

Abstract
Background and purpose:
The multidrug resistance of epilepsy may result from the overexpression of P-glycoprotein, but the mechanisms are unclear. We investigated whether the overexpression of P-glycoprotein in the brains of subjects with pharmacoresistant epilepsy resulted from both drug effects and seizure activity.
Experimental approach:
Kindled rats were developed by injecting a subconvulsive dose of pentylenetetrazole (33 mg·kg−1·day−1, i.p.) for 28 days. Groups were then treated with an oral dose of phenobarbital (45 mg·kg−1·day−1) for 40 days. In accord with behavioural observations, P-glycoprotein activity in brain was assessed using brain-to-plasma concentration ratios of rhodamine 123. P-glycoprotein levels in the brain regions were further evaluated using RT-PCR and Western blot analysis. The distribution of phenobarbital in the brain was assessed by measuring phenobarbital concentrations 1 h following its oral administration.
Key results:
The kindling significantly increased P-glycoprotein activity and expression. Good associations were found among P-glycoprotein activity, expression and phenobarbital concentration in the hippocampus. Short-term treatment with phenobarbital showed good anti-epileptic effect; the maximum effect occurred on day 14 when overexpression of P-glycoprotein was reversed. Continuous treatment with phenobarbital had a gradually reduced anti-epileptic effect and on day 40, phenobarbital exhibited no anti-epileptic effect; this was accompanied by both a re-enhancement of P-glycoprotein expression and decreased phenobarbital concentration in the hippocampus. P-glycoprotein function and expression were also increased in age-matched normal rats treated with phenobarbital.
Conclusions and implications:
The overexpression of P-glycoprotein in the brain of subjects with pharmacoresistant epilepsy is due to a combination of drug effects and epileptic seizures.
Keywords: P-glycoprotein, blood–brain barrier, pharmacoresistant epilepsy, phenobarbital, drug induction, anti-epileptic effect, rhodamin 123
Introduction
Epilepsy is one of the most frequent neurological disorders, affecting approximately 1–2% of the world population (Sander and Shorvon, 1987; Kosopoulos et al., 2002). Accumulating evidence has shown that 20–30% of patients suffering from epilepsy are pharmacoresistant (Cockerell et al., 1995; Lazarowski et al., 1999; Regesta et al., 1999; Dombrowski et al., 2001; Sisodiya et al., 2002). These patients do not respond a specific anti-epileptic drug (AED) are often refractory to other AEDs, even though these drugs act by different mechanisms (Löscher and Potschka, 2002). Despite numerous studies the real pathological mechanism of this drug resistance remains obscure (Wang et al., 2003; Bordet, 2004; Kwan and Brodie, 2004; Löscher and Schmidt, 2004; Zimprich et al., 2004). Decreased accumulation of the AED in the brain due to the overexpression of P-glycoprotein (P-GP) at the blood–brain barrier (BBB) is considered to be a cause of pharmacoresistant epilepsy. This hypothesis is supported by data showing that P-GP is overexpressed in endothelial cells of the brain microvessels of patients with pharmacoresistant epilepsy (Dombrowski et al., 2001; Rogawski, 2002; Sisodiya et al., 2002) and in the brain of kindled rats (Rizzi et al., 2002; Volk et al., 2004; 2005; Brandt et al., 2006; Liu et al., 2007).
However, it is still not clear whether the overexpression of P-GP in the epileptogenic brain tissue of patients with pharmacoresistant epilepsy is a consequence of epilepsy, uncontrolled seizures, chronic treatment with AEDs, or combinations of these factors. Several studies have demonstrated that experimentally induced seizures may result in the overexpression of P-GP in brain (Volk et al., 2004; 2005; Brandt et al., 2006; Liu et al., 2007). Results showing that AEDs, including phenobarbital (PB), carbamazepine, phenytoin, valproic acid and lamotrigine, increase P-GP expression and function in human tumour cell lines and porcine brain capillary endothelial cell lines are limited (Weiss et al., 2003; Eyal et al., 2006). In previous studies we demonstrated that chronic treatment with AEDs increased the expression of P-GP in rat brain and rat brain microvascular endothelial cells (Wen et al., 2008; Yang et al., 2008). Taken together, these results may give a hypothesis that overexpression of P-GP in the brains of patients with pharmacoresistant epilepsy may initially, at an early stage, result from the epileptic seizure and, at a later stage, be due to a combination of drug effects and epileptic seizure.
PB is recommended by the World Health Organization as a first-line approach for treating seizures in the developing world, and remains a popular choice in many developed countries (Brodie and Kwan, 2004). Several studies have shown that the transport of PB at the BBB is mediated by P-GP (Potschka et al., 2002; Yang et al., 2008). Hence, PB was used as a model drug in this study. The aim of this study was to verify our hypothesis using kindled rats induced by pentylenetetrazole (PTZ) as an animal model chronically treated with PB. The function of P-GP was determined by measuring the distribution of rhodamine 123 (Rho123). P-GP protein levels and mRNA levels were measured by Western blot and RT-PCR analysis respectively.
Methods
Animals
Male Sprague-Dawley rats, weighing 180–220 g, were purchased from Sino-British Sippr/BK Laboratory Animal Ltd. (Shanghai, China). The rats were housed under controlled environmental conditions (temperature, 23 ± 1°C; humidity, 55 ± 5%) and kept under a 12 h light/dark cycle; commercial food and water were freely available. The studies were approved by the Animal Ethics Committee of China Pharmaceutical University, and every effort was made to minimize stress to the animals.
Kindled model of epilepsy
Kindling was induced according to the method described previously (Suzuki et al., 2001). A subconvulsive dose of PTZ (33 mg·kg−1) was injected i.p. to rats at 09 h 00 min every day for up to 28 days. The convulsive activity was monitored for 30 min following this dose of PTZ. The intensity of the seizure response was scored according to the following scale (De Sarro et al., 1999): 0, no response; 1, mouth and facial jerks; 2, nodding or myoclonic body jerks; 3, forelimb clonus; 4, rearing, falling down, hindlimb clonus and forelimb tonus; and 5, tonic extension of hindlimb, status epileptic and/or death. The maximum response was recorded for each animal. Incidence of seizure and latency of seizure were also recorded. Only animals showing at least five consecutive stage 2 seizures or three consecutive stage 4 or 5 seizures were considered to be kindled rats and included in the study.
Behavioural observation of kindled rats treated with PB
The kindled rats were randomly divided into two groups. Group 1 served as the kindled control (PTZ-CT) and only received the vehicle. Group 2 (PTZ-PB) were given PB (45 mg·kg−1, p.o.) once a day for a given number of days. At 40 min after the oral dose, each rat received PTZ (33 mg·kg−1, i.p.). Seizure stage, seizure times and latency of seizure were recorded according to the scale described above.
Another batch of experimental rats was used to determine P-GP function and expression in the brain. The experimental procedure used was the same as that described above. At the same time, age-matched normal rats were also enrolled and randomly divided into two groups, assigned to be groups 3 and 4. Group 3 (PB-CT) was given PB (45 mg·kg−1, p.o.), once a day for a given number of days and group 4 (CT) received only the vehicle and served as an age-matched normal control. At a designated time based on behavioural observations during the PB treatment, some of the experimental rats were selected for assaying PB concentration, P-GP function and expression in their brains. A schematic illustration of the experimental design and the different groups is shown in Figure 1.
Figure 1.
