

Abstract
Our laboratory recently reported the identification of a peptide region, QVNI, within the prion domain of the yeast protein Ure2 that may act as an initiation point for fibril formation.1 This potential amyloid-forming region, which corresponds to residues 18–21 of Ure2, was initially identified by systematic cysteine scanning of the Ure2 prion domain. The point mutant R17C, and the corresponding octapeptide CQVNIGNR, were found to form fibrils rapidly under oxidative conditions due to the formation of a disulfide bond. Deletions within the QVNI sequence cause the fibril formation ability of R17C Ure2 to be inhibited. The aggregation propensity of this region is strongly modulated by its preceding residue: replacement of R17 with a hydrophobic residue promotes fibril formation in both full-length Ure2 and in the corresponding octapeptides. The wild-type octapeptide, RQVNIGNR, also forms fibrils, and is the shortest amyloid-forming peptide found for Ure2 to date. Interestingly, the wild-type octapeptide crystallizes readily and so provides a starting point towards obtaining high resolution structural information for the amyloid core of Ure2 fibrils.
Key words: yeast, prion, amyloid, cysteine scanning, disulphide bond, Ure2, Ure2p, peptide, crystal, initiation site
Unravelling Amyloid Structure
The association of amyloid-like fibrils with a number of human diseases2 as well as with other biological processes3 has provoked considerable research interest in understanding the mechanism by which fibrils form and how this process can be controlled. Obtaining detailed structural information about how the protein chains are arranged within amyloid fibrils has been the work of several decades, and only recently has significant progress been made.4–6 NMR has provided important insight into the fibrillar structure of the fungal prions Het-s7,8 and Sup35.9,10 Atomic resolution X-ray structural data was obtained first from microcrystals of a Sup35 peptide11 and this approach has since been extended to a range of amyloid peptides related to human diseases.12 Techniques such as solid-state NMR13,14 and H/D exchange combined with mass spectrometry15 have also been applied to Ure2; however, to date, these have yielded only limited structural information.5,6 The availability of activity assays for Ure2 has yielded insight into the overall architecture of the protein monomers within the fibrils, indicating that the globular GST-like C-domains have native structure16,17 and are arranged as dimers within the fibrillar arrays.18 However, the precise arrangement of the amyloid-like core is unknown and is a topic that has attracted some controversy.19,20 The slow rate of progress in defining the molecular structure of Ure2 fibrils may be attributed in part to several factors, including heterogeneity of the fibrils,21–24 the presence of a long flexible linker region between the N- and C-domains,14 and the lack of identifiable short amyloid-forming peptides which might provide a starting point to obtaining high-resolution structural data.1
Using Cysteine Scanning to Gain Insight into Amyloid Fibril Structure
Cysteine scanning, and introduction of disulphides and other linkers, are other approaches that have been used to gain insight into the structure of amyloids,25 including fibrils of Sup3526 and Ure2.27,28 Accessibility of cysteine residues26,29 or formation of disulphides or other linkages26–28 in preformed fibrils can provide insight into the packing structure of the monomeric polypeptide chains within the fibrils. However, the introduction of exogenous linkers, especially when combined with fluorescent or other labels, could potentially disrupt the packing within fibrils and so prevent the very interactions that are being probed by the technique. In general, introduction of disulphide-forming cysteine residues into amyloid-forming proteins has been found to inhibit formation of fibrils.21,26–28 Therefore, our discovery of a unique site within the Ure2 prion domain (R17) where the introduction of a disulphide bond dramatically increases the rate of fibril formation was particularly unexpected andinteresting.1 Importantly, the disulphide-containing fibrils are indistinguishable morphologically from the wild-type (WT) fibrils and are able to seed the WT protein to form