

Abstract
Background and purpose:
Alzheimer's disease (AD) is a multifactorial, neurodegenerative disease, which is in part caused by an impairment of synaptic function, probably mediated by oligomeric forms of amyloid-β (Aβ). While the Aβ pathology mainly affects the physiology of neurotransmission, neuronal decline is caused by excitotoxic cell death, which is mediated by the NMDA receptor. A comprehensive therapeutic approach should address both Aβ-induced synaptic deficits, as well as NMDA receptor-mediated neurodegeneration, via one molecular target. This study was designed to test whether calpain could be involved in both pathological pathways, which would offer a promising avenue for new treatments.
Experimental approach:
Application of the specific, water-soluble calpain inhibitor A-705253 was used to inhibit calpain in hippocampal slice cultures. We examined whether inhibition of calpain would prevent Aβ-induced deficits in neurotransmission in CA1, as well as NMDA-induced neuronal cell death.
Key results:
A-705253 dose-dependently prevented excitotoxicity-induced neurodegeneration at low nanomolar concentrations, determined by propidium iodide histochemistry. Inhibition of the NMDA receptor similarly protected from neuronal damage. Caspase staining indicated that calpain inhibition was protective by reducing apoptosis. Electrophysiological analysis revealed that inhibition of calpain by A-705253 also fully prevented Aβ oligomer-induced deficits in neurotransmission. The protective effect of calpain was compared to the clinically available NMDA receptor antagonist memantine, which was also effective in this model.
Conclusions and implications:
We suggest that inhibition of calpain exhibits a promising strategy to address several aspects of the pathology of AD that may go beyond the available therapeutic intervention by memantine.
Keywords: NMDA receptor, excitotoxicity, A-705253, oligomer, apoptosis, neuroprotection
Introduction
Alzheimer's disease (AD) is characterized by a number of particular neuroanatomical changes, including the deposition of amyloid-β (Aβ) and the intracellular formation of neurofibrillary tangles. Many publications have recently shown that Aβ in its soluble, oligomeric form interferes with synaptic function and plasticity of neurons. For example, synthetic Aβ oligomer preparations impair long-term potentiation (LTP) in vitro (Wang et al., 2002; 2004; Barghorn et al., 2005) and in vivo (Walsh et al., 2002). Injection of soluble Aβ in brain disturbs learning and memory (Cleary et al., 2005). Oligomeric Aβ is also found in brains of AD patients and amyloid precursor protein-over-expressing mice (Barghorn et al., 2005; Lesnéet al., 2006), and correlates with memory deficits (Lesnéet al., 2006). Besides the Aβ-induced synaptic pathology, AD is accompanied by shrinkage of brain regions that are involved in learning and memory, and of higher CNS functions. Several studies found neuronal loss in brains of AD patients, especially in areas like the nucleus basalis Meynert (Whitehouse et al., 1982), hippocampus (West et al., 1994; Rössler et al., 2002) and the entorhinal cortex (Gomez-Isla et al., 1996; Kordower et al., 2001). In some amyloid precursor protein-over-expressing transgenic animals, more than 50% of monoaminergic neurons are lost in subcortical regions (Liu et al., 2008), and it has been proposed that such cell loss is caused by an excitotoxic cascade involving the NMDA receptor (see Harkany et al., 2000). The most efficacious treatment of AD should therefore prevent both synaptic deficits caused by soluble Aβ, as well as neuronal decline.
There are indications that the Ca2+-dependent cysteine protease calpain may mediate both Aβ-induced impairment of synaptic vesicle release, as well as excitotoxic cell death: the former was shown by Kelly and Ferreira (2007), who provided evidence that Aβ oligomers prevent synaptic vesicle recycling in hippocampal neuron via dynamin 1 depletion, and that this process is mediated by calpain activation (Kelly et al., 2005). The role of calpain in cell death has been more extensively studied in various cell types, and it is now generally accepted that calpain is a central molecule in apoptotic pathways (Villa et al., 1998). Recently, calpain has also been suggested to contribute to excitotoxic processes in vitro (Chiu et al., 2005; Takano et al., 2005) and in vivo (Nimmrich et al., 2008b), indicating its involvement in neurodegenerative processes.
We have already described A-705253 as a highly specific low-molecular weight inhibitor of calpain (Lubisch et al., 2003). Unlike peptidic calpain inhibitors, A-705253 is selective against the proteasome, which also contributes to certain neurodegenerative diseases (Rubinsztein, 2006; Pan et al., 2008). A-705253 has recently been shown to successfully prevent oligomer-induced dynamin 1 cleavage in neurons (Sinjoanu et al., 2008), but it is still unclear whether this would result in a functional recovery of Aβ oligomer-induced synaptic disturbance. Using such a specific calpain inhibitor, we addressed the question whether inhibition of calpain would prevent both pathologies Aβ-induced synaptic deficits, as well as excitotoxic neuronal cell death, in the same in vitro model.