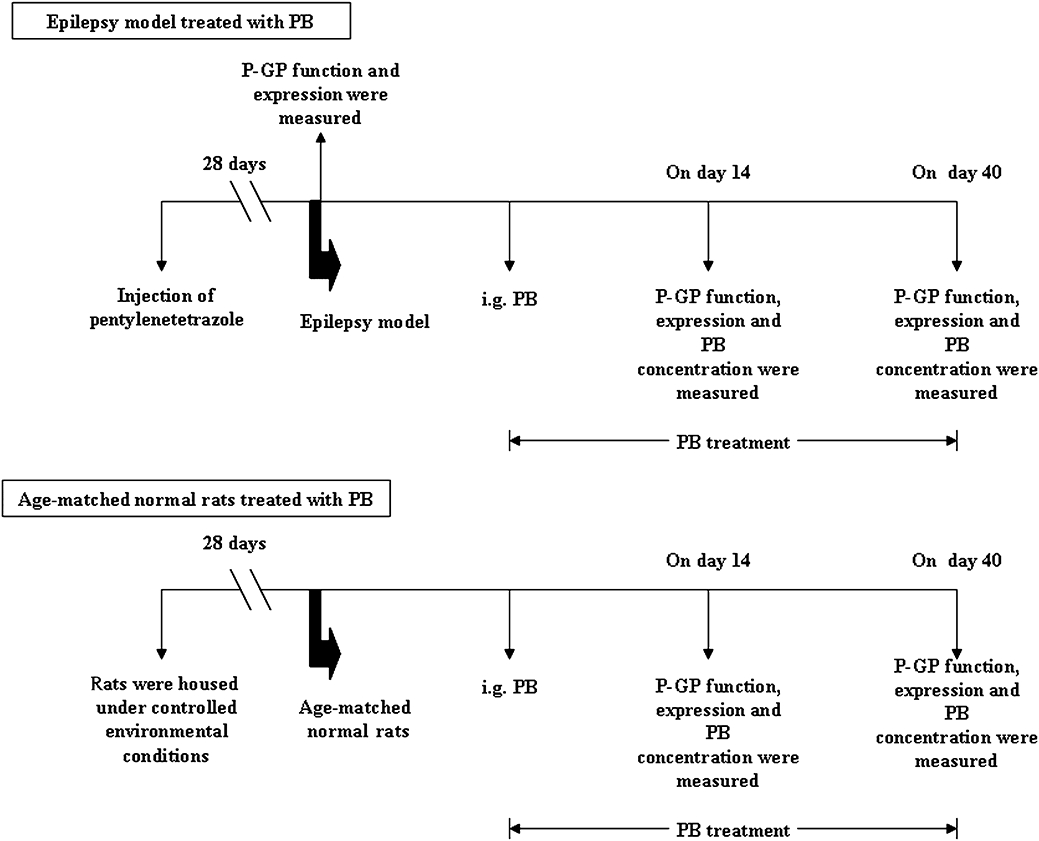
Schematic illustration of the experimental protocol and group distribution of the study. PB, phenobarbital.
Distribution of Rho123 and PB in brain
To evaluate the P-GP function or PB distribution in brain, rats immediately received Rho123 (0.2 mg·kg−1, i.v.) following oral administration of PB or vehicle. At 1 h after the injection of Rho123, rats were killed under light ether anaesthesia, blood was collected and plasma samples were obtained. The hippocampus and cerebral cortex were immediately removed and weighed. The plasma and brain samples were stored at −80°C for measuring PB and Rho123 concentration. The other brain tissues were used for RT-PCR and Western blot analysis respectively.
RT-PCR analysis
Total RNA was isolated from the hippocampus and cerebral cortex utilizing Trizol reagent according to the manufacturer's instructions. The purity of the RNA isolated was determined using UV absorption at 260 nm and 280 nm. The cDNA was synthesized from 2 µg of total RNA using oligo(dT)15 and M-MLV reverse transcriptase. PCR was performed on Gene Amp PCR System 9600. Primers used for the amplification of the cDNAs of interest were: for mdr1a gene (351 bp), forward 5′-GACGGAATTGATAATGTGGACA-3′ and reverse 5′-AAGGATCAGGAACAATAAA-3′; for mdr1b gene(351 bp), forward 5′-GCCCATCCTGTTTGACTG-3′ and reverse 5′-CGCTTCCTGGACGACCTT-3′; for glyceraldehydes phosphate dehydrogenase (GAPDH) (365 bp): forward 5′-GGTGCTGAGTATGTCGTGGAG-3′ and reverse 5′-ATGCAGGGATGATGTTCTGG-3′ (Sheng-Xing Sci-Tech Co. Nanjing China). After denaturation of the samples at 95°C for 5 min, the amplification was obtained by 30 cycles of 94°C for 30 s, 60°C for 30 s and 72°C for 1 min each. A final extension step at 72°C for 5 min was employed. PCR products were subjected to electrophoresis on 2% agarose gel and visualized by ethidium bromide staining. Densitometric quantification was recorded using gel image analysis system 3.3 (Jiangsu Jeda Science-Technology Co. Ltd, Nanjing, China). To normalize the data, the ratio between the densitometric quantification mdr1a/mdr1b gene and GAPDH gene was calculated by using the software Quantity One (Bio-Rad Laboratories, Richmond, CA, USA).
Western blot
Western blot analysis was used for assessing P-glycoprotein expression in rat brain according to a method described previously (Liu et al., 2008). Briefly, the brain tissues were homogenated and lysed in lysis buffer containing 10 mM Tris-HCl (pH 7.5), 1 mM EGTA, 1 mM MgCl2, 1 mM mercaptoethanol, 1% glycerol, protease inhibitor cocktail (1 mM dithiothreitol, 2 mM phenylmethylsulphonylfluoride. The lysate was incubated on ice for 30 min and centrifuged at 13 000× g for 10 min at 4°C. The supernatant was obtained as membrane fractions for Western blot. The protein concentration in the solution was measured by the Bio-Rad Protein Assay. An aliquot of tissue sample was diluted with an volume of 4 × sodium dodecyl sulphate (SDS) sample buffer containing 0.1 M Tris-HCl (pH 6.8), 4% SDS, 200 mM DTT, 20% glycerol, and 0.2% bromophenol blue. Proteins (25 µg per lane) were separated by electrophoresis on 8% SDS-polyacrylamide gel. After electrophoresis, the proteins were electrophoretically transferred to a nitrocellulose membrane. The membrane was blocked in PBS containing 0.1%Tween-20, PBST and 5% dried skim milk for 60 min at room temperature and washed three times for 15 min in PBST. Then the membrane was incubated with the primary monoclonal antibody C219, diluted 200-fold in PBST overnight at 4°C. After the membrane had been washed with PBST, it was incubated in the appropriate HRP-conjugated goat anti-mouse secondary antibody at room temperature for another 1 h and washed again three times in PBST. The transferred proteins were incubated with ECL substrate solution for 5 min according to the manufacturer's instructions and visualized with autoradiography X-film. The relative expressions were quantified densitometrically by using the quantity one software (Bio-Rad Laboratories, Richmond, CA, USA) and calculated according to the reference bands of β-actin (Boshide Biotech Co., Wuhan, China).
Drug assay
The concentrations of Rho123 in plasma and brain were measured by HPLC (Liu et al., 2007). The recoveries were higher than 85%; the linear ranges of Rho123 in plasma and brain were 3.12–200 ng·mL−1 and 0.32–10.4 ng·g−1 brain respectively. The relative standard deviations of the intra-day and inter-day concentrations in plasma and brain were both less than 10%. The concentrations of PB in plasma and brain tissues were also measured according to our previous method (Liu et al., 2007). The lowest limits of quantification of PB in plasma and brain were 1.56 µg·mL−1 and 0.2 µg·g−1 brain tissue respectively. The recoveries were higher than 80%; the linear ranges of PB in plasma and brain were 1.56–100 µg·mL−1 and 0.2–62.5 µg·g−1 respectively. The relative standard deviations of the intra-day and inter-day levels were less than 10%.
Data analysis
Results are expressed as mean ± SD. The overall differences between groups were determined by one-way anova. If analysis was significant, the differences between groups were estimated using Student–Newman–Keuls multiple comparison post hoc test. A P-value of less than 0.05 indicated a significant difference.
Materials
PTZ, Rho123 and the lysis buffer were purchased from Sigma Chemical Co. (St. Louis, MO, USA). PB was purchased from New Asiatic Pharmaceutical Co., Ltd. (Shanghai, China). The Bio-Rad Protein Assay was obtained from Bio-Rad Laboratories (Richmond, CA, USA); ECL substrate solution from Cell Signaling (USA); Trizol reagent from Invitrogen Co. (USA). All other reagents were commercially available and were of analytical grade. PTZ was dissolved in physiological saline. PB was suspended in solution of 0.25% carboxymethylcellulose sodium (CMC-Na). Rho123 was dissolved in 0.01 M phosphate-buffered saline (PBS, pH 7.4) before use.
Results
Behavioural observations
The development of PTZ-induced kindling was observed (Figure 2A). The animals treated with repeated subconvulsive dose of PTZ (33 mg·kg−1·day−1, i.p.) for 28 injections went gradually through the stages. Almost all rats became kindled rats that showed at least five consecutive stage 2 seizures or three consecutive stage 4 or 5 seizures. The kindled rats were highly sensitive to sound. Even a mild applause could induce a seizure in them. Only fully kindled rats were chosen for the following experiments.
Figure 2.
