

Abstract
Background and purpose:
Neuronal ion channels are key targets of general anaesthetics and alcohol, and binding of these drugs to pre-existing and relatively specific sites is thought to alter channel gating. However, the underlying molecular mechanisms of this action are still poorly understood. Here, we investigated the neuronal Shaw2 voltage-gated K+ (Kv) channel to ask whether the inhalational anaesthetic halothane and n-alcohols share a binding site near the activation gate of the channel.
Experimental approach:
Focusing on activation gate mutations that affect channel modulation by n-alcohols, we investigated n-alcohol-sensitive and n-alcohol-resistant Kv channels heterologously expressed in Xenopus oocytes to probe the functional modulation by externally applied halothane using two-electrode voltage clamping and a gas-tight perfusion system.
Key results:
Shaw2 Kv channels are reversibly inhibited by halothane in a dose-dependent and saturable manner (K0.5= 400 µM; nH= 1.2). Also, discrete mutations in the channel's S4S5 linker are sufficient to reduce or confer inhibition by halothane (Shaw2-T330L and Kv3.4-G371I/T378A respectively). Furthermore, a point mutation in the S6 segment of Shaw2 (P410A) converted the halothane-induced inhibition into halothane-induced potentiation. Lastly, the inhibition resulting from the co-application of n-butanol and halothane is consistent with the presence of overlapping binding sites for these drugs and weak binding cooperativity.
Conclusions and implications:
These observations strongly support a molecular model of a general anaesthetic binding site in the Shaw2 Kv channel. This site may involve the amphiphilic interface between the S4S5 linker and the S6 segment, which plays a pivotal role in Kv channel activation.
Keywords: general anaesthesia, anaesthetic binding site, potassium channel, S4S5 linker, BKCa channel activation gating, global kinetic modelling
Introduction
The notion of relatively specific alcohol and inhalational anaesthetic binding sites in ion channels and other proteins is generally regarded as the most viable hypothesis to explain the relevant actions of these drugs in the nervous system (Dilger, 2002; Hemmings et al., 2005; Franks, 2006, 2008; Forman and Chin, 2008; Harris et al., 2008; Urban, 2008). Among several candidates, the ion channel targets (nomenclature follows Alexander et al., 2008) include ionotropic receptors (GABAA, glycine, 5-HT, NMDA, etc.) and K+ channels (GIRK, BKCa, two-pore and Shaw2). The presumed occupancy of these sites induces inhibition or potentiation of ion channel function, and multiple studies have identified discrete regions, which may contribute to putative alcohol and inhalational anaesthetic binding sites within ion channel proteins (Franks, 2008; Harris et al., 2008). However, a direct link between the modulation of a particular ion channel and the clinical end points of anaesthesia and alcohol intoxication is not firmly established. Nevertheless, the study of ion channels as targets of alcohol and general anaesthetics has helped to determine the basic molecular features of the putative binding sites, and formulate sound hypotheses about the mechanisms of action. Because alcohol and general anaesthetics are typically ‘low-affinity’ drugs sharing pharmacological and physical–chemical properties, a key question is whether a common mechanism governs the modulation ion channels by alcohol and inhalational anaesthetics. Similar amphiphilic interactions are presumably involved in the binding of these drugs to proteins (Eckenhoff and Johansson, 1997; Kruse et al., 2003; Urban, 2008), and therefore, whether the binding sites overlap or co-exist within the same ion channel molecule is a relevant problem.
From the biophysical and molecular perspectives, one of the best understood K+ channel targets of n-alcohols is the Shaw2 channel from Drosophila melanogaster. This channel is related to voltage-gated K+ channels (Kv channels) in the Shaker superfamily, and is selectively modulated (inhibited and potentiated) by n-alcohols through putative amphiphilic interactions with discrete components of the activation gate (Shahidullah et al., 2003; Covarrubias et al., 2005; Bhattacharji et al., 2006). Critical determinants of the putative binding site include the cytoplasmic S4S5 linker and the S6-b segment. Structural models based on current crystal structures of Kv channels suggest that the n-alcohol site in the Shaw2 channels may reside in the interface between these regions, which typically adopt α-helical structures (Covarrubias et al., 2005). In light of this body of knowledge, we asked whether the inhalational anaesthetic halothane may also modulate the Shaw2 channel by interacting with the putative n-alcohol site. To answer this question, we expressed wild-type and mutant Shaw2 channels in Xenopus oocytes, and applied two-electrode voltage clamping to investigate the resulting K+ currents in the absence and presence of halothane and a typical n-alcohol, 1-butanol. The results strongly support the presence of a shared binding site and mechanism of action for n-alcohols and halothane in the Shaw2 channel. We discuss the implications of these findings in terms of the general structural features and amphiphilic character of related protein sites in other ion channels and soluble proteins. In light of recent studies suggesting relevant modulation of other voltage-gated ion channels by general anaesthetics (Na+ and HCN channels) (Shiraishi and Harris, 2004; Chen et al., 2005; Hemmings et al., 2005; Ying et al., 2006; Horishita and Harris, 2008; Chen et al., 2009), studying the Shaw2 channel gains further relevance.
Methods
Preparation of oocytes
All animal care and experimental procedures involving Xenopus laevis frogs were carried out according to a protocol approved by the IACUC of Thomas Jefferson University. The Shaw2 and Kv3.4 cDNAs were maintained as reported previously (Covarrubias et al., 1995; Harris et al., 2000; 2003; Bhattacharji et al., 2006). The mutagenesis kit QuickChange (Stratagene, La Jolla, CA, USA) was used to create all mutations, which were verified by automated DNA sequencing (Kimmel Cancer Center, Thomas Jefferson University). For expression in Xenopus oocytes, mRNA was synthesized in vitro as described previously (Harris et al., 2000; 2003;).
To enhance resolution, the Shaw2-F335A mutant was used for the pharmacological characterization of the halothane modulation (Figures 1, 2B and 5). This mutant expresses more robustly than the Shaw2 wild-type, and exhibits mostly unaltered biophysical and pharmacological properties (Harris et al., 2003; Shahidullah et al., 2003). However, the Hill coefficient for 1-butanol is increased (Supporting Information Fig. S1B).