We here provide data that calpain inhibition fully prevents Aβ oligomer-triggered disturbed neurotransmission in hippocampal slice cultures. It also dose-dependently prevents excitotoxic neurodegeneration, indicating that inhibition of calpain may represent a promising avenue for the development of both symptomatic, as well as disease-modifying treatments of AD.
Methods
Preparation of slice cultures
All animal care and experimental procedures were according to the guidelines of the AAALAC commission, and were approved by the government of Rhineland Platinate.
For excitotoxicity studies
Male Wistar rats (7–9 days old) were used for the experiment. The animals were killed, and the brains were removed. Slice cultures were prepared as interphase cultures according to a modified protocol of Stoppini et al. (1991). Briefly, hippocampi were isolated, and transverse hippocampal slices (350 µM thickness) were prepared by using a McIlwain tissue chopper (Mickle Laboratory Engineering Co., Guildford, UK). The slices were placed on membrane inserts (0.4 µM Millicell-CM culture plate inserts, Cat # PICMORG 50, Millipore, Billerica, MA, USA) in six-well plates. Cultures were kept at 37°C with 5% CO2. The slices were cultured for the first 2 days in 1 mL of tissue culture medium consisting of 75% culture medium HME 03 (Cell Concepts, Umkirch, Germany), including 10 mg·mL−1 gentamycin (Biochrom, Berlin, Germany), 0.5% glutamine (Biochrom) and 25% horse serum (Gibco, Carlsbad, CA, USA), pH 7.4. From day 4, the slices were cultured in Neurobasal medium (Gibco) with 0.5% B27 supplement (Gibco) at 33°C.
For electrophysiological studies
Hippocampal slice cultures were prepared from 9- to 10-day-old Wistar rats (Janvier, Genest St.Ile, France). Hippocampi were isolated, and transverse hippocampal slices (400 µM thickness) were prepared by using a tissue chopper (Mickle Laboratory Engineering, Gomshall, UK) and cultured on millicell-CM membranes (Millipore, Billerica, MA, USA) in high-potassium medium [40% basal medium Eagle (BME) with Earle's salts, 25% horse serum, 25% Earle's balanced salt solution, 1 mM Glutamax I, 28 mM glucose, 10% 250 mM Na–HEPES in BME (all chemicals from Invitrogen, Paisley, UK, except Na–HEPES from Sigma-Aldrich, Steinheim, Germany)] at 34°C, 5% CO2 for 3 days, then in Neurobasal A medium (96.4% Neurobasal A medium, 2% B 27 supplement, 1 mM l-glutamine; all from Invitrogen, Paisley, UK), 25 mM d-glucose (Sigma-Aldrich).
Propidium iodide (PI) pre-selection
PI uptake is indicative of membrane injury, and correlates well with cell death (Macklis and Madison, 1990). At day 11 of culturing, 2 µM PI (10 µg·mL−1, Sigma) was added for 12 h, and the slices were pre-selected by fluorescence analysis to ensure that no anatomical damage had taken place. Only slices that did not show PI fluorescence were selected for the experiment. PI was removed after pre-selection by changing the culture medium.
Induction of excitotoxicity
At day 12 of culturing, slices were pretreated with A-705253 at the respective concentration, with MK-801 (dizocilpine maleate; 10 µM) as reference compound, or with vehicle only. Then, glutamate was added at a final concentration of 15 mM in the presence of the compounds. During this period, the culture medium of the treated slices and the negative control that did not receive glutamate contained 40 mM HEPES to buffer the excess acid introduced by the addition of glutamate. After this period, the culture medium was exchanged and the slices were cultured in the presence of the respective compounds until PI staining.
PI analysis
Twenty-two hours after the addition of glutamate, organotypic cultures were stained with PI for 2 h. PI fluorescence was elicited at 546 nm and recorded at >610 nm by an inverted fluorescence microscope (Nikon, Düsseldorf, Germany). Images were captured using a CCD camera (Visitron Systems, Puchheim, Germany), stored and subsequently analysed, without knowledge of the treatments, using an image analysis software (LUCIA M, Nikon). For quantification of neuronal damage, the percentage of CA1-CA3 area expressing PI fluorescence above background level was calculated in relation to the total area of each organotypic culture. The region of interest (the somatic and dendritic regions of areas CA1, CA2 and CA3) was determined manually. The amount of PI fluorescence was determined densitometrically after transforming the red values into grey values. In order to compensate for variations in PI fluorescence between different experiments, the values of the slice cultures treated under identical conditions were normalized against standard damage. Standard damage was obtained as the mean percentage of damage in organotypic slice cultures subjected to glutamate with no compound applied.