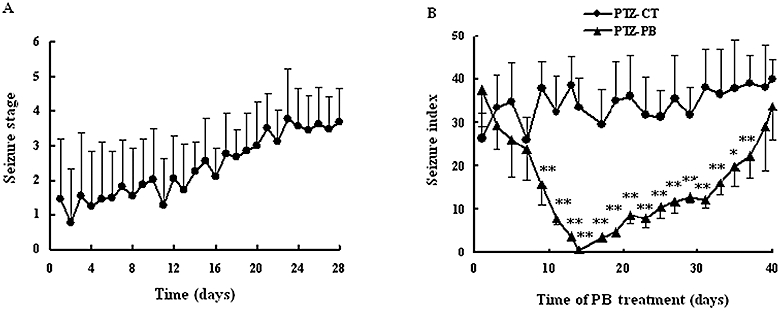
(A) Development of PTZ (33 mg·kg−1·day−1, i.p.)-induced seizure to the fully kindled state. Data are mean ± SD (n= 10). (B) Anti-epileptic activity of PB during PB treatment. PTZ was given to kindled rats 40 min after oral administration of PB (45 mg·kg−1·day−1). Seizure stage and times were observed. Seizure indexes of PTZ-kindled model group (PTZ-CT rats) and PB treatment group (PTZ-PB rats) are presented. Index of seizure was defined as seizure stages × seizure times. Data are mean ± SD (n= 5). *P < 0.05, **P < 0.01 versus PTZ-CT rats using Student's t-test. PB, phenobarbital; PTZ, pentylenetetrazole.
The anti-epileptic effects of PB on PTZ-kindled rats were observed during PB treatment (Figure 2B). Severity, seizure times and latency of seizure were used as indicies of seizures. Significant inter-individual variations were observed for the severity (seizure stage) and seizure times in the same group. The index of seizure was defined as seizure stage multiplied by seizure time. It was found that at an early stage, PB treatment gradually decreased the index of seizure and prolonged the latency of seizure. On day 14 of PB treatment, a maximal anti-epileptic effect of PB was observed (P < 0.01 vs. PTZ-CT group). The intensity of the seizure was decreased to 0 or 1. However, continuous PB treatment had a gradually weakened anti-epileptic effect. On day 40 of PB treatment, the rats again showed at least five consecutive stage 2 seizures or three consecutive stage 4 or 5 seizures, and the index of seizure was similar to that of the PTZ-CT rats (P > 0.05 vs. PTZ-CT group).
Accordingly, experimental rats before and on day 14 and 40 of PB treatment were selected for evaluating P-GP function and expression in brain.
Function of P-GP in brain
Concentrations of Rho123, a typical substrate of P-GP, in plasma and brain tissues of experimental rats were measured 1 h after i.v. administration of Rho123 and the ratios of brain-to-plasma were calculated for evaluating P-GP function in brain. It was found that kindling did not affect Rho123 concentrations in plasma (Figure 3A), but significantly decreased the concentrations of Rho123 in both the hippocampus and cerebral cortex, resulting in a lower ratio of brain-to-plasma (P < 0.01, Figure 3B,C). PB treatment slightly altered Rho123 concentration in plasma of treated rats. But 14 day PB treatment significantly reversed the decreased Rho123 concentration in the hippocampus induced by kindling (2.45 ± 0.69 ng·g−1 tissue in PTZ-PB rats vs. 1.60 ± 0.29 ng·g−1 tissue in PTZ-CT rats, P < 0.01). However, continuous PB treatment further decreased the concentration of Rho 123 in the hippocampus to that of PTZ-CT rats. In contrast, PB treatment did not modify the Rho123 concentration in the cerebral cortex of PTZ-kindled rats. In age-matched normal rats (PB-CT rats), both 14 day and 40 day PB treatments significantly (P < 0.01) decreased the concentration of Rho123 in the hippocampus and cerebral cortex as compared with age-matched control rats (CT), resulting in lower brain-to-plasma concentration ratios (Figure 3B,C).
Figure 3.
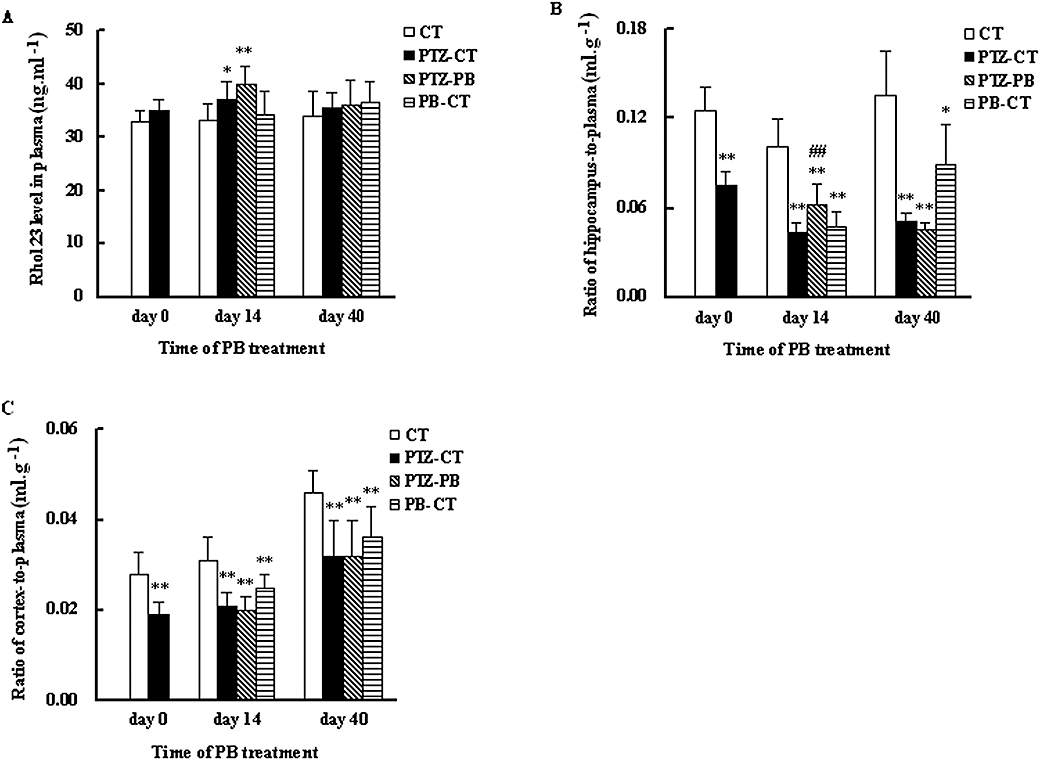
Effects of kindling and PB treatment on Rho123 distribution in plasma (A), cerebral cortex (B) and hippocampus (C). The treated kindled rats (PTZ-PB rats) and age-matched treated rats (PB-CT rats) were given PB (45 mg·kg−1·day−1, p.o.) for 14 or 40 consecutive days. Age-matched control rats (CT rats) and untreated kindled rats (PTZ-CT rats) only received 0.25% CMC-Na. The concentrations of Rho123 in plasma, cerebral cortex and hippocampus were measured 1 h after injection of Rho123 (0.2 mg·kg−1, i.v.). Each value is presented as mean ± SD (n= 8). *P < 0.05, **P < 0.01 versus CT rats and ##P < 0.01 versus PTZ-CT rats using one-way anova following by Student–Newman–Keuls multiple comparison post hoc test. PB, phenobarbital; PTZ, pentylenetetrazole; Rho123, rhodamine 123.
Distribution of PB in the brain
The PB levels in plasma and brain were simultaneously measured, 1 h after oral administration of PB on day 14 and day 40 of PB treatment respectively (Table 1). It was found that PB concentration in hippocampus was lower than that in cortex, which is in line with the Rho123 results. Compared with 14 day PB treatment, although 40 day PB treatment showed a trend to increase plasma concentration of PB (P > 0.05), 40 day PB treatment significantly decreased PB concentrations in hippocampus (P < 0.01) and slightly decreased PB concentration in cerebral cortex (P > 0.05). In age-matched normal rats, 40 day PB treatment also decreased PB concentration in both the hippocampus and cortex as compared with 14 day PB treatment (P < 0.05).
Table 1.
Effects of kindling and PB treatment on distribution of PB in plasma, cerebral cortex and hippocampus
|
PTZ-PB |
PB-CT |
|||
|---|---|---|---|---|
| 14 day treatment | 40 day treatment | 14 day treatment | 40 day treatment | |
| Plasma, µg·mL−1 | 72.19 ± 6.98 | 77.04 ± 5.16 | 64.84 ± 11.04 | 66.50 ± 16.52 |
| Cortex, µg·g−1 tissue | 23.44 ± 3.58 | 20.53 ± 3.19 | 23.45 ± 4.89 | 16.74 ± 5.00* |
| Cortex/plasma, mL·g−1 | 0.32 ± 0.06 | 0.27 ± 0.04* | 0.36 ± 0.04 | 0.25 ± 0.04** |
| Hippocampus, µg·g−1 tissue | 52.26 ± 9.57 | 35.93 ± 5.56** | 32.06 ± 5.59 | 26.21 ± 5.12* |
| Hippocampus/plasma, mL·g−1 | 0.72 ± 0.11 | 0.47 ± 0.08** | 0.50 ± 0.06 | 0.39 ± 0.10* |
The treated kindled rats (PTZ-PB rats) and age-matched treated rats (PB-CT rats) were orally given PB (45 mg·kg−1·day−1, i.g.) for consecutive 14 or 40 days. At 1 h after PB administration (i.g.), PB concentrations in plasma, cerebral cortex, and hippocampus were measured. Each value is presented as mean ± SD (n= 8).