fibrils, suggesting that the presence of the disulphide bond constrains the polypeptide chains in a way that promotes fibril formation and is compatible with the WT fibril structure.1 The R17C mutation was also identified in a previous in vivo screen for prion inducing mutations in Ure2: this mutant was functional and was found to increase the prion induction rate, though only when combined with other mutations in the N and/or C domains.30 In our in vitro study, R17C had no effect on the fibril formation rate under reducing conditions1 i.e., under conditions that mirror the reducing environment of the cytosol, and so our results obtained in vitro are consistent with the previous observations in vivo.30
Understanding the Driving Force for Amyloid Formation
In order to understand the mechanism of fibril formation, one of the pressing issues is to identify the driving force, or the trigger, for this reaction i.e., the first event during protein conformational change from the soluble state to amyloid fibrils. This information might allow us to control the initiation of amyloid formation in both prion diseases and other neurodegenerative diseases, such as Alzeimer and Parkinson. The “amyloid stretch hypothesis” proposes that a short peptide stretch bearing a highly amyloidogenic motif might supply most of the driving force needed to trigger the self-catalytic assembly of a protein into amyloid fibrils.31 This hypothesis is consistent with the finding that many amyloidogenic polypeptides contain shorter sequence regions that readily form amyloid;12,32,33 further, the insertion of an amyloidogenic peptide stretch into a non-amyloid-related protein can induce the protein to form amyloid.34 Currently available computational algorithms for predicting amyloid-forming regions have shown good agreement with experiment, at least for hydrophobic amyloid-forming sequences.33,35,36 The yeast prion proteins including Sup35, Ure2, Rnq1 and Swil1 are different in the fact that they lack continuous stretches of hydrophobic residues, but instead are rich in Gln and Asn,37 analogous to the Gln-rich sequences seen in some disease-related human proteins such as the Huntingtin protein.38 Hydrogen bonding between these polar residues is suggested to form the basis for their tendency to aggregate.11,39
Defining a Potential Amyloid Stretch within Ure2
In our study1 we systematically mutated every residue within the first 70 residues of the Ure2 prion domain that was neither a Gln nor an Asn residue, producing a total of 35 mutants (Fig. 1). Each of these point mutants of the full-length Ure2 protein was expressed in E. coli, purified and then incubated under oxidizing conditions (i.e., in the presence of 3 mM H2O2). Under these conditions, R17C formed fibrils significantly more rapidly than the WT protein (i.e., within hours instead of days when allowed to stand at 8°C), and was the only cysteine mutant that formed fibrils on a measurable time scale. (Reduced R17C or R17S formed fibrils on a similar timescale to WT). The fibrils of R17C were examined by electron microscopy and far-UV CD, and were found to be morphologically and structurally indistinguishable from WT fibrils, before and after proteinase K digestion. Further, the fibrils of R17C could seed the formation of WT fibrils, suggesting that the packing within the two types of fibrils is identical.
Figure 1.

The prion domain sequence of Ure2. The Gln/Asn-rich region of the 354-residue Ure2 protein extends from residues 1–89.43 The residues mutated to Cys and tested for amyloid-forming ability under oxidizing conditions1 are underlined. R17 is shown in bold.
Having established that linking the polypeptide chains with a disulphide bond at site 17 dramatically facilitates fibril formation of Ure2, we then made a series of deletions in the surrounding residues, in order to identify which residues within Ure2 are critical for fibril formation.1 We narrowed the crucial sequence region down to a 4-residue stretch immediately following on from site 17, QVNI (residues 18–21), as deletion of any one of these residues completely removed the ability of oxidized R17C to form fibrils, whereas even quite substantial deletions in other parts of the prion domain (e.g., Δ1-16, Δ22, Δ23, Δ24-42 or Δ42-81) still allowed fibril formation of R17C.