Caspase staining
After PI fluorescence determination, the slice cultures were fixed in 4% formaldehyde (Merck, Darmstadt, Germany) solution. Paraformaldehyde was dissolved in 0.1 M phosphate-buffered saline. After cryosectioning (20 µm), sections were washed in Tris-buffered saline (TBS) with 1% Triton X-100 (TBS/Triton) and subsequently incubated for 30 min in TBS/Triton with 10% fetal calf serum (FCS). Then, the antibody against the activated caspase 3 was applied (rabbit anti-mouse/human caspase 3A, R&D Systems, Minneapolis, MN, USA; AF835; Lot # 314061; dilution 1:1000 in TBS/Triton/FCS), and the slices were incubated in a moistened chamber at 4°C for 48 h. After the incubation period, the slices were washed with TBS/Triton, incubated with the secondary antibody (goat anti-rabbit-biotin, Dianova, Hamburg, Germany, dilution 1:500 in TBS/Triton/FCS) for 1 h in a moist chamber at room temperature, washed again and incubated for 1 h with ABC kit solution (Vector Laboratories, Burlingame, CA, USA) in a moist chamber at room temperature. After staining with diaminobenzidine for 2 min, the slices were air-dried on the slides and counterstained with Nissl stain. The quantification of the immunohistochemistry (single labelling; cell counting) was performed in areas CA1, CA3 and dentate gyrus (DG) using Zeiss Axiovision microscopy image analysis software. Data are given in cells/mm2.
Electrophysiological recordings
The slices were cultured for 15–25 days before recording. For oligomer experiments, 82 nM Aβ (1–42) globulomer (prepared according to Barghorn et al., 2005) was applied to slice culture medium 24 h before recording. In the compound group, 100 nM A-705253 or memantine was co-applied. Globulomer ultrafiltrate was used as control. For the calpain control experiment, the slices were maintained in culture for 16–18 days, and 100 nM A-705253 or vehicle was applied 24 h before recording. Shortly before recording slices were placed in a Haas interface recording chamber (Harvard Apparatus, Hugstetten, Germany) and continuously perfused with artificial cerebrospinal fluid (aCSF; composition in mM: NaCl, 118; KCl, 5; MgSO4, 2; CaCl2, 2.5; KH2PO4, 1.24; NaHCO3, 25.6; glucose, 10; pH 7.4; Barghorn et al. 2005), and allowed to equilibrate for at least 60 min at 32°C. The Schaffer collateral was then stimulated with bipolar pulses (0.1 ms/phase) using a 0.5 M bipolar tungsten electrode (WPI; Sarasota, FL, USA), and extracellular field potential (fEPSP) amplitudes were recorded with glass electrodes filled with aCSF (0.7–1.1 Megaohm, GC150F-15, Harvard Apparatus, Hugstetten, Germany). Signals were digitized using a power CED 1401 (Cambridge Electronic Design Ltd, Cambridge, UK) and analysed using Signal 2.14 (Cambridge Electronic Design).
Data analysis
Data are shown as means ± SEM. Significance of treatments in the electrophysiological assays (input/output relations) was determined by two-way anova. Statistical analysis of PI signal was assessed by Kruskal–Wallis and Dunn's multiple comparison test. Caspase-3 activation was analysed by the Mann–Whitney U-test.
Materials
A-705253 was synthesized at Abbott. Memantine was obtained from Sequoia, Pangbourne, UK, glutamate from Sigma-Aldrich and MK801 from Tocris, Bristol, UK.
Results
Inhibition of calpain prevents excitotoxic cell death in hippocampal slice cultures in a dose-dependent manner
In this study, the efficacy of A-705253 on neuronal survival was tested in organotypic slice cultures after a 45 min challenge with 15 mM glutamate. Glutamate application strongly induced neuronal cell death, as assessed by PI staining. The compound significantly reduced neuronal damage at concentrations from 10 nM to 10 µm, but not at lower concentrations (0.1 nM and 1 nM; Figure 1A). Maximal protection was reached at 10 nM (Figure 1B). The NMDA receptor antagonist MK-801 (10 µM) was used as positive control and was also effective, indicating that the damage elicited by glutamate was mostly dependent on the activation of NMDA receptors.
Figure 1.