P < 0.05,
P < 0.01 versus the 14th day using Student's t-test.
PB, phenobarbital; PTZ, pentylenetetrazole.
P-GP expression in brain
Western blot was used to investigate further the levels of P-GP in the brain regions of interest. The results revealed a band of 170 kDa, corresponding to P-GP. It was found that kindling induced by 28 injections of PTZ significantly (P < 0.01) increased the levels of P-GP in the hippocampus and cerebral cortex, to 147.2 ± 23.9% and 144.1 ± 6.07% of CT rats respectively (Figure 4). This effect of PB on P-GP levels in the brain was dependent on the region and time. In the hippocampus of PTZ-kindled rats, 14 day PB treatment reversed the increase in P-GP levels induced by kindling, resulting in a decrease of 28.3% compared with PTZ-CT rats. However, 40 day PB treatment further re-enhanced the P-GP level, which was found to be similar to that of PTZ-CT rats (Figure 4B). In the cerebral cortex, neither the 14 day or 40 day PB treatment modified the increase in P-GP (Figure 4D). In age-matched normal rats, both the 14 and 40 day PB treatments significantly increased the levels of P-GP in the hippocampus and cerebral cortex (Figure 4). The alterations of P-GP levels were in line with the Rho 123 concentrations in the brain.
Figure 4.
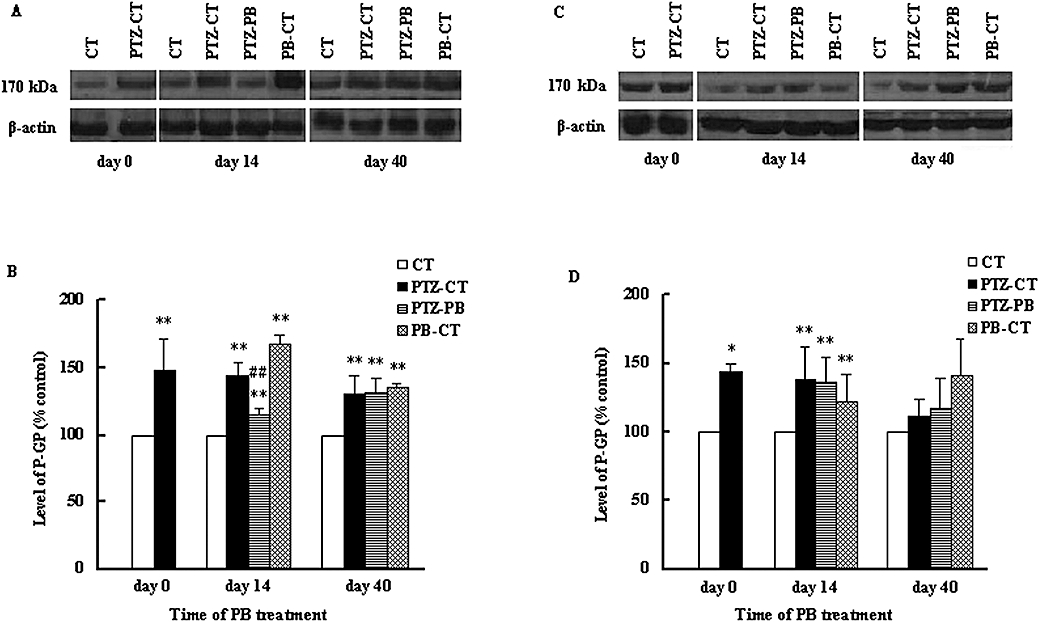
Effects of kindling and PB treatment on protein levels of P-GP in hippocampus and cerebral cortex. The treated kindled rats (PTZ-PB rats) and age-matched treated rats (PB-CT rats) were given PB (45 mg·kg−1·day−1, p.o.) for 14 or 40 consecutive days. Age-matched control rats (CT rats) and untreated kindled rats (PTZ-CT rats) only received 0.25% CMC-Na. Representative Western blot of P-GP in hippocampus (A) and cortex (C) and ratio of relative staining intensity for P-GP in hippocampus (B) and cortex (D) are shown. Each band corresponding to 170 kDa was observed. Data are presented as mean ± SD (n= 4). *P < 0.05, **P < 0.01 versus CT rats and ##P < 0.05 versus PTZ-CT rats using anova statistics following by Student–Newman–Keuls multiple comparison post hoc test. P-GP, P-glycoprotein; PB, phenobarbital; PTZ, pentylenetetrazole.
P-GP mRNA
In rodents, P-GP is encoded by two genes, mdr1a and mdr1b, so the expression of both genes was examined in this study using RT-PCR analysis. It was found that the effect of PB treatment on mdr1a/1b was also dependent on time, region and species; 28 injections of PTZ significantly increased the levels of mdr1a/1b mRNA in the hippocampus and cerebral cortex (P < 0.01, Figure 5). In the hippocampus of PTZ-kindled rats, 14 day PB treatment restored the levels of mdr1a/1b mRNA to those of age-matched control rats (CT). However, 40 day PB treatment increased mdr1a/1b mRNA levels again, to even higher levels than those of PTZ-CT rats (Figure 5). In the cortex of PTZ-kindled rats, 14 day PB treatment only partly reversed the increase in mdr1a/1b mRNA levels induced by kindling. PB treatment for 40 days still decreased the levels of mdr1a mRNA, but increased mdr1b mRNA levels as compared with that of PTZ-CT rats (Figure 6).
Figure 5.
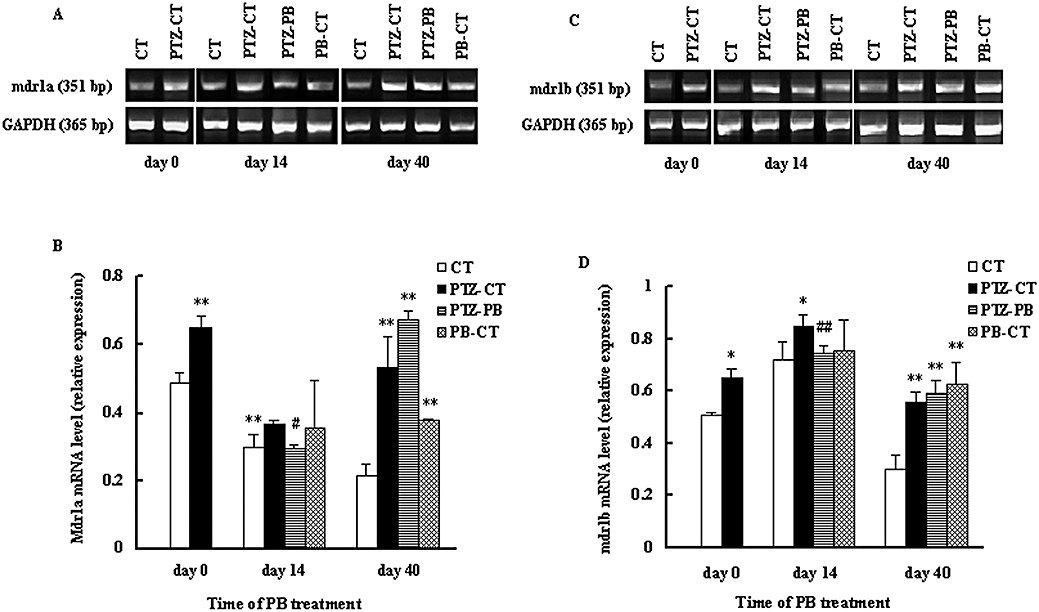
Effects of kindling and PB treatment on P-GP mdr1a/mdr1b mRNA levels in the hippocampus. The treated kindled rats (PTZ-PB rats) and age-matched treated rats (PB-CT rats) were given PB (45 mg·kg−1·day−1, p.o.) for 14 or 40 consecutive days. Age-matched control rats (CT rats) and untreated kindled rats (PTZ-CT rats) only received 0.25% CMC-Na. Expression of P-GP mdr1a (A) and mdr1b (C) mRNA in the hippocampus and relative staining intensity for mdr1a (B) and mdr1b (D) mRNA in hippocampus are shown. Data are presented as mean ± SD (n= 4). *P < 0.05, **P < 0.01 versus CT rats and #P < 0.05, ##P < 0.05 versus PTZ-CT rats using anova statistics following by Student–Newman–Keuls multiple comparison post hoc test. PTZ, pentylenetetrazole. P-GP, P-glycoprotein; PB, phenobarbital; PTZ, pentylenetetrazole.