Figure 1.

Dose dependence of the inhibition of Shaw2 Kv channels by halothane. (A) Whole-oocyte Shaw2-F335 currents evoked by step depolarizations from −100 to +60 mV delivered at 30 s intervals. Several superimposed traces are shown before adding halothane, in the presence of 0.46 mM halothane and after washout (C, H and W respectively). The corresponding time-course of the experiment is shown below the traces. The horizontal black bar indicates the exposure of the oocyte to the indicated concentration of halothane. (B) Semilogarithmic halothane dose–response curve. Each point shown is the mean of 3–12 independent determinations. The Hill equation best-fit parameters are shown inside the graph, and the inset displays the dose–response curve on linear scales.
Figure 2.
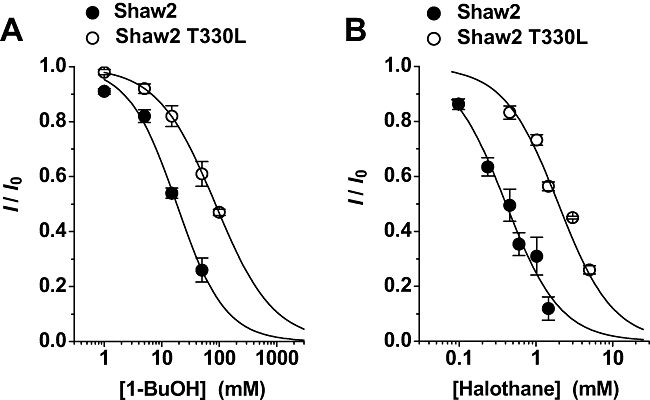
Threonine 330 in the S4S5 linker of the Shaw2 channel is a determinant of 1-butanol (1-BuOH) and halothane action. (A) and (B) Semilogarithmic dose–response curve for 1-butanol and halothane. Each graph displays the effect of the Shaw2 T330L mutation on the parameters of the dose–response curves. Individual symbols in (A) represent the means of two to six (Shaw2) and three to four (Shaw2 T330L) independent determinations, and the symbols in (B) represent the means of 3–12 (Shaw2, replotted from Figure 1) and four to eight (Shaw2 T330L) independent determinations. The Hill equation best-fit parameters in (A) were: K0.5 = 18 mM, nH = 1.1 (Shaw2) and K0.5 = 86 mM, nH = 0.9 (Shaw2 T330L); and the best-fit parameters in (B) were: K0.5 = 0.4 mM, nH = 1.2 (Shaw2) and K0.5 = 2 mM, nH = 1.2 (Shaw2 T330L). The observed rightward shifts induced by the T330L mutation correspond to virtually identical binding free energy changes (ΔΔG = 920 and 940 cal·mol−1 in (A) and (B) respectively).
Figure 5.

Competition between halothane and 1-butanol (BuOH) for overlapping binding sites in the Shaw2 channel. All complementary competition experiments were conducted and analysed as explained under Methods and Results. (A) Halothane dose–response curves in the presence of various fixed concentrations of 1-butanol as indicated. In each curve, symbols are the means of 3–12 (0 mM BuOH), 3–16 (5 mM BuOH), 5–26 (18 mM BuOH) and 3–14 (30 mM BuOH) independent determinations. (B) Dose–response curves for 1-butanol in the presence of various fixed concentrations of halothane as indicated in the graph. In each curve, symbols are the means of 2–5 (0 µM halothane), 3–10 (100 µM halothane), 3–13 (400 µM halothane) and 3–14 (1000 µM halothane) independent determinations. In both graphs, the error bars represent the standard deviations of the samples. The solid lines in (A) and (B) are the best global fits assuming Scheme 3. The best-fit values of the equilibrium dissociation constants and the corresponding χ2 of the global fits are shown with the scheme below the graphs.
Electrophysiology
Two to five days after the injection of the appropriate mRNA into defolliculated oocytes, the two-electrode voltage clamp method was used to record the expressed whole-oocyte currents in normal extracellular bath solution according to established procedures (Covarrubias et al., 1995; Jerng et al., 1999; Harris et al., 2000; 2003;). The program pClamp 8–9 (Axon Instruments and Molecular Devices, Sunnyvale, CA, USA) was used for acquisition, data reduction and initial analysis of the recorded currents. Generally, macroscopic currents were low-passed filtered at 0.5–1 kHz and digitized at 1–2 kHz. Leak current was subtracted off-line by assuming a linear leak. All recordings were obtained at room temperature (23 ± 1°C).
Halothane and isoflurane were delivered through Teflon tubing directly into the recording chamber via a 50 mL Hamilton Gastight Syringe (Hamilton Company, Reno, NV, USA), and 1-butanol was delivered into the recording chamber by gravity as reported previously (Harris et al., 2000; 2003; Bhattacharji et al., 2006). For competition experiments, the Shaw2 current level in control bath solution (no drug) was first established, and, subsequently, the oocyte was equilibrated with a given concentration of 1-butanol (0, 5, 18 and 30 mM). Then, still in the presence of 1-butanol and once a new reduced and stable level of current was reached, the oocyte was additionally exposed to a given concentration of halothane (typically, 98, 240, 460 or 1020 µM). The converse complementary experiment was conducted in the same manner, except that individual oocytes were first equilibrated with various doses of halothane (0, 100, 400 and 1000 µM), and subsequently, still in the presence of halothane, challenged with increasing doses of 1-butanol (1, 5, 10, 25 and 50 mM).