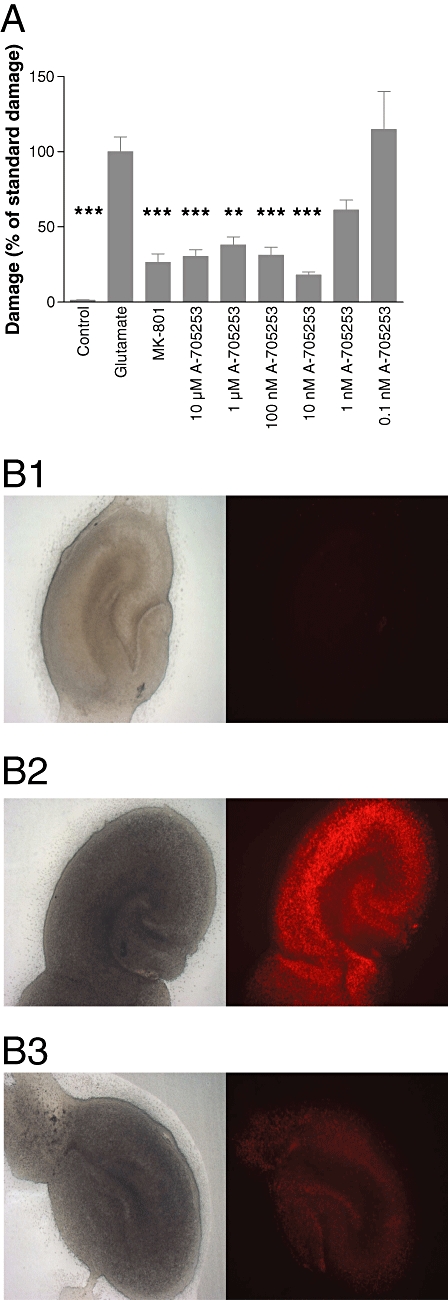
Effect of A-705253 on glutamate-induced damage assessed with PI staining. Application of A-705253 2 h before until 24 h after glutamate-induced excitotoxic insult concentration-dependently reduced damage in organotypic slice cultures. (A) Damage was assessed with PI staining 24 h after the insult. Control: n= 22 slices; glutamate: n= 26 slices; MK-801 (10 µM): n= 24 slices; A-705253 (10 µM): n= 25 slices; A-705253 (1 µM): n= 25 slices; A-705253 (100 nM): n= 25 slices; A-705253 (10 nM): n= 16 slices; A-705253 (1 nM): n= 7 slices; A-705253 (0.1 nM): n= 7 slices; ***P < 0.001; **P < 0.01; Kruskal–Wallis test and Dunn's multiple comparison test. (B) Typical slice without glutamate (control; B1), after treatment with glutamate (B2) and after treatment with glutamate and 10 nM A-705253 (B3). Left panel shows slices in phase contrast, right panel immunofluorescence after PI.
Inhibition of calpain prevents apoptotic cell death in hippocampal slice cultures
Calpain has been shown to contribute to apoptotic cell death. To evaluate whether the cell death induced in this assay was driven by apoptosis, and apoptotic cell death could be prevented by calpain inhibition, we tested slice cultures for caspase-3 activity, a molecular marker for apoptosis (Figure 2). Glutamate challenge strongly induced caspase-3 activation in CA3, CA1 and the DG, as detected immunohistochemically, indicating that cell death detected by PI was indeed apoptotic. Inhibition of calpain with A-705253 (10 nM–10 µM) significantly prevented caspase-3 activation, indicating that the protective effect of calpain was likely to be due to the prevention of apoptosis.
Figure 2.
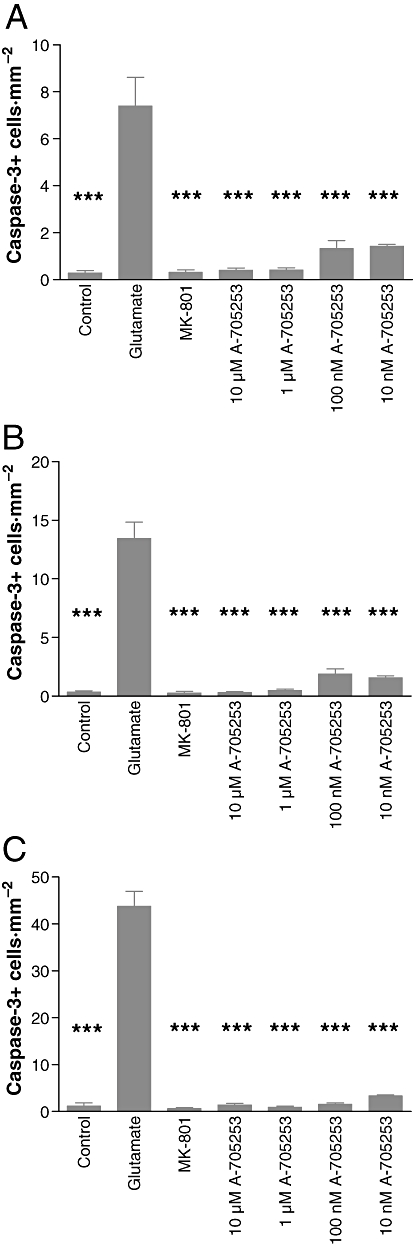
Effect of A-705253 on caspase-3 activation in hippocampal slice cultures. Application of A-705253, 2 h before until 24 h after excitotoxic insult induced by glutamate concentration-dependently reduced caspase-3 staining in organotypic slice cultures. The figure indicates the mean number of activated caspase-3 positive cells of eight different slices per condition in CA1 (A; ***P < 0.001; Mann–Whitney U-test) CA3 (B; ***P < 0.001; Mann–Whitney U-test) or in the DG (C; ***P < 0.001; Mann–Whitney U-test).
Inhibition of calpain prevents Aβ oligomer-induced deficits in synaptic transmission
For the analysis of neurotransmission, we recorded fEPSPs in the CA1 region of hippocampal slice cultures. Basal synaptic transmission was assessed by stimulation of the Schaffer collateral at increasing intensities, and simultaneous recording of the fEPSP responses in stratum pyramidale (input/output relation). For induction of synaptic deficits, we used the previously described Aβ oligomer preparation, Aβ (1–42) globulomer (Barghorn et al., 2005). We used an ultrafiltrate of this preparation as vehicle control. Synaptic activity was significantly reduced in slice cultures incubated for 24 h with 82 nM Aβ (1-42) globulomer. Co-application of 100 nM A-705253 completely prevented such deficit (Figure 3A).