Figure 6.
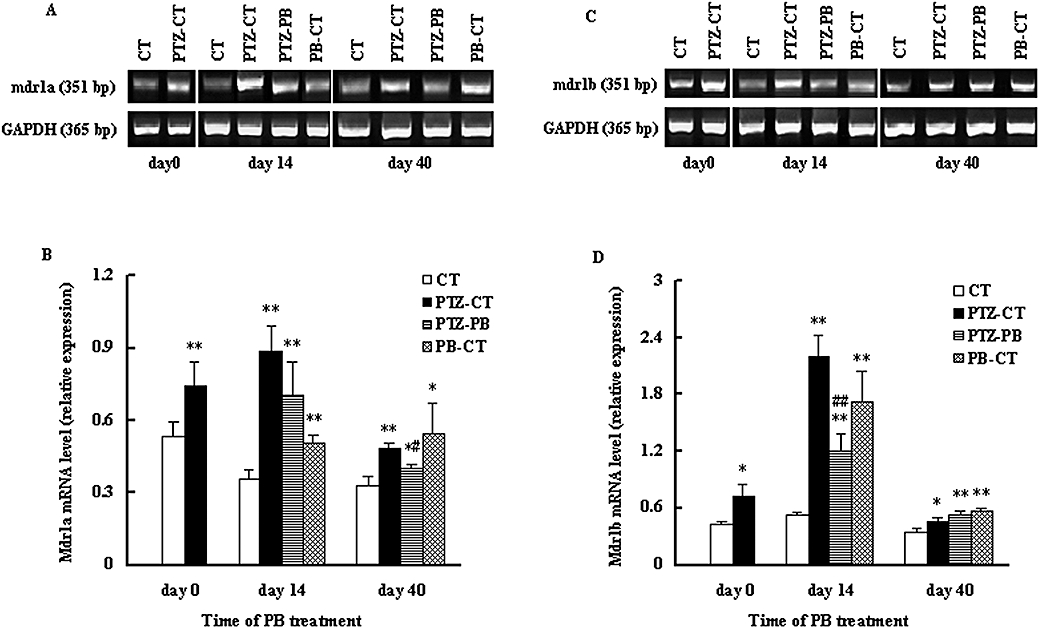
Effects of kindling and PB treatment on P-GP mdr1a/mdr1b mRNA levels in the cerebral cortex. The treated kindled rats (PTZ-PB rats) and age-matched treated rats (PB-CT rats) were given PB (45 mg·kg−1·day−1, p.o.) for 14 or 40 consecutive days. Age-matched control rats (CT rats) and untreated kindled rats (PTZ-CT rats) only received 0.25% CMC-Na. Expression of P-GP mdr1a (A) and mdr1b (C) mRNA in the cerebral cortex and relative staining intensity for mdr1a (B) and mdr1b (D) mRNA in hippocampus are shown. Data are presented as mean ± SD (n= 4). *P < 0.05, **P < 0.01 versus CT rats and #P < 0.05, ##P < 0.05 versus PTZ-CT rats using anova statistics following by Student–Newman–Keuls multiple comparison post hoc test. CMC-Na, carboxymethylcellulose sodium; P-GP, P-glycoprotein; PB, phenobarbital; PTZ, pentylenetetrazole.
In age-matched normal rats, the effect of PB exposure on the levels of mdr1a/1b mRNA was also dependent on region and time. Both the 14 and 40 day PB treatments increased mdr1a/1b mRNA levels in cerebral cortex (PB-CT rats) (Figure 6). Only the 40 day PB treatment induced higher levels of mdr1a/1b mRNA in the hippocampus (Figure 5).
Discussion and conclusions
It is generally assumed that seizures can increase the expression of efflux transporter genes and proteins, an assumption supported by results from both clinical investigations and animal models (Kwan et al., 2005; Löscher and Potschka, 2005a,b; Liu et al., 2007). Accumulating evidence demonstrated that AEDs may increase P-GP expression and function in brain (Weiss et al., 2003; Eyal et al., 2006; Wen et al., 2008; Yang et al., 2008). Several reports have also shown that P-GP inhibitors can enhance the anti-epileptic effect of PB (Brandt et al., 2006; Liu et al., 2007), which indicates the important role of P-GP in pharmacoresistant epilepsy. The present results further supported this view. However, a recent report (Bankstahl and Löscher, 2008) has shown that during the early phase (1.5–24 h) following kindling induced by pilocarpine or electrical stimulation of the basolateral amygdale, the expressions of P-GP in the brain of rats were not altered and P-GP inhibitors did not enhance the anti-epileptic effect of an AED, but significant increases in the expression of P-GP were observed 48 h following kindling. These results indicate that the increase in P-GP induced by kindling is time-dependent. Therefore, our result was not contrast to Bankstahl's report. Animal models of epilepsy in the present study were induced by repeated dose of PTZ for 28 days and PB was given at 40 min before PTZ administration. While in Bankstahl's study, kindled rats were induced by pilocarpine or electrical stimulation of the basolateral amygdale, and AEDs were given to rats at 10–90 min after onset of status epilepticus induced by pilocarpine or 5–480 min after onset of status epilepticus induced by electrical stimulation of the basolateral amygdale.
The present study focused on the hypothesis that overexpression of P-GP in the brain of subjects with pharmacoresistant epilepsy results from the combined effects of epileptic seizure and AED. At first, the anti-epileptic effect of PB (Figure 2B) was observed using PTZ-kindled rats. It was found that at an early stage of PB treatment, PB exhibited a good anti-epileptic effect; the maximal effect occurred on day 14 of PB treatment. However, continuous PB treatment exhibited a gradually weakened anti-epileptic effect. On day 40 of PB treatment, PB no longer exhibited an anti-epileptic effect, which indicated the development of pharmacoresistant epilepsy. It has been reported that the anti-epileptic effect of PB is related to its levels in the brain (Nagatomo et al., 1996). The cerebral cortex and hippocampus, which have often been implicated in pharmacoresistant types of epilepsy (Regesta et al., 1999), were selected as brain regions for investigation in the present study. One hour after the administration of PB, its concentrations in plasma and these brain regions were measured. At this time point, PB treatment did not alter the concentration of PB in plasma, but the 40 day PB treatment significantly decreased the PB concentration in the hippocampus, which was in agreement with weakness of PB anti-epileptic effect. These results suggest that the decrease level of PB in the brain may be one of the reasons for its diminished anti-epileptic effect. However, the finding that, in the hippocampus, the bioavailability of PB only decreased by 31% following 40 days of treatment cannot fully explain the present finding; the almost complete disappearance of the anti-epileptic effect of PB at this time point. Also, in our study the measurement of PB concentration in the brain 1 h after its administration does not reflect the overall concentration-time course. Further studies are needed to determine the overall concentration-time course of PB using new technology, such as microdialysis or radioautography. In addition, other factors may be involved in the development of pharmacoresistant epilepsy. Intrinsic (genetic) and acquired (disease-related) alterations to the structure and/or functionality of AED targets in epileptogenic brain regions may lead to reduced drug effects (Schmidt and Löscher, 2009). It has been shown that NKCC1 expression in the hippocampus of patients with refractory epilepsy increased, impairing the inhibitory capacity of GABA and resulting in hyperexcitability (Sen et al., 2007). It has also been suggested that a dysfunctional adenosine kinase system is involved in pharmacoresistant epilepsy (Boison, 2006), and this is supported by findings in experimental animals (Rebola et al., 2003; 2005;) and patients (Glass et al., 1996). A decrease in the expression of GABAA receptors in the hippocampus of chronic pharmacoresistant patients has also been observed (Wolf et al., 1994) and animal experiments have shown that this alteration of GABAA receptors is associated with resistance to the anti-epileptic effects of PB (Volk et al., 2006). Taken together, these results demonstrate that the development of pharmacoresistant epilepsy is a complex process.
Several studies have shown that the transport of many AEDs including PB across the BBB is mediated by P-GP (Löscher and Potschka, 2005c; Liu et al., 2007; Yang et al., 2008). Our experiments were designed to investigate whether the decrease in PB concentration in brain tissues results from an increase in the expression and function of P-GP. P-GP function and protein levels as well as mdr1a/1b mRNA levels in the specified brain regions were measured at three time points, the day before PB treatment (kindling development), day 14 of PB treatment (maximal anti-epileptic effect observed) and day 40 of PB treatment (development of pharmacoresistant epilepsy).