We then characterized a series of short peptides derived from the R17C Ure2 sequence, each containing the CQVNI sequence.1 This pentapeptide had extremely low solubility and no fibrils could be formed from it, but the hexa-(CQVNIG), hepta-(CQVNIGN) and octa-(CQVNIGNR) peptides could all form fibrils; the octapeptide, as the most soluble, was characterized further. This octapaptide formed fibrils within minutes under oxidizing conditions, and like full-length R17C, was also able to seed fibril formation of WT full-length Ure2. In order to further understand the potential role of site 17 in controlling fibril formation for Ure2, a series of substitutions were made in the octapeptide. We found that when the cysteine residue of the octapeptide (equivalent to site R17C in Ure2) was substituted with a hydrophobic residue (V, L, I, F or M), the octapeptide readily formed fibrils (i.e., within days when incubated with agitation at 8°C at a peptide concentration of 1–4 mg/ml), and these fibrils were likewise able to seed fibril formation of WT Ure2. In contrast, while the WT octapeptide (RQVNIGNR) was eventually able to form fibrils under these conditions, this required a much longer incubation time (i.e., several months). Interestingly, however, the WT octapeptide, which is predicted to be an intact β-strand in a model for Ure2 fibrils,40 readily formed crystals when incubated at higher concentration overnight (Fig. 2). Therefore the identification of this peptide provides a starting point for further structural characterization of Ure2 fibrils.
Figure 2.
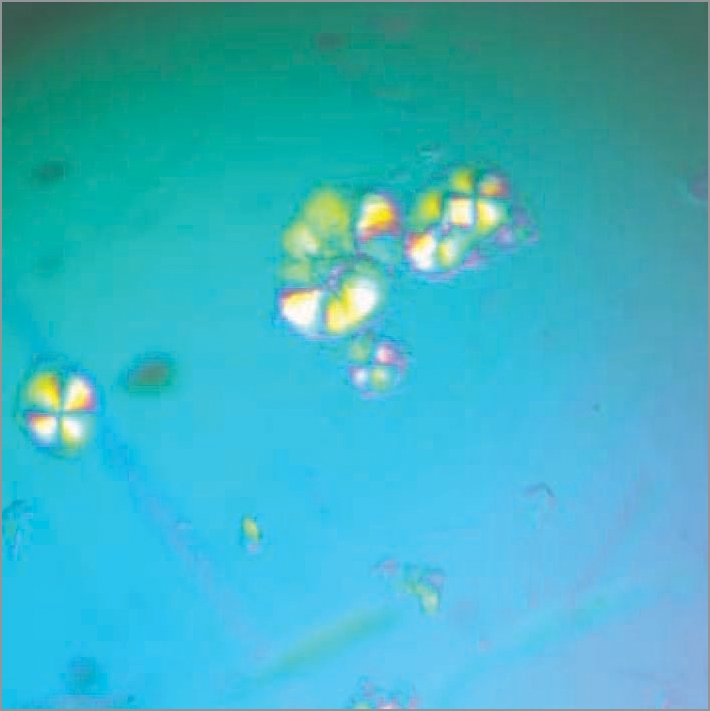
Crystals of the wild-type Ure2 octapeptide RQVNINIGNRNR viewed under the polarized microscope. The crystals were formed by incubating the octapeptide solution (10 mg/ml in Tris buffer pH 8.4 with 200 mM NaCl) at 4°C overnight.
Our peptide studies suggest that the charged residue, Arg, at site 17 of Ure2 actually reduces dramatically the amyloid forming propensity of the adjacent QVNI sequence.1 In order to confirm a similar role for this residue in the full-length protein, we introduced the equivalent hydrophobic substitutions at site 17. It was found that replacement of R17 with a hydrophobic residue increased the rate of fibril formation for Ure2, whereas substitution with another charged residue (e.g., Lys) inhibited fibril formation to a similar extent as the WT Arg. Deletion of residue 17, which brings a Leu residue adjacent to the QVNI sequence, had a similar effect as the other hydrophobic substitutions in promoting fibril formation.