Figure 3.
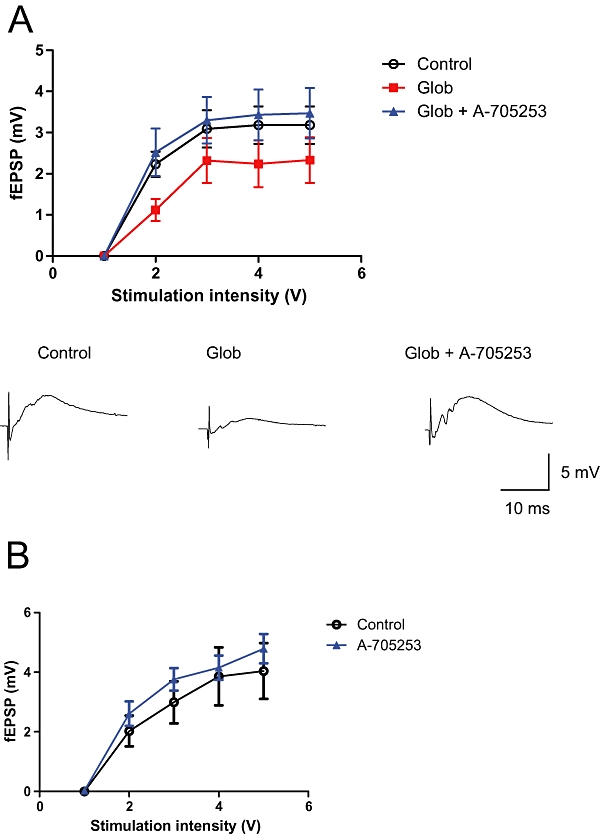
Effect of A-705253 on Aβ oligomer-induced deficits in synaptic transmission. Strength of synaptic transmission is illustrated by the input/output relation in hippocampal slice cultures after stimulation of the Schaffer collateral. A 24 h application of 82 nM globulomer significantly reduces glutamatergic transmission in CA1 (P= 0.012, two-way anova). Co-application of 100 nM A-705253 completely prevents this deficit (P < 0.001, two-way anova). Upper panel: mean values from control slice cultures (n= 7), slice cultures with globulomer (n= 8) and those with globulomer + A-705253 (n= 8). Data points are shown as means ± SEM. Lower panel: representative traces at stimulation intensity 3V. (B) A-705253 does not affect the input/output relation (P= 0.017, two-way anova). Data shown are mean (±SEM) values from control slice cultures (n= 14) and those treated with A-705253 (n= 14).
Inhibition of calpain does not alter synaptic activity
We then analysed whether calpain inhibition alone could have increased neuronal activity in CA1, and thus simulate a preventive effect of Aβ pathology. We therefore applied the same concentration of A-705253 (100 nM) to slice cultures without parallel incubation of Aβ, and recorded synaptic activity. There was no significant change in the input/output relation as compared to control slice cultures after application of the calpain inhibitor (Figure 3B), indicating that calpain inhibition itself does not modify glutamatergic neurotransmission in CA1.
Memantine does not prevent Aβ oligomer-induced deficits in synaptic transmission
We wanted to examine whether the NMDA receptor antagonist memantine, which is used clinically for the treatment of AD, would prevent Aβ oligomer-induced synaptic deficits. Therefore, we co-applied memantine, at a concentration effective in other in vitro studies (10 µM; Volbracht et al., 2006), with Aβ (1–42) globulomer to hippocampal slice cultures and recorded basal synaptic transmission in CA1. In parallel, we recorded from slice cultures that were only treated with Aβ (1–42) globulomer, or with vehicle alone. Aβ (1–42) globulomer application significantly suppressed glutamatergic synaptic transmission, as determined by an input/output curve. Co-application of 10 µM memantine did not prevent this impairment (Figure 4C). However, when memantine was applied at 10-fold lower concentration (1 µM), it significantly reversed the globulomer-induced suppression of glutamatergic transmission (Figure 4A). Although there was a slight tendency for an improvement also at 3 µM memantine, it did not reach the level of significance (Figure 4B). We then analysed whether the apparent reversal at 1 µM memantine was due to a direct effect of the compound on the input–output relation. Memantine did not affect neurotransmission, when applied alone (Figure 4D).
Figure 4.