Rho123 has been extensively used as an index of P-GP mediated transport in in vitro and in vivo studies (Kageyama et al., 2006; Turncliff et al., 2006; Barta et al., 2008; Nishimura et al., 2008; Pires et al., 2009; Tanaka et al., 2009) and was selected as a marker for evaluating P-GP function in our experiments. In addition to P-GP, other ABC efflux transporters such as members of the multidrug resistance protein (MRP) family and breast cancer resistance protein (BCRP) have been shown to contribute to BBB function (Löscher and Potschka, 2005c) and may be involved in the transport of Rho123. However, other studies have indicated that MRP1 makes a minimal contribution to Rho123 efflux (Dogan et al., 2004) and Rho123 is not transported by BCRP (Alqawi et al., 2004); there is no evidence that MRP2 and MRP4 are involved in the transport of Rho123 across the BBB. In the present study, compared with age-matched control rats, a lower concentration of Rho123 was found in the specified brain regions of kindled rats following 28 injections of PTZ, and this was associated with higher levels of P-GP protein and mdr1a/1b mRNA. Short-term exposure (14 day treatment) of PB reversed the increase in P-GP activity (lower brain-to-plasma ratio of Rho123), levels of P-GP protein and mdr1a/1b mRNA induced by kindling in the hippocampus; this was accompanied by an alleviation of the symptoms of epilepsy. The finding that short-term exposure of PB prevented the overexpression of P-GP induced by kindling indicates that it is unlikely that chronic PTZ has a toxic action. Hence, P-GP overexpression induced during kindling is probably due to a direct effect of the presence of convulsions but not to the action of PTZ. It is reasonable therefore to deduce that at an early stage of epilepsy the transporter-modifying factor is a consequence of the convulsive state itself, but long-term exposure (40 day PB treatment) to PB may result in an increase in P-GP activity, levels of P-GP protein and mdr1a/1b mRNA levels again, and this is accompanied by lower levels of PB and an attenuation of the anti-epileptic effect of PB. These results further support clinical findings demonstrating that an overexpression of P-GP occurs in the brain of patients with pharmacoresistant epilepsy (Lazarowski et al., 1999; Dombrowski et al., 2001; Sisodiya et al., 2002). PB treatment also increased P-GP expression and function in age-matched normal rats, which shows that the effects of PB have an important role in the development of pharmacoresistant epilepsy. These results are in agreement with those from our previous report (Wen et al., 2008).
Seegers et al. (2002) found that an 11 or 7 day exposure of AEDs did not affect P-GP expression, but it is possible that this exposure time is too short to compare to clinical practice. We found that, after 40 days of PB treatment, the ratio of hippocampus/plasma in the non-kindled, treated group was higher than that in the kindled, treated group, which indicates that P-GP function in the non-kindled, treated group was weaker than that in the kindled, treated group. All these results demonstrate that overexpression of P-GP in the brain at an early stage, is caused by the epileptic seizure and at the later stage results from a combination of the effects of the seizures and the drug; the latter enhances the development of pharmacoresistant epilepsy.
The present results also showed that concentrations of Rho123, as well as mdr1 mRNA in the brain were region-dependent. In age-matched control rats, the concentrations of Rho123 in the hippocampus were significantly higher than those in the cerebral cortex, indicating that the expression of P-GP in the cortex is higher than that in the hippocampus. The higher level of mdr1a mRNA in the cortex supports this speculation. Kindling may increase the levels of P-GP protein and mdr1a/1b mRNA in specific brain regions. The effect of PB exposure on function and expression was dependent on treatment time, region and species. In the hippocampus, a 14 day PB treatment restored the levels of mdr1a/1b mRNA induced by kindling to levels of age-matched control rats. In contrast, after a 40 day PB treatment the overexpression of mdr1a/1b mRNA induced by kindling was further enhanced. The higher levels of mdr1a/1b and P-GP protein were associated with an increase in P-GP function (lower brain-to-plasma concentration ratio of Rho123). In the cortex, the 14 day PB treatment only modified the levels of mdr1a/1b mRNA and the 40 day PB treatment still did not reverse the decrease in mdr1a, but increased the levels of mdr1b mRNA, as compared with PTZ-CT rats. This alteration of mdr1a/1b levels was not associated with P-GP protein levels, or P-GP function. We also found that the increase in mdr1a/1b concentrations in the cortex induced by kindling following 28 injections of PTZ was larger than that in the hippocampus, but alterations in P-GP protein levels in the cortex were similar to those in the hippocampus.
It was well known that under physiological conditions, P-GP is predominantly expressed at capillary endothelial cells, as well as at parenchymal and perivascular astrocytes at low levels (Aquilante et al., 2000). In addition to these three cell types, seizures also induce the expression of P-GP in neurones (Volk et al., 2005). Using mdr1a-knock-out mice, Schinkel et al. (1994) proposed that alterations in mdr1b mRNA might compensate for changes in mdr1a mRNA, but this would not explain the present findings. In addition to their common action as a drug efflux transporter, individual mdr1 isoforms appear to have a specific function. Products of the mdr1a have been proposed to regulate cell volume by influencing swelling-activated chloride currents via a protein kinase C sensitive phosphorylation site on P-GP (Bond et al., 1998), whereas mdr1b has been shown to be involved in apoptotic mechanisms (Lecureur et al., 2001) and cellular stress (Zhou and Kuo, 1998; Ziemann et al., 1999). However, these findings are still controversial. Other studies have suggested that expression of the mdr1 contributes to neuroprotection because a link was found between loss of proapoptotic protein p53 and expression of mdr1a/mdr1b (Bush and Li, 2002; Marroni et al., 2003). The exact location of each mdr1 isoform in the normal brain has not been fully elucidated, although mdr1a appears to be preferentially expressed in microvessel endothelium in the brain (Demeule et al., 2001) and the mdr1b is mainly expressed in the cerebral cells (astrocytes or neurones) (Ballerin et al., 2002). Both kindling and long-term PB treatment may simultaneously increase the expression of these two genes, which indicates that overexpression of P-GP in the brain results from the concurrent effects of mdr1a and mdr1b, with mdr1a contributing more than mdr1b.
The RT-PCR results indicate that mdr1a/mdr1b changes in the hippocampus were highly correlated with the changes in P-GP function and expression induced by epilepsy and PB. The 40 day PB treatment again decreased the hippocampus-to-plasma concentration ratios of Rho123 and this was accompanied by an increase in mdr1a/mdr1b mRNA levels and a decrease in PB concentrations in the hippocampus. In contrast, mdr1a/mdr1b changes in the cortex were less associated with the changes in P-GP function and expression under the same conditions, which indicates that lesions of the hippocampus and the concentration of therapeutic drug in the hippocampus is closely associated with epilepsy. The hippocampus, amygdala, and parahippocampal region are components of the mesial temporal lobe. The hippocampus is thought to be a key structure for the facilitation of temporal lobe epilepsy, the most prevalent of all epilepsies (Lowenstein, 1996; Wasterlain et al., 1996; Ben-Ari and Cossart, 2000). The surgical removal of the hippocampus and its surrounding structures has been found to result in a decrease or complete cessation of seizures in human patients, which validates the epileptogenicity of the hippocampus and its role in epileptogenesis (Falconer and Serafetinides, 1963; Rasmussen, 1983; King et al., 1986; Clusmann et al., 2002; Hardy et al., 2003; Wieser et al., 2003).
In conclusion, our data strongly suggest that the overexpression of P-GP during the development of pharmacoresistant epilepsy is much more complex than previously thought and that the role of the hippocampus in the development of pharmacoresistant epilepsy is more important than that of the cerebral cortex. Early PB treatment reversed the increase in P-GP function and expression induced by kindling, but long-term PB treatment enhanced its overexpression. All the results indicate that the overexpression of P-GP in the brain is induced by the epileptic seizures during the early stages and by the combined effects of chronic drug treatment and epilepsy at the later stages.
Acknowledgments
The work was supported by the Project of ‘333’, Jiangsu Province (No. Y092009).
Glossary
Abbreviations:
- AED
anti-epileptic drug
- BBB
blood–brain barrier
- CMC-Na
carboxymethylcellulose sodium
- P-GP
P-glycoprotein
- PB
phenobarbital
- PTZ
pentylenetetrazole
- Rho123
rhodamine 123
Conflict of interest
None.