Data analysis
Curve fitting, data display and analysis were carried out in Origin 7.5 and OriginPro 8.0 (OriginLab, Northampton, MA, USA). Unless stated otherwise, all results are reported as mean ± SEM. One-way anova was used to evaluate the statistical significance of apparent differences. Dose–response curves were characterized as described previously (Covarrubias et al., 1995; Harris et al., 2000; Shahidullah et al., 2003). Briefly, the normalized equilibrium dose–inhibition curves (the inhibited current I relative to the control current I0 observed in the absence of drug) were empirically described by assuming this form of the Hill equation:
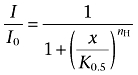 |
where x is the drug concentration, K0.5 is the drug concentration that induces 50% inhibition and nH is the index of cooperativity or Hill coefficient. To characterize the interactions between halothane and 1-butanol at equilibrium, we made assumptions based on the outcomes of empirical analyses (Results), and applied non-linear curve fitting in OriginPro 8.0 to test the following explicit schemes:
Scheme 1
 |
Scheme 2
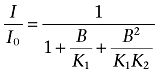 |
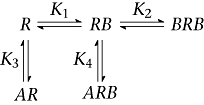
Scheme 3
 |
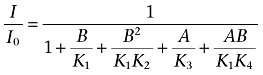 |
R, A and B represent the channel as the drug receptor, halothane and 1-butanol, respectively; and K, K1, K2, K3 and K4 are the associated equilibrium dissociation constants. The corresponding equations below the respective scheme calculate the free receptor concentration (R0) with respect to the concentrations of all species (R) in the scheme at equilibrium (R0/R=I/I0), where A and B are the concentrations of halothane and 1-butanol respectively. Thus, all bound species contribute to the pharmacological response. Quantitative global modelling assuming Scheme 3 evaluated all data points in a complementary competition experiment simultaneously (Figure 5). Typical data sets included >20 independent data points distributed in four distinct dose–response curves.
Materials
Halothane and isoflurane were purchased from Halocarbon Laboratories (River Edge, NJ, USA), and dilutions were prepared fresh before an experiment. Using a Hamilton Gastight Syringe, 3 mL of halothane was thoroughly mixed with 37 mL of ND-96 (recording bath solution, see above) in a gastight glass screw-top vial (penetrable Tuf-Bond Teflon septa, Thermo Scientific, Rockford, IL, USA), and allowed to equilibrate overnight at room temperature to a final concentration of 17.5 mM. The resulting stock solution and all necessary dilutions were held sealed in gastight vials. Halothane concentrations were determined by HPLC (Dr Qing Chen Meng, Department of Anaesthesiology and Critical Care, University of Pennsylvania). HPLC-grade 1-butanol was purchased from Fisher Scientific (Hampton, NH, USA). Isoflurane dilutions were prepared and handled similarly. All 1-butanol dilutions were also made fresh before an experiment.
Results
Fast, reversible and saturable inhibition of Shaw2 channels by halothane
An earlier report suggested that the inhalational anaesthetic halothane may directly modulate Shaw2 channels (Covarrubias and Rubin, 1993). However, no further studies have been conducted to support and characterize this finding. To investigate whether halothane may interact with a discrete site in Shaw2 channels, we carried out a systematic study of the interaction between halothane and Shaw2 channels heterologously expressed in Xenopus oocytes. Pre-equilibrated solutions of halothane were prepared as described, and concentrations were determined by HPLC. To test the modulation of the Shaw2 channels by halothane, outward currents were evoked by repetitive 400 ms step depolarizations to +60 mV (30 s, start-to-start). Once the current remained at a stable level in the control bath solution, the oocyte was exposed to halothane by perfusing the bath with a solution containing the desired concentration, and the exposure was continued until the current reached a new stable level (Figure 1). Halothane (0.46 mM) rapidly inhibited the current by ∼50% (within the start-to-start interval), and this response was also rapidly reversible upon washout (Figure 1A and B). The inhibition by isoflurane is qualitatively similar to that induced by halothane (Supporting Information Fig. S2). Furthermore, the inhibition of Shaw2 channels by halothane was concentration dependent and exhibited saturable behaviour (Figure 1C). A halothane concentration of ∼1 MAC (0.25 mM) inhibited the current by ∼35%, and the dose–response curve was well described by the Hill equation (Methods) with the following best-fit parameters: K0.5= 0.4 mM and nH= 1.2. These results indicate that Shaw2 channels were inhibited by relevant concentrations of halothane and were consistent with the presence of a discrete protein site for inhalational anaesthetics. Thus, the inhibition of Shaw2 channels by halothane and n-alcohols is qualitatively similar, albeit halothane is a more potent general anaesthetic (Covarrubias et al., 1995). Based on our earlier studies, we hypothesized that these agents share a site involving the S4S5 linker and S6-b segment of the Shaw2 channel. To test this hypothesis, we investigated two key aspects of the modulation: (i) the molecular determinants; and (ii) the interaction between halothane and 1-butanol.
Modulations by halothane and n-alcohols share molecular determinants in Shaw2 channels
Previously, we established that the S4S5 linker and the S6 segment are key determinants of the modulation of Shaw2 channels by n-alcohols (Covarrubias et al., 1995; 2005; Harris et al., 2000; 2003; Shahidullah et al., 2003; Bhattacharji et al., 2006). To investigate whether the inhibition of Shaw2 channels by halothane also involves these regions, we tested mutations in the S4S5 linker and the S6 segment. At the C-terminal end of the S4S5 linker, threonine 330 (T330) is found in the Shaw2 protein, while leucine occupies the equivalent position (L382) in the Kv3.4 channel, which is resistant to n-alcohols (Covarrubias et al., 1995; Harris et al., 2000). Demonstrating the particular importance of T330, the Shaw2-T330L mutant exhibits rightward-shifted dose–response curves for halothane and 1-butanol (Figure 2), but displays normal voltage-dependent activation and kinetics (Supporting Information Fig. S3). Another mutation, T330G, produced qualitatively similar results (data not shown). Thus, in agreement with the hypothesis and previous results, point mutations at a critical site in the S4S5 linker of the Shaw2 channel are sufficient to decrease its inhibition by general anaesthetics. Notably, the K0.5 for both drugs is increased ∼5-fold by the T330L mutation (Figure 2), indicating a similar energetic effect on halothane and 1-butanol binding (Figure 2 legend).