Taken together, these results suggest that residue R17 in Ure2 acts as a gate-keeper residue for the downstream potential amyloid stretch QVNI, limiting its propensity to form fibrils. This is consistent with the likelihood that the prion state of Ure2 only conveys an advantage to the organism under rare and limited circumstances, and so it is likely that the Ure2 sequence has evolved to limit its tendency to switch to the prion state.5,41,42 The dramatic increase in the ability of the protein to form fibrils when disulphide cross-linked at site 17 suggests that this site and its downstream residues may act as an initiation point for assembly of the polypeptide chains into Ure2 fibrils, and that tethering at site 17 brings the polypeptide chains together in a manner that is not only compatible with the WT Ure2 fibril structure, but promotes its formation, thus mimicking the trigger for fibril formation in this protein.1
Acknowledgements
Work in the Perrett laboratory is supported by grants from the National Natural Science Foundation of China (30620130109, 30670428, 30870482), the Chinese Ministry of Science and Technology (2006CB500703, 2006CB910903) and the Chinese Academy of Sciences (KSCX2-YW-R-119). We thank members of the Perrett laboratory for critical reading of the manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/10670
References
- 1.Fei L, Perrett S. Disulfide Bond Formation Significantly Accelerates the Assembly of Ure2p Fibrils because of the Proximity of a Potential Amyloid Stretch. J Biol Chem. 2009;284:11134–11141. doi: 10.1074/jbc.M809673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Protein misfolding, functional amyloid and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid-from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Rambaran RN, Serpell LC. Amyloid fibrils: abnormal protein assembly. Prion. 2008;2:112–117. doi: 10.4161/pri.2.3.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrett S, Jones GW. Insights into the mechanism of prion propagation. Curr Opin Struct Biol. 2008;18:52–59. doi: 10.1016/j.sbi.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Baxa U. Structural basis of infectious and non-infectious amyloids. Curr Alzheimer Res. 2008;5:308–318. doi: 10.2174/156720508784533367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, et al. Correlation of structural elements and infectivity of the HET-s prion. Nature. 2005;435:844–848. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 9.Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc Natl Acad Sci USA. 2006;103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 11.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 13.Chan JC, Oyler NA, Yau WM, Tycko R. Parallel beta-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 2005;44:10669–10680. doi: 10.1021/bi050724t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baxa U, Wickner RB, Steven AC, Anderson DE, Marekov LN, Yau WM, Tycko R. Characterization of beta-sheet structure in Ure2p1-89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry. 2007;46:13149–131462. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- 15.Redeker V, Halgand F, Le Caer JP, Bousset L, Laprevote O, Melki R. Hydrogen/deuterium exchange mass spectrometric analysis of conformational changes accompanying the assembly of the yeast prion Ure2p into protein fibrils. J Mol Biol. 2007;369:1113–11125. doi: 10.1016/j.jmb.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 16.Bai M, Zhou JM, Perrett S. The yeast prion protein Ure2 shows glutathione peroxidase activity in both native and fibrillar forms. J Biol Chem. 2004;279:50025–50030. doi: 10.1074/jbc.M406612200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang ZR, Bai M, Wang XY, Zhou JM, Perrett S. “Restoration” of glutathione transferase activity by single-site mutation of the yeast prion protein Ure2. J Mol Biol 20. 2008;384:641–651. doi: 10.1016/j.jmb.2008.09.047. [DOI] [PubMed] [Google Scholar]
- 18.Zhang ZR, Perrett S. Novel glutaredoxin activity of the yeast prion protein Ure2 reveals a native-like dimer within fibrils. J Biol Chem. 2009;284:14058–14067. doi: 10.1074/jbc.M901189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bousset L, Briki F, Doucet J, Melki R. The native-like conformation of Ure2p in fibrils assembled under physiologically relevant conditions switches to an amyloid-like conformation upon heat-treatment of the fibrils. J Struct Biol. 2003;141:132–142. doi: 10.1016/s1047-8477(02)00606-8. [DOI] [PubMed] [Google Scholar]