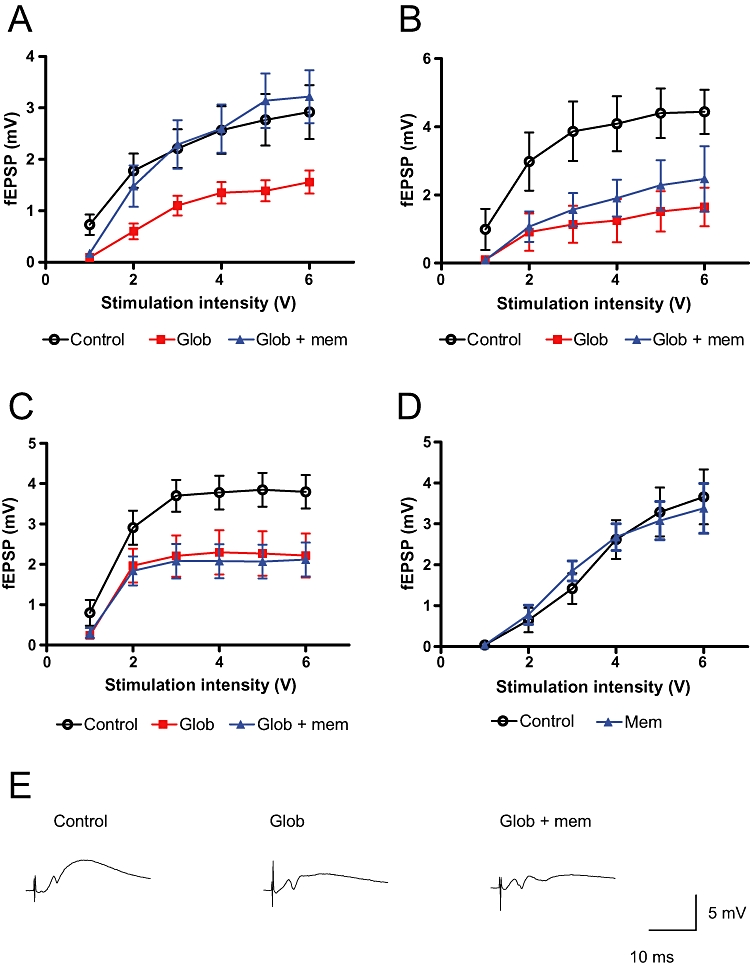
Effect of memantine on Aβ oligomer-induced deficits in synaptic transmission. Strength of synaptic transmission is illustrated by the input/output relation in hippocampal slice cultures after stimulation of the Schaffer collateral. (A–D) Mean values from control slice cultures, globulomer-treated slice cultures and slice cultures treated with globulomer and memantine. (A) 24 h application of 82 nM globulomer significantly reduced glutamatergic neurotransmission in CA1 (P < 0.001; two-way anova). Co-application of 1 µM memantine fully prevented this deficit (P < 0.001; two-way anova; n= 8 slice cultures/group). (B) 24 h application of 82 nM globulomer significantly reduced glutamatergic neurotransmission in CA1 (P < 0.001; two-way anova). Co-application of 3 µM memantine did not prevent this deficit (P= 0.247; two-way anova; n= 5–8 slice cultures per group). (C) 24 h application of 82 nM globulomer significantly reduced glutamatergic neurotransmission in CA1 (P < 0.001; two-way anova). Co-application of 10 µM memantine did not prevent this deficit (P= 0.622; two-way anova; n= 11–12 slice cultures/group). (D) Memantine did not have any effect on the input–output relation when applied for 24 h at 1 µM (n= 10 slice cultures/group). Data points are shown as means ± SEM. Representative sample traces (at 3 V stimulation intensity) for the input–output curve in C are shown in E.
Discussion and conclusions
A major challenge for the development of treatment strategies for AD is in the nature of this heterogenous disease: AD not only combines several pathologies, most prominently the over-expression of Aβ and the hyperphosphorylation of tau protein. It also involves physically and temporally spaced cascades, which are usually activated by different proteins. The progression of the disease is finally accompanied by massive brain shrinkage, which is at least in part due to progressive loss of cells and reduced synaptic processes in various brain regions involved in cognition (e.g. Whitehouse et al., 1982; West et al., 1994; Gomez-Isla et al., 1996).
Our studies were encouraged by the finding that calpain, a Ca2+-dependent cysteine protease, may contribute to several of the described pathologies. First, calpain cleaves various proteins of the cascades of neuronal cell death (Wu et al., 2004; Hou et al., 2006). Xu et al. (2007) provided data showing that calpain-mediated truncation of mGluR1α is required for excitotoxicity. Cleavage of p35, the precursor of p25 (an activator of cdk5), by calpain mediates degeneration of cortical neurons in vitro (Kim et al., 2007). Interestingly, the cleavage product of p35 also activates cdk5, a kinase that hyperphosphorylates tau protein, leading to the formation of neurofibrillary tangles (Noble et al., 2003). The recent finding that calpain may mediate the Aβ-induced dysfunction of neurotransmission (Kelly et al., 2005) indicates that inhibition of calpain may interfere with several of the proposed pathways during AD.
This study analyses the contribution of calpain to two pathological processes relevant to AD and possibly other neurodegenerative disorders. Both processes, Aβ-induced synaptic dysfunction as well as NMDA receptor-mediated neuronal cell death, were elicited in rat hippocampal slice cultures and damage measured electrophysiologically or by immunohistochemistry. Application of the non-peptidic, low-molecular weight calpain inhibitor A-705253 completely prevented synaptic impairment as well as loss of neuronal cells. This dual mode of protection offers a promising strategy for the development of therapeutics against AD.