Given that the Kv3.4 S4S5 linker may exhibit a relatively low α-helical propensity (Bhattacharji et al., 2006), the corresponding converse mutation in Kv3.4 (L382T) alone failed to increase the sensitivity of the channel to 1-butanol (not shown). Thus, we asked whether other discrete S4S5 linker mutations in the n-alcohol-resistant Kv3.4 channel may conversely confer modulation by halothane. In previous studies, we observed that the double mutation G371I/T378A in the Kv3.4 protein confers substantial inhibition of the channel by n-alcohols (Covarrubias et al., 1995; Harris et al., 2000). These mutations partially convert the S4S5 linker of Kv3.4 into that of Shaw2 and promote α-helicity in this linker (Bhattacharji et al., 2006). We found that wild-type Kv3.4 channel is inhibited only 2 and 8% by 0.46 and 1 mM halothane, respectively (Figure 3). By contrast, resembling its increase in sensitivity to n-alcohols, the double mutant Kv3.4 G371I/T378A exhibits significantly greater inhibition by halothane (18 and 36% at 0.46 and 1 mM, respectively) (Figure 3). As previously, we also tested a mutant lacking the inactivation domain (first 28 residues of the channel subunit) to probe the significance of fast inactivation on the insensitivity of the Kv3.4 channel to halothane (Harris et al., 2000; Bhattacharji et al., 2006). The ΔN(2-28)-Kv3.4 mutant exhibited a halothane sensitivity marginally stronger than that of wild-type Kv3.4 and little or no dose dependence (∼10% inhibition; Figure 3). ΔN(2-28)-Kv3.4 G371I/T378A, however, induced currents that were too small for further investigation. Nevertheless, it is clear that the G371I/T378A mutation in the S4S5 linker enhances the inhibition of the inactivating Kv3.4 channel by halothane substantially, which supports the proposed hypothesis.
Figure 3.

Discrete mutations in the S4S5 linker of the Kv3.4 channel confer modulation by halothane. (A–C) Whole-oocyte Kv3.4 currents evoked by step depolarizations from −100 to +60 mV delivered at 30 s intervals. Several superimposed traces are shown before adding halothane and in the presence of 1 mM halothane (C and H respectively). (D) and (E) Bar graphs summarizing the mean values of the fractional inhibition of wild-type Kv3.4 and Kv3.4 G371I/T378A, and ΔN-Kv3.4 by 0.46 and 1 mM halothane. From left to right, the number of independent determinations was: 5, 4, 4 (D) and 5, 3, 3 (E). Relative to Kv3.4 WT, the double Kv3.4 mutant increases the halothane-induced inhibition by ∼9-fold and ∼4.5-fold at 0.46 and 1 mM halothane respectively. This enhanced inhibition of the Kv3.4 G371I/T378A channel is statistically significant at low and high doses of halothane (*P= 4 × 10−5 and <1 × 10−6 respectively). The small inhibition of the ΔN-Kv3.4 channel (∼10%) exhibits no apparent dose dependence (P= 0.5), but is slightly stronger than that of the Kv3.4 WT (†P= 2.5 × 10−3 and 4.3 × 10−4, at 0.46 and 1 mM halothane respectively).
The P410A mutation in the distal region of the S6 segment is known to switch the response of Shaw2 channels to n-alcohols from inhibition to potentiation (Harris et al., 2003). Thus, the shared-site hypothesis predicts that halothane should also potentiate the outward currents induced by the Shaw2 P410A mutant. Accordingly, and in sharp contrast to the consistent inhibitory responses of the Shaw2 channel to halothane, application of the inhalational anaesthetic to the Shaw2 P410A mutant induced substantial dose-dependent and reversible potentiation of the outward currents (Figure 4). Overall, the results from the mutational and functional analyses strongly suggest that n-alcohols and halothane share molecular determinants within regions typically involved in activation gating of Kv channels.
Figure 4.

Conversion of the halothane-induced inhibition into potentiation by a single substitution in the S6 PVPV motif of the Shaw2 channel. (A) Whole-oocyte Shaw2 P410A currents evoked by step depolarizations from −100 to +60 mV delivered at 30 s intervals. Several superimposed traces are shown before adding halothane, in the presence of 0.46 or 1 mM halothane and after washout (C, H and W respectively). (B) Bar graph summarizing the mean % change induced by two concentrations of halothane (0.46 or 1 mM). Negative and positive changes correspond to inhibition and potentiation respectively. From left to right, the number of independent determinations was: 6, 8, 8 and 9.
Halothane and n-alcohols compete for shared sites in the Shaw2 channel
To test whether n-alcohols and halothane may interact with overlapping sites in the Shaw2 channel, we conducted competition experiments under two separate, but complementary conditions, and applied modelling and constrained global curve fitting to analyse the results. Shaw2 channels expressed in Xenopus oocytes were either equilibrated with constant subsaturating doses of 1-butanol, and titrated at equilibrium with variable doses of halothane (Figure 5A), or they were equilibrated with constant subsaturating doses of halothane and titrated at equilibrium with variable doses of 1-butanol (Figure 5B). Control dose–response curves (no equilibration with constant subsaturating doses) were included in each data set to constrain the modelling and account for oocyte-dependent seasonal variations. In both instances, increasing doses of halothane or 1-butanol induced additional inhibition of the Shaw2 current, which could result from direct competition between the two drugs for free sites in the channel. To test this possibility more systematically, we applied the following quantitative modelling strategy. Because the empirical analysis of the halothane dose–response curve indicated an nH∼1 (Figure 1; Supporting Information Fig. S1A), we hypothesized that the inhibition induced by halothane may result from binding to a single site (Scheme 1). Accordingly, Scheme 1 yielded similar excellent best-fits to two separate data sets (Supporting Information Fig. S1C). By contrast, the empirical analysis indicated that the inhibition of Shaw2 channels by 1-butanol is more commonly associated with an nH ranging between 1.5 and 2 (Supporting Information Fig. S1B), and therefore, we hypothesized that the inhibition induced by 1-butanol may result from cooperative sequential binding to two sites (Scheme 2; Supporting Information Fig. S1D). Supporting this hypothesis, Scheme 2 also yielded excellent best-fits to two separate data sets with modestly different K0.5 (Supporting Information Fig. S1B; K1 > K2).