- 20.Baxa U, Cheng N, Winkler DC, Chiu TK, Davies DR, Sharma D, et al. Filaments of the Ure2p prion protein have a cross-beta core structure. J Struct Biol. 2005;150:170–179. doi: 10.1016/j.jsb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 21.Bousset L, Thomson NH, Radford SE, Melki R. The yeast prion Ure2p retains its native alpha-helical conformation upon assembly into protein fibrils in vitro. EMBO J. 2002;21:2903–29011. doi: 10.1093/emboj/cdf303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Y, Li H, Zhu L, Zhou JM, Perrett S. Amyloid nucleation and hierarchical assembly of Ure2p fibrils. Role of asparagine/glutamine repeat and nonrepeat regions of the prion domains. J Biol Chem. 2004;279:3361–3369. doi: 10.1074/jbc.M310494200. [DOI] [PubMed] [Google Scholar]
- 23.Catharino S, Buchner J, Walter S. Characterization of oligomeric species in the fibrillization pathway of the yeast prion Ure2p. Biol Chem. 2005;386:633–641. doi: 10.1515/BC.2005.074. [DOI] [PubMed] [Google Scholar]
- 24.Ranson N, Stromer T, Bousset L, Melki R, Serpell LC. Insights into the architecture of the Ure2p yeast protein assemblies from helical twisted fibrils. Protein Sci. 2006;15:2481–2487. doi: 10.1110/ps.062215206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shivaprasad S, Wetzel R. Analysis of amyloid fibril structure by scanning cysteine mutagenesis. Methods Enzymol. 2006;413:182–198. doi: 10.1016/S0076-6879(06)13010-4. [DOI] [PubMed] [Google Scholar]
- 26.Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fay N, Redeker V, Savistchenko J, Dubois S, Bousset L, Melki R. Structure of the prion Ure2p in protein fibrils assembled in vitro. J Biol Chem. 2005;280:37149–37158. doi: 10.1074/jbc.M506917200. [DOI] [PubMed] [Google Scholar]
- 28.Fayard B, Fay N, David G, Doucet J, Melki R. Packing of the prion Ure2p in protein fibrils probed by fluorescence X-ray near-edge structure spectroscopy at sulfur K-edge. J Mol Biol. 2006;356:843–839. doi: 10.1016/j.jmb.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 29.Shivaprasad S, Wetzel R. Scanning cysteine mutagenesis analysis of Abeta-(1-40) amyloid fibrils. J Biol Chem. 2006;281:993–1000. doi: 10.1074/jbc.M505091200. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Bellot E, Guillemet E, Cullin C. The yeast prion [URE3] can be greatly induced by a functional mutated URE2 allele. EMBO J. 2000;19:3215–3222. doi: 10.1093/emboj/19.13.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastor MT, Esteras-Chopo A, Serrano L. Hacking the code of amyloid formation: the amyloid stretch hypothesis. Prion. 2007;1:9–14. doi: 10.4161/pri.1.1.4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, et al. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J Mol Biol. 2000;295:1055–1071. doi: 10.1006/jmbi.1999.3422. [DOI] [PubMed] [Google Scholar]
- 33.Ivanova MI, Thompson MJ, Eisenberg D. A systematic screen of beta(2)-microglobulin and insulin for amyloid-like segments. Proc Natl Acad Sci USA. 2006;103:4079–4082. doi: 10.1073/pnas.0511298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esteras-Chopo A, Serrano L, Lopez de la Paz M. The amyloid stretch hypothesis: recruiting proteins toward the dark side. Proc Natl Acad Sci USA. 2005;102:16672–16677. doi: 10.1073/pnas.0505905102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- 36.Cerda-Costa N, Esteras-Chopo A, Aviles FX, Serrano L, Villegas V. Early kinetics of amyloid fibril formation reveals conformational reorganisation of initial aggregates. J Mol Biol. 2007;366:1351–1363. doi: 10.1016/j.jmb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Crow E, Du Z, Li L. New insights into prion biology from the novel [SWI+] system. Prion. 2008;2:141–144. doi: 10.4161/pri.2.4.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- 39.Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc Natl Acad Sci USA. 2002;99:5596–5600. doi: 10.1073/pnas.042681599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kajava AV, Baxa U, Wickner RB, Steven AC. A model for Ure2p prion filaments and other amyloids: the parallel superpleated beta-structure. Proc Natl Acad Sci USA. 2004;101:7885–7890. doi: 10.1073/pnas.0402427101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 42.Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol. 2007;5:611–618. doi: 10.1038/nrmicro1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perrett S, Freeman SJ, Butler PJ, Fersht AR. Equilibrium folding properties of the yeast prion protein determinant Ure2. J Mol Biol. 1999;290:331–345. doi: 10.1006/jmbi.1999.2872. [DOI] [PubMed] [Google Scholar]