References
- Alqawi O, Bates S, Georges E. Arginine482 to threonine mutation in the breast cancer resistance protein ABCG2 inhibits rhodamine 123 transport while increasing binding. Biochem J. 2004;382:711–716. doi: 10.1042/BJ20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilante CL, Letrent SP, Pollack GM, Brouwer KL. Increased brain P-glycoprotein in morphine tolerant rats. Life Sci. 2000;66:47–51. doi: 10.1016/s0024-3205(99)00599-8. [DOI] [PubMed] [Google Scholar]
- Ballerin P, Di IP, Ciccarelli R, Nargi E, D'Alimonte I, Traversa U, et al. Glial cells express multiple ATP binding cassette proteins, which are involved in ATP release. Neuroreport. 2002;13:1789–1792. doi: 10.1097/00001756-200210070-00019. [DOI] [PubMed] [Google Scholar]
- Bankstahl JP, Löscher W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res. 2008;82(1):70–85. doi: 10.1016/j.eplepsyres.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Barta CA, Sachs-Barrable K, Feng F, Wasan KM. Effects of monoglycerides on P-glycoprotein: modulation of the activity and expression in Caco-2 cell monolayers. Mol Pharm. 2008;5:863–875. doi: 10.1021/mp800050q. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27(12):652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Bond TD, Valverde MA, Higgins CF. Protein kinase C phosphorylation disengages human and mouse-1a P-glycoproteins from influencing the rate of activation of swelling activated chloride currents. J Physiol. 1998;508:333–340. doi: 10.1111/j.1469-7793.1998.333bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordet R. Drug-resistant partial epilepsy: pharmacological criteria. Rev Neurol (Paris) 2004;160:36–42. [PubMed] [Google Scholar]
- Brandt C, Bethmann K, Gastens AM, Löscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006;24:202–211. doi: 10.1016/j.nbd.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Kwan P. Phenobarbital: a drug for the 21st century? Epilepsy Behav. 2004;5:802–803. doi: 10.1016/j.yebeh.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Bush JA, Li G. Regulation of the p53-deficient mouse model. Carcinogenesis. 2002;23:1603–1607. doi: 10.1093/carcin/23.10.1603. [DOI] [PubMed] [Google Scholar]
- Clusmann H, Schramm J, Kral T, Helmstaedter C, Ostertun B, Fimmers R, et al. Prognostic factors and outcome after different types of resection for temporal lobe epilepsy. J Neurosurg. 2002;97:1131–1141. doi: 10.3171/jns.2002.97.5.1131. [DOI] [PubMed] [Google Scholar]
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet. 1995;346:140–144. doi: 10.1016/s0140-6736(95)91208-8. [DOI] [PubMed] [Google Scholar]
- De Sarro A, Naccari F, De Sarro G. Enhanced susceptibility of pentylenetetrazole kindled mice to quinolone effects. Int J Antimicrob Agents. 1999;12:239–244. doi: 10.1016/s0924-8579(99)00067-9. [DOI] [PubMed] [Google Scholar]
- Demeule M, Labelle M, Régina A, Berthelet F, Béliveau R. Isolation of endothelial cells from brain, lung, and kidney: expression of the multidrug resistance P-glycoprotein isoforms. Biochem Biophys Res Commun. 2001;281:827–834. doi: 10.1006/bbrc.2001.4312. [DOI] [PubMed] [Google Scholar]
- Dogan AL, Legrand O, Faussat AM, Perrot JY, Marie JP. Evaluation and comparison of MRP1 activity with three fluorescent dyes and three modulators in leukemic cell lines. Leuk Res. 2004;28:619–622. doi: 10.1016/j.leukres.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Dombrowski SM, Desai SY, Marroni M, Cucullo L, Goodrich K, Bingaman W, et al. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42:1501–1506. doi: 10.1046/j.1528-1157.2001.12301.x. [DOI] [PubMed] [Google Scholar]
- Eyal S, Lamb JG, Smith-Yockman M, Yagen B, Fibach E, Altschuler Y, et al. The antiepileptic and anticancer agent, valproic acid, induces P-glycoprotein in human tumour cell lines and in rat liver. Br J Pharmacol. 2006;149:250–260. doi: 10.1038/sj.bjp.0706830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer MA, Serafetinides EA. A follow-up study of surgery in temporal lobe epilepsy. J Neurosurg Psychiatry. 1963;26:154–165. doi: 10.1136/jnnp.26.2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Faull RLM, Bullock JY, Jansen K, Mee EW, Walker EB, et al. Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res. 1996;710:56–68. doi: 10.1016/0006-8993(95)01313-x. [DOI] [PubMed] [Google Scholar]
- Hardy SG, Miller JW, Holmes MD, Born DE, Ojemann GA, Dodrill CB, et al. Factors predicting outcome of surgery for intractable epilepsy with pathologically verified mesial temporal sclerosis. Epilepsia. 2003;44:565–568. doi: 10.1046/j.1528-1157.2003.39202.x. [DOI] [PubMed] [Google Scholar]
- Kageyama M, Fukushima K, Togawa T, Fujimoto K, Taki M, Nishimura A, et al. Relationship between excretion clearance of rhodamine 123 and P-glycoprotein (Pgp) expression induced by representative Pgp inducers. Biol Pharm Bull. 2006;29:779–784. doi: 10.1248/bpb.29.779. [DOI] [PubMed] [Google Scholar]
- King DW, Flanigin HF, Gallagher BB, So EL, Murvin AJ, Smith DB, et al. Temporal lobectomy for partial complex seizures: evaluation, results and 1-year follow-up. Neurology. 1986;36:334–339. doi: 10.1212/wnl.36.3.334. [DOI] [PubMed] [Google Scholar]
- Kosopoulos IA, van Merode T, Kessels FG, de Krom MC, Knottnerus JA. Systematic review and meta-analysis of incidence studies of epilepsy and unprovoked seizures. Epilepsia. 2002;43:1402–1409. doi: 10.1046/j.1528-1157.2002.t01-1-26901.x. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Drug treatment of epilepsy: when does it fail and how to optimize its use? CNS Spectr. 2004;9:110–119. doi: 10.1017/s1092852900008476. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Potential role of drug transporters in the pathogenesis of medically intractable epilepsy. Epilepsia. 2005;46:224–235. doi: 10.1111/j.0013-9580.2005.31904.x. [DOI] [PubMed] [Google Scholar]
- Lazarowski A, Sevlever G, Taratuto A, Massaro M, Rabinowicz A. Tuberous sclerosis associated with MDR1 gene expression and drug-resistant epilepsy. Pediatr Neurol. 1999;21:731–734. doi: 10.1016/s0887-8994(99)00074-0. [DOI] [PubMed] [Google Scholar]
- Lecureur V, Thottassery JV, Sun D, Schuetz EG, Lahti J, Zambetti GP, et al. Mdr1b facilitates p53-mediated cell death and p53 is required for mdr1b upregulation in vivo. Oncogene. 2001;20:303–313. doi: 10.1038/sj.onc.1204065. [DOI] [PubMed] [Google Scholar]
- Liu HY, Liu XD, Jia L, Liu YC, Yang HW, Wang GJ, et al. Insulin therapy restores impaired function and expression of P-glycoprotein barrier of experimental diabetes. Biochem Pharmacol. 2008;75:1649–1658. doi: 10.1016/j.bcp.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Liu XD, Yang ZH, Yang JS, Yang HW. Increased P-glycoprotein expression and decreased phenobarbital distribution in the brain of pentylenetetrazole-kindled rats. Neuropharmacology. 2007;53:657–663. doi: 10.1016/j.neuropharm.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J Pharmacol Exp Ther. 2002;301:7–14. doi: 10.1124/jpet.301.1.7. [DOI] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005a;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005b;76:22–76. doi: 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRX. 2005c;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs: the search for new targets. Epilepsy Res. 2004;60:77–159. doi: 10.1016/j.eplepsyres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH. Recent advances related to basic mechanisms of epileptogenesis. Epilepsy Res Suppl. 1996;11:45–60. [PubMed] [Google Scholar]
- Marroni M, Agrawal ML, Kight K, Hallene KL, Hossain M, Cucullo L. Relationship between expression of multiple drug resistance proteins and p53 tumor suppressor gene proteins in human brain astrocytes. Neuroscience. 2003;121:605–617. doi: 10.1016/s0306-4522(03)00515-3. [DOI] [PubMed] [Google Scholar]