To characterize the complementary competition experiments, we combined Schemes 1 and 2, and formulated a new working hypothesis (Scheme 3). Scheme 3 assumes a strict competition between halothane and 1-butanol for the unoccupied (R) and mono-liganded channels (BR), and allows formation of the ternary complex BRA with steric interactions. 1-Butanol cannot bind to a second site if halothane binds first forming RA, and cannot dissociate from the ternary complex until halothane dissociates. To carry out global fitting of all curves within a data set, we initialized the adjustable parameters by using the best-fit values of the equilibrium dissociation constants obtained from separate 1-butanol or halothane dose–response curves (above; K1, K2 and K3 in Scheme 3). Then, to constrain the fits, these values were either fixed, or two of them were allowed to vary during minimization to optimize the fit. K4 was typically a free parameter, but was initialized assuming K4=K3. Generally, this strategy produced global fits that were in very good overall agreement with the results of the complementary competition experiments (Figure 5). There are, however, some modest quantitative differences between the best-fit equilibrium dissociation constants from both data sets (Figure 5), namely, K1= 38 and 20 mM, and K4= 0.5 and 0.11 mM, which may reflect seasonal variability, as indicated by variation in the corresponding control 1-butanol dose–response curves (Supporting Information Fig. S1B). Nevertheless, Scheme 3 is a relatively simple viable working hypothesis that combines positive binding cooperativity, strict competition and steric hindrance to explain the crosstalk between halothane and 1-butanol at the general anaesthetic site(s) of the Shaw2 channel.
Although extensive kinetic modelling is outside the scope of this study, we also considered the schemes shown below.
Scheme 4
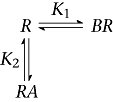 |
Scheme 5
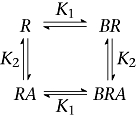 |
Scheme 4 assumes a mutually exclusive interaction with a binding site, and Scheme 5 additionally allows the formation of a ternary complex and may include binding cooperativity. Scheme 4 was able to describe the results in Figure 5A globally, but failed to account for the results of the complementary experiment. Scheme 5 also failed to return acceptable complementary global fits.
Discussion
The knowledge generated to date strongly supports the notion of an amphiphilic n-alcohol binding site involving the activation gate of the Shaw2 channel (Covarrubias and Rubin, 1993; Covarrubias et al., 1995, 2005; Harris et al., 2000, 2003; Shahidullah et al., 2003; Bhattacharji et al., 2006). Thus, given the apparent physical–chemical and molecular features of this site, it is relevant to ask whether inhalational general anaesthetics may also interact with it. Answering this question would open new opportunities to investigate the molecular mechanisms governing the actions of inhalational anaesthetics at the level of a discrete binding site in an ion channel with a relatively well-defined gating mechanism. The data reported here indicate convincingly that: (i) the modulations of Shaw2 channels by n-alcohols and halothane share molecular determinants in the S4S5 linker and the S6 segment; and (ii) n-alcohols and halothane interact competitively with overlapping sites and may co-exist in at the general anaesthetic binding site of the Shaw2 channel. Therefore, pharmacological, mutational and computational approaches converge to provide compelling evidence of a pharmacologically relevant, discrete site for inhalational anaesthetics in the Shaw2 channel. As proposed before for n-alcohols, this site may involve components of the channel's activation machinery.
Functional and molecular evidence of an inhalational anaesthetic site in Shaw2 channels
To support the presence of a relevant inhalational anaesthetic site in the Shaw2 channel, several critical criteria must be met, including: (i) pharmacological dose–response properties consistent with the presence of a relevant discrete site; (ii) selective perturbation of the anaesthetic's modulatory actions by discrete mutations in functionally relevant regions of the channel protein; and (iii) evidence of competitive interactions among general anaesthetics with similar pharmacological effects on the channel.
To evaluate these criteria, we characterized first the equilibrium dose–response relation of halothane, and determined that this compound inhibited the Shaw2 current rapidly and reversibly in a saturable dose-dependent manner that was described empirically by the Hill equation (Figure 1). Importantly, this analysis showed that halothane was a relatively potent modulator, which inhibited Shaw2 channels substantially at pharmacologically relevant concentrations (∼35% at ∼1 MAC). In light of these observations and our earlier studies with n-alcohols, the new data clearly suggest that Shaw2 channels bear a halothane site with pharmacological features that indicate a relevant interaction. Accordingly, the anaesthetic and inhibitory potencies of n-alcohols and halothane are directly correlated with a unity slope (Supporting Information Fig. S4). Furthermore, by testing critical amino acid substitutions within regions involved in activation gating of the channel (S4S5 linker and distal S6; Figures 2–4), we showed the parallel effects of discrete mutations on 1-butanol and halothane modulations (inhibition and potentiation). T330 is especially interesting because it corresponds to the T57 in the homologous n-alcohol binding site of the LUSH protein (Kruse et al., 2003). T57 is involved in critical H-bonding with n-alcohols (Thode et al., 2008).
Lastly, we assumed Schemes 1–3 and subjected the results of the complementary competition experiments to a quantitative global modelling strategy (Figure 5). By allowing strict competition for two sites that interact cooperatively when 1-butanol binds and steric hindrance introduced by halothane, Scheme 3 is an economic explanation of the interactions between 1-butanol and halothane at overlapping binding sites in the Shaw2 channel. In terms of a possible physical model, Schemes 1–3 suggest a difference between the modes of inhibition of Shaw2 channels by halothane and 1-butanol. Whereas binding of one halothane molecule to one subunit may be sufficient to inhibit channel gating (Figure 6), the inhibition by 1-butanol exhibits modest positive cooperativity involving binding to two sites, possibly in two subunits. Whether or not the latter is a feature exacerbated by the F335A mutation in Shaw2 channels with enhanced expression will require further investigation. Furthermore, the formation of ternary complexes (BRA= 1-butanol : channel : halothane) suggests that two anaesthetic molecules may occupy overlapping sites in a subunit. This overlap is in turn responsible for the apparent steric hindrance, which prevents binding of 1-butanol if halothane binds first and does not allow 1-butanol dissociation until halothane unbinds. Further studies will be necessary to test this model further. For instance, whether the overlap is present or not may depend on the n-alcohol's chain length.
Figure 6.
