro LcV, Alvarenga L, Souza MClBVd, Souza TMLd, Ferreira VF. Bioorg Med Chem Lett. 2006;16:1010–1013. doi: 10.1016/j.bmcl.2005.10.111. [DOI] [PubMed] [Google Scholar]
- 24.Hadida Ruah SS, Hazlewood AR, Grootenhuis PDJ, Van Goor FF, Singh AK, Zhou J, McCartney J. 2006002421. WO. 2005
- 25.Cruz Adl, Elguero J, Goya P, Martínez A, Pfleiderer W. Tetrahedron. 1992;48(29):6135–6150. [Google Scholar]
- 26.Mphahlele MJ, Fernandes MA, El-Nahas AM, Ottosson H, Ndlovu SM, Sithole HM, Dladla BS, Waal DD. J Chem Soc, Perkin Trans 2. 2002;(12):2159–2164. [Google Scholar]
- 27.Geometry optimization for the enol 23b and keto 23b were performed using PM3 with the Gaussian 03 program. The keto 23b form was relatively 2.9 kcal/mol more stable than the enol 23b.
- 28.Volle JN, Mävers U, Schlosser M. Eur J Org Chem. 2008;(14):2430–2438. [Google Scholar]
- 29.Madrid PB, Wilson NT, DeRisi JL, Guy RK. J Comb Chem. 2004;6(3):437–442. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.N-ethylated 7 was prepared by the treatment of 7 with ethyl iodide and potassium carbonate in DMF.
- 31.Kohlbrenner WE, Wideburg N, Weigl D, Saldivar A, Chu DT. Antimicrob Agents Chemother. 1992;36(1):81–86. doi: 10.1128/aac.36.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wentland MP, Lesher GY, Reuman M, Gruett MD, Singh B, Aldous SC, Dorff PH, Rake JB, Coughlin SA. J Med Chem. 1993;36(19):2801–2809. doi: 10.1021/jm00071a010. [DOI] [PubMed] [Google Scholar]
- 33.Albertini S, Chetelat AA, Miller B, Muster W, Pujadas E, Strobel R, Gocke E. Mutagenesis. 1995;10(4):343–351. doi: 10.1093/mutage/10.4.343. [DOI] [PubMed] [Google Scholar]
- 34.Divo AA, Sartorelli AC, Patton CL, Bia FJ. Antimicrob Agents Chemother. 1988;32:1182–1186. doi: 10.1128/aac.32.8.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubar F, Anquetin G, Pradines B, Dive D, Khalife J, Biot C. J Med Chem. 2009;52:7954–7957. doi: 10.1021/jm901357n. [DOI] [PubMed] [Google Scholar]
- 36.Trager W, Jensen JB. Science. 1976;193(4254):673–5. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 37.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Antimicrob Agents Chemother. 2004;48(5):1803–6. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mallari JP, Shelat AA, Obrien T, Caffrey CR, Kosinski A, Connelly M, Harbut M, Greenbaum D, McKerrow JH, Guy RK. J Med Chem. 2008;51(3):545–552. doi: 10.1021/jm070760l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.