Calpain inhibition prevents NMDA receptor-mediated neuronal cell death
We have previously shown that calpain inhibition by A-705253 was neuroprotecitve in vivo in NMDA-induced degeneration of the nucleus basalis magnocellularis of Meynert in rat (Nimmrich et al., 2008b). The relevance of our findings is also strengthened by other reports showing that excitotoxic cell death can be prevented by calpain inhibition in vivo (Chiu et al., 2005; Takano et al., 2005). Although the nature of cellular degeneration in AD is not well understood, there are indications that excessive calcium influx due to an overstimulation of the NMDA receptor may underlie such cellular decline (Lipton and Rosenberg, 1994; Harkany et al., 2000). To prove the relevance of our in vitro study, we controlled our excitotoxicity experiment for NMDA receptor dependency, and treated slice cultures in parallel with MK-801, an NMDA receptor antagonist. Such treatment completely prevented glutamate-induced cell death, indicating that the cell death model in this study fully depends on NMDA receptor activation. Although this suggests that calpain inhibition interferes with NMDA receptor-dependent cascades, the physiological function of the NMDA receptor is not altered by the same concentration of A-705253 in hippocampal slices. This was shown in a previous study where calpain inhibition using A-705253 at neuroprotective concentrations did not impair NMDA-mediated LTP (Nimmrich et al., 2008b). The fact that it nevertheless protects from neurodegenerative processes, and hence may prevent cognitive decline, could open promising avenues for the generation of therapeutics against neurodegeneration-associated cognitive deficits.
Inhibition of calpain prevents Aβ-induced synaptic dysfunction
Many publications have recently explored the mechanism of Aβ-induced synaptic impairment. However, the exact mechanism is not entirely understood. Several authors suggested that oligomeric Aβ may affect NMDA receptor functioning, thereby altering synaptic function and plasticity (Kelly and Ferreira, 2006; Shankar et al., 2007). We recently provided evidence that one oligomer preparation, Aβ (1–42) globulomer, specifically interferes with the presynaptic P/Q calcium channel (Nimmrich et al., 2008a). Here, we show that calpain inhibition can prevent globulomer-induced synaptic dysfunction. This supports recent findings of Kelly et al. (2005), which suggested that Aβ oligomer-induced synaptic dysfunction is due to an increased dynamin cleavage by calpain. As a consequence, vesicle recycling is impaired, leading to a reduced pool of readily releasable vesicles (Kelly and Ferreira, 2007). Recently, the compound used here, A-705253, was shown to inhibit Aβ oligomer-induced dynamin cleavage in hippocampal neurons (Sinjoanu et al., 2008). Our study suggests that this translates into functional recovery from oligomer-induced deficits. Theoretically, our observations could have been obscured by a direct effect of A-705253 on synaptic activity. We therefore controlled for this possibility, and showed that calpain inhibition alone did not alter synaptic transmission. The observed effect is thus more likely to be due to a prevention of the synaptic deficit.
Memantine is an NMDA receptor antagonist, which is used clinically for the treatment of AD. The mechanism of action of memantine has been extensively studied, and it has been shown to protect from excitotoxic neurodegeneration (Volbracht et al., 2006). While memantine is effective in excitotoxicity models, it has not been evaluated whether memantine would also be able to prevent the physiological alterations mediated by the Aβ oligomer. For this reason, we tested memantine also in our model of Aβ-oligomer-induced suppression of glutamateric transmission. Memantine was completely ineffective at a concentration usually used in other in vitro models (10 µM). However, it significantly reversed the globulomer-induced deficits at a lower concentration (1 µM). This indicates that the globulomer induced an NMDA receptor-related pathological cascade, and that calpain may be protective against Aβ-triggered pathological events involving the NMDA receptor.
The calpain inhibitor A-705253 was shown to be specific and water soluble (Lubisch et al., 2003). The IC50 for A-705253 previously reported (Nimmrich et al., 2008b) is also in the nanomolar range (0.41 µM), albeit not completely identical to the IC50 in the toxicity assay described here. A caveat in the determination of calpain activation is the fact that such activation can only be indirectly measured, for example, by spectrin degradation products. However, it is not certain whether such readouts correctly reflect the IC50 of calpain inhibition in any given assay, and they are certainly not fully comparable to the IC50 determined in other systems. However, unlike peptidic calpain inhibitors, A-705253 does not affect the proteasome complex, which contributes, in a complex with ubiquitin, to some neurodegenerative diseases (Rubinsztein, 2006; McCray and Taylor, 2008; Pan et al., 2008). A-705253 effectively prevented both cell death as well as Aβ oligomer-induced synaptic deficits at nanomolar concentrations. Thus, it is likely that the protective effect was due to inhibition of calpain and not caused by other mechanisms. Nevertheless, the development of further specific compounds that could be used in conjunction with A-705253 is highly desirable for future studies.