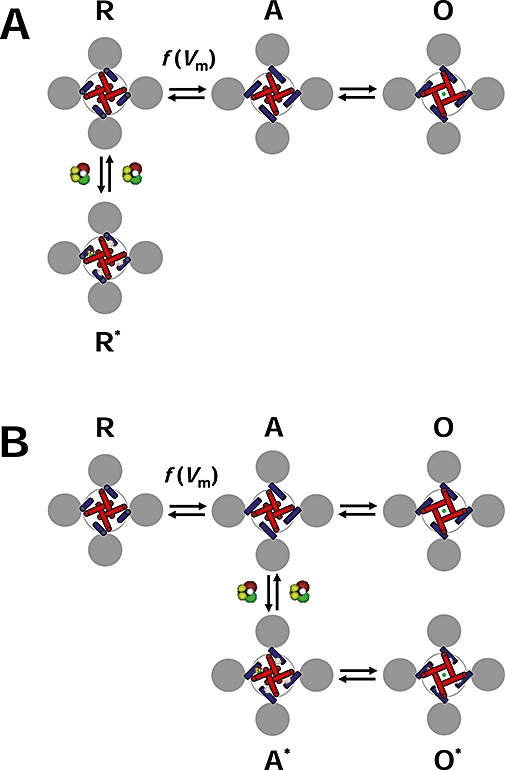
Diagram of the allosteric modulation of Shaw2 channels by halothane. (A) Working hypothesis of the inhibition of the Shaw2 channel by halothane. The drawing represents the tetrameric Shaw2 channel viewed from cytoplasmic side. The grey circles represent the voltage sensing domains (VSDs), the blue cylinders represent the S4S5 linkers that connect the VSDs to the pore domain (PD) and the red cylinders represent the S6 helix bundle that forms the activation gate of the K+ channel in the PD. The symbols R, A and O label three distinct conformations of the channel: resting-closed, pre-open activated and open respectively. Note that the voltage-dependent movement of the VSDs displaces the S4S5 linkers to activate the channel; and consequently, the S6 helix bundle undergoes a concerted conformational change to open the channel. The green circle represents K+ in the pore. Whereas channel activation is governed by voltage-dependent rate constants [f(Vm)], the final concerted opening step may only depend weakly on the membrane potential. To explain the inhibition of the Shaw2 channel by halothane, we hypothesize that it binds to the resting closed conformation R preferentially and stabilizes it (R*). The interfaces between the S4S5 linker and the distal S6 segment are the putative binding sites. Binding to one site is sufficient to stabilize the resting closed conformation. (B) Working hypothesis of the potentiation of the Shaw2 P410A channel by halothane. This diagram uses the same conventions as described above. When the P410A mutation in the PVPV motif reduces the kink in distal segment of S6, halothane may bind to the pre-open activated conformation (A), and thereby stabilizes it (A*). Consequently, channel opening (O*) is more likely than channel deactivation, and the current is potentiated. Although the local structural change induced by the P410A mutation may have changed the relationship between the S4S5 linker and distal S6, the interface between these regions may still constitute the binding site for halothane.
An allosteric model of general anaesthetic action in Shaw2 Kv channels
According to canonical models of Kv channel activation, a direct interaction between the S4S5 linker and the distal segment of S6 is necessary to couple the movement of the S4 voltage sensor to the opening of the internal activation gate (Lu et al., 2002; Tristani-Firouzi et al., 2002; Long et al., 2005b; Labro et al., 2008). Based on earlier work (Introduction) and the results from this study, we hypothesize that n-alcohols and inhalational anaesthetics bind to the amphiphilic interface between the S4S5 linker and the distal segment of S6 when the Shaw2 channel is in the resting closed conformation. Thus, these agents inhibit the channel by ‘jamming’ the electromechanical coupling between the voltage sensor and the activation gate, and thereby stabilizing the resting closed conformation of the channel (Figure 6A). This is a feasible scenario because inhalational anaesthetics and n-alcohols are thought to bind to pre-existing protein cavities or interfaces, which stabilize the targeted conformation of the protein (Eckenhoff and Johansson, 1997; Hemmings et al., 2005; Bucci et al., 2006; Franks, 2006; Urban, 2008).
To explain the potentiation of the Shaw2 P410A mutant by inhalational anaesthetics, a key consideration is the local structural effect of the mutation. The crystal structure of a Kv channel shows that prolines in the PVPV motif introduce a kink in the S6 segment (Long et al., 2005a; 2007;) (Figure 6A), and the P410A mutation may straighten the S6 segment (Figure 6B). In this situation, halothane may preferentially bind to the activated pre-open conformation of the channel. Halothane binding stabilizes this conformation and, consequently, channel opening is preferred over channel deactivation (Figure 6B). Potentiation of the Shaw2 current by halothane is the manifestation of this mechanism.
These working hypotheses are mainly based on the results from the mutational and functional analyses. Thus, a word of caution is necessary. Given the critical location of the amino acid substitutions that affect the modulation by halothane and n-alcohols, the mutations may impact channel conformations and gating, and thereby influence the channel's sensitivities to the drugs. From the available information, this possibility cannot be ruled out. However, we have shown previously that many activation gate mutations in Shaw2 do not appear to affect gating significantly, but they still affect the modulation by n-alcohols and halothane profoundly (Figures 2 and 4; Supporting Information Fig. S3) (Harris et al., 2000, 2003). Therefore, the tested residues are likely to make a clear contribution to the binding of general anaesthetic.
Inhalational anaesthetic sites in ion channels and model proteins
Allosteric modulation of Cys-loop neurotransmitter-gated receptors (GABA, glycine, 5-HT and acetylcholine) by inhalational anaesthetics and n-alcohols depends on homologous molecular determinants in the channel subunits (Hemmings et al., 2005; Franks, 2006, 2008; Harris et al., 2008). Therefore, it is hypothesized that these drugs occupy overlapping sites involving similar interactions and utilizing similar mechanisms of action. However, these relationships are less clear for other putative targets of inhalational anaesthetics and n-alcohols, such as two-pore K+ channels, NMDA receptor channels and GIRK channels. We propose that the halothane site in the activation gate of Shaw2 Kv channels is analogous to that of Cys-loop neurotransmitter-gated receptors, albeit, in contrast to GABA receptors, it may only accommodate small- and medium-size n-alcohols (Covarrubias et al., 1995). Moreover, similar sites may exist in topologically related voltage-gated ion channels (Na+ and HCN channels) that are also modulated by relevant concentrations of general anaesthetics (Shiraishi and Harris, 2004; Chen et al., 2005; 2009; Hemmings et al., 2005; Ying et al., 2006; Horishita and Harris, 2008). Gating of these channels also involves the S4S5 linker (McPhee et al., 1998; Chen et al., 2001; Prole and Yellen, 2006), which, as in Shaw2 channels, may be targeted by general anaesthetics.