- Nagatomo I, Akasaki Y, Nagase F, Nomaguchi M, Takigawa M. Relationships between convulsive seizures and serum and brain concentrations of phenobarbital and zonisamide in mutant inbred strain EL mouse. Brain Res. 1996;731:190–198. doi: 10.1016/0006-8993(96)82386-9. [DOI] [PubMed] [Google Scholar]
- Nishimura A, Honda N, Sugioka N, Takada K, Shibata N. Evaluation of carbamazepine pharmacokinetic profiles in mice with kainic acid-induced acute seizures. Biol Pharm Bull. 2008;31:2302–2308. doi: 10.1248/bpb.31.2302. [DOI] [PubMed] [Google Scholar]
- Pires MM, Emmert D, Hrycyna CA, Chmielewski J. Inhibition of P-glycoprotein-mediated paclitaxel resistance by reversibly linked quinine homodimers. Mol Pharmacol. 2009;75:92–100. doi: 10.1124/mol.108.050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potschka H, Fedrowitz M, Löscher W. P-glycoprotein-mediated efflux of phenobarbital, lamotrigine and felbamate at the blood-brain barrier: evidence from microdialysis experiments in rats. Neurosci Lett. 2002;327:173–176. doi: 10.1016/s0304-3940(02)00423-8. [DOI] [PubMed] [Google Scholar]
- Rasmussen TB. Surgical treatment of complex partial seizures: results, lessons and problems. Epilepsia. 1983;24(Suppl. 1):S65–S76. doi: 10.1111/j.1528-1157.1983.tb04645.x. [DOI] [PubMed] [Google Scholar]
- Rebola N, Coelho JE, Costenla AR, Lopes LV, Parada A, Oliveira CR, et al. Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. Eur J Neurosci. 2003;18(4):820–828. doi: 10.1046/j.1460-9568.2003.02815.x. [DOI] [PubMed] [Google Scholar]
- Rebola N, Porciúncula LO, Lopes LV, Oliveira CR, Soares-da-Silva P, Cunha RA. Long-term effect of convulsive behavior on the density of adenosine A1 and A 2A receptors in the rat cerebral cortex. Epilepsia. 2005;46(Suppl. 5):159–165. doi: 10.1111/j.1528-1167.2005.01026.x. [DOI] [PubMed] [Google Scholar]
- Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res. 1999;34:109–122. doi: 10.1016/s0920-1211(98)00106-5. [DOI] [PubMed] [Google Scholar]
- Rizzi M, Caccia S, Giuso G, Richichi C, Gorter JA, Aronica E, et al. Limbic seizures induce P-glycoprotein in rodent brain: functional implications for pharmacoresistance. J Neurosci. 2002;22:5833–5839. doi: 10.1523/JNEUROSCI.22-14-05833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. Does P-glycoprotein play a role in pharmacoresistance to antiepileptic drugs? Epilepsy Behav. 2002;3:493–495. doi: 10.1016/s1525-5050(02)00557-7. [DOI] [PubMed] [Google Scholar]
- Sander JW, Shorvon SD. Incidence and prevalence studies in epilepsy and their methodological problems: a review. J Neurol Neurosurg Psychiatry. 1987;50:829–839. doi: 10.1136/jnnp.50.7.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Löscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr. 2009;9(2):47–52. doi: 10.1111/j.1535-7511.2008.01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seegers U, Potschka H, Löscher W. Lack of effects of prolonged treatment with phenobarbital or phenytoin on the expression of P-glycoprotein in various rat brain regions. Eur J Pharmacol. 2002;451:149–155. doi: 10.1016/s0014-2999(02)02235-5. [DOI] [PubMed] [Google Scholar]
- Sen A, Martinian L, Nikolic M, Walker MC, Thom M, Sisodiya SM. Increased NKCC1 expression in refractory human epilepsy. Epilepsy Res. 2007;74:220–227. doi: 10.1016/j.eplepsyres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Sisodiya SM, Lin WR, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain. 2002;125:22–31. doi: 10.1093/brain/awf002. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Omura S, Ohashi Y, Kawai M, Iwata Y, Tani K, et al. FK506 facilitates chemical kindling induced by pentylenetetrazole in rats. Epilepsy Res. 2001;46:279–282. doi: 10.1016/s0920-1211(01)00284-4. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Masuda M, Nakajima K, Ido N, Ohtsuka T, Nishida M, et al. P-glycoprotein function in peripheral T lymphocyte subsets of myasthenia gravis patients: clinical implications and influence of glucocorticoid administration. Int Immunopharmacol. 2009;9:284–290. doi: 10.1016/j.intimp.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Turncliff RZ, Tian X, Brouwer KL. Effect of culture conditions on the expression and function of Bsep, Mrp2, and Mdr1a/b in sandwich-cultured rat hepatocytes. Biochem Pharmacol. 2006;71:1520–1529. doi: 10.1016/j.bcp.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Volk HA, Arabadzisz D, Fritschy JM, Brandt C, Bethmann K, Löscher W. Antiepileptic drug resistant rats differ from drug responsive rats in hippocampal neurodegeneration and GABAA-receptor ligand-binding in a model of temporal lobe epilepsy. Neurobiol Dis. 2006;21:633–646. doi: 10.1016/j.nbd.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Volk HA, Potschka H, Löscher W. Increase expression of the multidrug transporter P-glycoprotein in limbic brain regions after amygdala-kindled seizures in rats. Epilepsy Res. 2004;58:67–79. doi: 10.1016/j.eplepsyres.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Volk HA, Potschka H, Löscher W. Immunohistochemical localization of P-glycoprotein in rat brain and detection of its increased expression by seizure are sensitive to fixation and staining variables. J Histochem Cytochem. 2005;34:517–531. doi: 10.1369/jhc.4A6451.2005. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhou D, Wang B, Li H, Chai H, Zhou Q, et al. A kindling model of pharmacoresistant temporal lobe epilepsy in Sprague-Dawley rats induced by Coriaria lactone and its possible mechanism. Epilepsia. 2003;44:475–488. doi: 10.1046/j.1528-1157.2003.32502.x. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Shirasaka Y, Mazarati AM, Spigelman I. Chronic epilepsy with damage restricted to the hippocampus: possible mechanisms. Epilepsy Res. 1996;26:255–265. doi: 10.1016/s0920-1211(96)00058-7. [DOI] [PubMed] [Google Scholar]
- Weiss J, Kerpen CJ, Lindenmaier H, Dormann SM, Haefeli WE. Interaction of antiepileptic drugs with human P-glycoprotein in vitro. J Pharmacol Exp Ther. 2003;307:262–267. doi: 10.1124/jpet.103.054197. [DOI] [PubMed] [Google Scholar]
- Wen T, Liu YC, Yang HW, Liu HY, Liu XD, Wang GJ, et al. Effect of 21-day exposure of phenobarbital, carbamazepine and phenytoin on P-glycoprotein expression and activity in the rat brain. J Neurol Sci. 2008;270:99–106. doi: 10.1016/j.jns.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Wieser HG, Ortega M, Friedman A, Yonekawa Y. Long-term seizure outcomes following amygdalohippocampectomy. J Neurosurg. 2003;98:751–763. doi: 10.3171/jns.2003.98.4.0751. [DOI] [PubMed] [Google Scholar]
- Wolf HK, Spänle M, Müller MB, Elger CE, Schramm J, Wiestler OD. Hippocampal loss of the GABAA receptor α1 subunit in patients with chronic pharmacoresistant epilepsies. Acta Neuropathol. 1994;88(4):313–319. doi: 10.1007/BF00310375. [DOI] [PubMed] [Google Scholar]
- Yang HW, Liu HY, Liu X, Zhang DM, Liu XD, Wang GJ, et al. Increased P-glycoprotein function and level after long-term exposure of four antiepileptic drugs to rat brain microvascular endothelial cells in vitro. Neurosci Lett. 2008;434:299–303. doi: 10.1016/j.neulet.2008.01.071. [DOI] [PubMed] [Google Scholar]
- Zhou G, Kuo MT. Wild-type p53-mediated induction of rat mdr1b expression by the anticancer drug daunorubicin. J Biol Chem. 1998;273:15387–15394. doi: 10.1074/jbc.273.25.15387. [DOI] [PubMed] [Google Scholar]
- Ziemann C, Burkle A, Kahl GF, Hirsch-Ernst KI. Reactive oxygen species participate in mdr1b mRNA and P-glycoprotein overexpression in primary rat hepatocyte cultures. Carcinogenesis. 1999;20:407–414. doi: 10.1093/carcin/20.3.407. [DOI] [PubMed] [Google Scholar]
- Zimprich F, Sunder-Plassmann R, Stogmann E, Gleiss A, Dal-Bianco A, Zimprich A, et al. Association of an ABCB1 gene haplotype with pharmacoresistance in temporal lobe epilepsy. Neurology. 2004;63:955–958. doi: 10.1212/01.wnl.0000141021.42763.f6. [DOI] [PubMed] [Google Scholar]