Taken together, the results of this study showed that inhibition of calpain was sufficient to prevent two components of AD pathology in vitro: NMDA receptor-dependent excitotoxic neuronal degeneration and Aβ oligomer-induced disturbed neurotransmission. The calpain inhibitor was effective at concentrations previously shown to allow normal NMDA receptor function. Our results thus strengthen the hypothesis that calpain links several pathways relevant to the development of AD, and we suggest that inhibition of calpain may be an attractive therapeutic alternative to currently available therapeutic interventions. Further studies should reveal whether calpain inhibition will also prevent cognitive decline in chronic animal models of AD.
Acknowledgments
The authors would like to thank Tanja Georgi, Michael Bahr and Cornelia Garz for technical assistance.
Glossary
Abbreviations:
- A-705253
(N-(1-benzyl-2-carbamoyl-2-oxoethyl)-2-[E-2-(4-diethlyaminomethylphenyl) ethen-1-yl]benzamide
- Aβ
amyloid-β
- AD
Alzheimer's disease
- fEPSP
field excitatory postsynaptic potential
- PI
propidium iodide
- TBS
Tris-buffered saline
Conflict of interest
The authors state no conflict of interest.
References
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, et al. Globular amyloid beta-peptide oligomer – a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Chiu K, Lam TT, Li WWY, Caprioli J, Kwong JMK. Calpain and N-methyl-d-aspartate (NMDA)-induced excitotoxicity in rat retinas. Brain Res. 2005;1046:207–215. doi: 10.1016/j.brainres.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Abrahám I, Timmerman W, Laskay G, Tóth B, Sasvári M, et al. Beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci. 2000;12:2735–2745. doi: 10.1046/j.1460-9568.2000.00164.x. [DOI] [PubMed] [Google Scholar]
- Hou ST, Jiang SX, Desbois A, Huang D, Kelly J, Tessier L, et al. Calpain-cleaved collapsing response mediator-protein 3 induces neuronal death after glutamate toxicity and cerebral ischemia. J Neurosci. 2006;26:2241–2249. doi: 10.1523/JNEUROSCI.4485-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. Beta-amyloid-induced dynamin degradation is mediated by N-methyl-d-aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience. 2007;147:60–70. doi: 10.1016/j.neuroscience.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BL, Vassar R, Ferreira A. Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons. J Biol Chem. 2005;280:31746–31753. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Oh SJ, Park SH, Kang HJ, Won MH, Kang TC, et al. Neuronal loss in primary long-term cortical culture involves neurodegeneration-like cell death via calpain and p35 processing, but not developmental apoptosis or aging. Exp Mol Med. 2007;39:14–26. doi: 10.1038/emm.2007.3. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennett D, et al. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49:202–213. [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, et al. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubisch W, Beckenbach E, Bopp S, Hofmann H-P, Kartal A, Kästel C, et al. Benzoylalanine-derived ketoamines carrying venylbenzyl amino residues: discovery of potent water-soluble calpain inhibitors with oral bioavailability. J Med Chem. 2003;46:2404–2412. doi: 10.1021/jm0210717. [DOI] [PubMed] [Google Scholar]
- Macklis JD, Madison RD. Progressive incorporation of propidium iodide in cultured mouse neurons correlates with declining electrophysiological status: a fluorescence scale of membrane integrity. J Neurosci Methods. 1990;31:43–46. doi: 10.1016/0165-0270(90)90007-3. [DOI] [PubMed] [Google Scholar]
- McCray BA, Taylor JP. The role of autophagy in age-related neurodegeneration. Neurosignals. 2008;16:75–84. doi: 10.1159/000109761. [DOI] [PubMed] [Google Scholar]
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, et al. Amyloid β oligomers (Aβ 1–42 globulomer) suppress spontaneous synaptic activity by inhibition of P/Q calcium currents. J Neurosci. 2008a;28:788–797. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmrich V, Szabo R, Nyakas C, Granic C, Reymann KG, Schröder UH, et al. Inhibition of calpain prevents N-methyl-d-aspartate-induced degeneration of the nucleus basalis and associated behavioral dysfunction. J Pharmacol Exp Ther. 2008b;327:343–352. doi: 10.1124/jpet.108.142679. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- Rössler M, Zarski R, Bohl J, Ohm TG. Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer's disease. Acta Neuropathol. 2002;103:363–369. doi: 10.1007/s00401-001-0475-7. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signalling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinjoanu RC, Kleinschmidt S, Bitner RS, Brioni JD, Moeller A, Ferreira A. The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons. Neurochem Int. 2008;53:79–88. doi: 10.1016/j.neuint.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Meth. 1991;37:173–183. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, et al. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- Villa PG, Henzel WJ, Sensenbrenner M, Henderson CE, Pettmann B. Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J Cell Sci. 1998;111:713–722. doi: 10.1242/jcs.111.6.713. [DOI] [PubMed] [Google Scholar]
- Volbracht C, va Beek J, Zhu C, Blomgren K, Leist M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci. 2006;23:2611–2622. doi: 10.1111/j.1460-9568.2006.04787.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potentially inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, DeLong MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, et al. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-mediated mGluR1alpha truncation: a kex step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]