The crystal structures of soluble model proteins bound to n-alcohols and general anaesthetics (intravenous and inhaled) have revealed molecular features and interactions, which are probably conserved in ion channels modulated by relevant concentrations of these drugs (Kruse et al., 2003; Liu et al., 2005; Franks, 2006, 2008; Strzalka et al., 2009; Vedula et al., 2009). Thus, the n-alcohol/halothane site in Shaw2 channels is likely to be a pre-existing water-filled cavity/interface formed by adjacent α-helical segments. In this amphiphilic site, a hydrophobic effect and weak polar interactions (van der Waals forces and H-bonds) govern drug binding (Shahidullah et al., 2003). The results of this study and previously published observations from our laboratory would strongly support this proposal.
Acknowledgments
This work was supported by a research grant from the National Institutes of Health, USA (R01 AA010615). Mr Nathan Klett was supported in part by a Summer Research Scholarship from Thomas Jefferson University. We thank Dr T. Ratanadilok Na-Phuket for his critical reading of the manuscript. Also, we thank Ms Annika Fitzpatrick for assisting with the construction of 1-butanol dose–response curves from Shaw2-F335A channels, Mr Brian Urbani for harvesting Xenopus oocytes, and all members of the Covarrubias lab for their constructive feedback and support.
Glossary
Abbreviations:
- BKCa
large conductance Ca++-activated K+ channel
- GIRK
G-protein-gated inward rectifying K+ channel
- HCN
hyperpolarization-activated, cyclic nucleotide-regulated cationic channel
- HPLC
high-performance liquid chromatography
- MAC
minimum alveolar concentration
- NMDA
N-methyl-d-aspartate
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Halothane and 1-BuOH dose–response curves of Shaw2 channels (wild-type and Shaw2 F335A). These curves were obtained as explained in the main body of the article (Methods and Results). (A) Halothane dose–inhibition relations of Shaw2 F335A. Filled and hollow symbols represent two data sets recorded at different times. The black and red lines are the best empirical Hill equation fits with the parameters (K0.5 and nH) indicated in the graph. Symbols and error bars correspond to mean ± SD (n = 3–14). (B) 1-BuOH dose–inhibition relations of Shaw2 F335A (filled and hollow circles) and wild-type Shaw2 (hollow diamonds). The two separate plots from Shaw2 F335A were taken at different times. Thus, the apparent shift may reflect oocyte seasonal variations, which have a greater impact on 1-BuOH sensitivity. The solid (red) and dashed (black) lines represent the best empirical Hill equation fits with the parameters indicated in the graph. Symbols and error bars correspond to mean ± SD (n = 2–29). The K0.5 and the nH are within the range of values previously reported by our group (Covarrubias et al., 1995; Harris et al., 2000; Shahidullah et al., 2003; Bhattacharji et al., 2006). (C) and (D) depict the same Shaw2 F335A results plotted in (A) and (B). In these plots, however, the solid lines are best-fits to explicit state diagrams depicted below the corresponding graphs. The best-fit parameters are indicated in the graphs.
Figure S2 Reversible inhibition of Shaw2 F335A by isoflurane. This experiment was conducted as described in the main text of the article and Figure 1 legend. Note that isoflurane also inhibits the Shaw2 channel effectively and in a manner similar to that induced by halothane.
Figure S3 Normalized whole-oocyte current–voltage relations of Shaw2 wild-type (WT) and two Shaw2 S4S5 linker mutants. The plots were normalized to the current amplitude at +70 mV. The symbols and bars correspond to the mean ± SE (n = 5, each channel type). The inset depicts typical families of current traces evoked by step depolarizations (−90 to +70 mV, 10 mV intervals). Note that the mutations do not significantly affect the kinetics and voltage dependence of the currents. However, both mutations decrease the inhibitory potency of the anaesthetics (main text).
Figure S4 Correlation between the anaesthetic and inhibitory potencies of n-alcohols and halothane. The inhibitory potencies refer to the modulation of the Shaw2 channel by these anaesthetics. The plotted values were obtained from previously published results and this study (Covarrubias et al., 1995; Shahidullah et al., 2003; Franks, 2006; Franks and Lieb, 1994). The line corresponds to the best-fit linear regression; the slope and r2 are indicated in the graph. Note the strong correlation between the anaesthetic potency and the potency of the drugs as inhibitors of the Shaw2 channel.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2008;153:2S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharji A, Kaplan B, Harris T, Qu X, Germann MW, Covarrubias M. The concerted contribution of the S4-S5 linker and the S6 segment to the modulation of a Kv channel by 1-alkanols. Mol Pharmacol. 2006;70:1542–1554. doi: 10.1124/mol.106.026187. [DOI] [PubMed] [Google Scholar]
- Bucci BK, Kruse SW, Thode AB, Alvarado SM, Jones DN. Effect of n-alcohols on the structure and stability of the Drosophila odorant binding protein LUSH. Biochem. 2006;45:1693–1701. doi: 10.1021/bi0516576. [DOI] [PubMed] [Google Scholar]
- Chen J, Mitcheson JS, Tristani-Firouzi M, Lin M, Sanguinetti MC. The S4-S5 linker couples voltage sensing and activation of pacemaker channels. Proc Natl Acad Sci USA. 2001;98:11277–11282. doi: 10.1073/pnas.201250598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shu S, Bayliss DA. Suppression of ih contributes to propofol-induced inhibition of mouse cortical pyramidal neurons. J Neurophysiol. 2005;94:3872–3883. doi: 10.1152/jn.00389.2005. [DOI] [PubMed] [Google Scholar]
- Chen X, Shu S, Kennedy DP, Willcox SC, Bayliss DA. Subunit-specific effects of isoflurane on neuronal Ih in HCN1 knockout mice. J Neurophysiol. 2009;101:129–140. doi: 10.1152/jn.01352.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Rubin E. Ethanol selectively blocks a noninactivating K+ current expressed in Xenopus oocytes. Proc Natl Acad Sci USA. 1993;90:6957–6960. doi: 10.1073/pnas.90.15.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Vyas TB, Escobar L, Wei A. Alcohols inhibit a cloned potassium channel at a discrete saturable site. Insights into the molecular basis of general anesthesia. J Biol Chem. 1995;270:19408–19416. doi: 10.1074/jbc.270.33.19408. [DOI] [PubMed] [Google Scholar]
- Covarrubias M, Bhattacharji A, Harris T, Kaplan B, Germann MW. Alcohol and anesthetic action at the gate of a voltage-dependent K+ channel. In: Mashimo T, Ogli K, Uchida I, editors. Basic and Systemic Mechanisms of Anesthesia. Amsterdam, The Netherlands: Elsevier B.V.; 2005. pp. 55–60. [Google Scholar]
- Dilger JP. The effects of general anaesthetics on ligand-gated ion channels. Br J Anaesth. 2002;89:41–51. doi: 10.1093/bja/aef161. [DOI] [PubMed] [Google Scholar]
- Eckenhoff RG, Johansson JS. Molecular interactions between inhaled anesthetics and proteins. Pharmacol Rev. 1997;49:343–367. [PubMed] [Google Scholar]
- Forman SA, Chin VA. General anesthetics and molecular mechanisms of unconsciousness. Int Anesthesiol Clin. 2008;46:43–53. doi: 10.1097/AIA.0b013e3181755da5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. Molecular targets underlying general anaesthesia. Br J Pharmacol. 2006;147(Suppl 1):S72–S81. doi: 10.1038/sj.bjp.0706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- Harris T, Shahidullah M, Ellingson JS, Covarrubias M. General anesthetic action at an internal protein site involving the S4–S5 cytoplasmic loop of a neuronal K+ channel. J Biol Chem. 2000;275:4928–4936. doi: 10.1074/jbc.275.7.4928. [DOI] [PubMed] [Google Scholar]
- Harris T, Graber AR, Covarrubias M. Allosteric modulation of a neuronal K+ channel by 1-alkanols is linked to a key residue in the activation gate. Am J Physiol Cell Physiol. 2003;285:C788–C796. doi: 10.1152/ajpcell.00113.2003. [DOI] [PubMed] [Google Scholar]
- Harris RA, Trudell JR, Mihic SJ. Ethanol's molecular targets. Sci Signal. 2008;1:re7. doi: 10.1126/scisignal.128re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Horishita T, Harris RA. n-Alcohols inhibit voltage-gated Na+ channels expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2008;326:270–277. doi: 10.1124/jpet.108.138370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Shahidullah M, Covarrubias M. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J Gen Physiol. 1999;113:641–660. doi: 10.1085/jgp.113.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse SW, Zhao R, Smith DP, Jones DN. Structure of a specific alcohol-binding site defined by the odorant binding protein LUSH from Drosophila melanogaster. Nat Struct Biol. 2003;10:694–700. doi: 10.1038/nsb960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro AJ, Raes AL, Grottesi A, Van Hoorick D, Sansom MS, Snyders DJ. Kv channel gating requires a compatible S4–S5 linker and bottom part of S6, constrained by non-interacting residues. J Gen Physiol. 2008;132:667–680. doi: 10.1085/jgp.200810048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Loll PJ, Eckenhoff RG. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005;19:567–576. doi: 10.1096/fj.04-3171com. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005a;309:897–902. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005b;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Lu Z, Klem AM, Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J Gen Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for the S4–S5 intracellular loop in domain IV of the sodium channel alpha-subunit in fast inactivation. J Biol Chem. 1998;273:1121–1129. doi: 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- Prole DL, Yellen G. Reversal of HCN channel voltage dependence via bridging of the S4–S5 linker and post-S6. J Gen Physiol. 2006;128:273–282. doi: 10.1085/jgp.200609590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Harris T, Germann MW, Covarrubias M. Molecular features of an alcohol binding site in a neuronal potassium channel. Biochemistry. 2003;42:11243–11252. doi: 10.1021/bi034738f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi M, Harris RA. Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J Pharmacol Exp Ther. 2004;309:987–994. doi: 10.1124/jpet.103.064063. [DOI] [PubMed] [Google Scholar]
- Strzalka J, Liu J, Tronin A, Churbanova IY, Johansson JS, Blasie JK. Mechanism of interaction between the general anesthetic halothane and a model ion channel protein, I: structural investigations via X-ray reflectivity from Langmuir monolayers. Biophys J. 2009;96:4164–4175. doi: 10.1016/j.bpj.2009.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thode AB, Kruse SW, Nix JC, Jones DN. The role of multiple hydrogen-bonding groups in specific alcohol binding sites in proteins: insights from structural studies of LUSH. J Mol Biol. 2008;376:1360–1376. doi: 10.1016/j.jmb.2007.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tristani-Firouzi M, Chen J, Sanguinetti MC. Interactions between S4–S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J Biol Chem. 2002;277:18994–19000. doi: 10.1074/jbc.M200410200. [DOI] [PubMed] [Google Scholar]
- Urban BW. The Site of Anesthetic Action. Handbook of Experimental Pharmacology. Berlin/Heidelberg, Germany: Springer Berlin Heidelberg; 2008. pp. 3–29. [DOI] [PubMed] [Google Scholar]
- Vedula LS, Brannigan G, Economou NJ, Xi J, Hall MA, Liu R, et al. A unitary anesthetic-binding site at high resolution. J Biol Chem. 2009;284:24176–24184. doi: 10.1074/jbc.M109.017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Abbas SY, Harrison NL, Goldstein PA. Propofol block of I(h) contributes to the suppression of neuronal excitability and rhythmic burst firing in thalamocortical neurons. Eur J Neurosci. 2006;23:465–480. doi: 10.1111/j.1460-9568.2005.04587.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.