

Abstract
In eukaryotic cells amyloid aggregates may incorporate various functionally unrelated proteins. In mammalian diseases this may cause amyloid toxicity, while in yeast this could contribute to prion phenotypes. Insolubility of amyloids in the presence of strong ionic detergents, such as SDS or sarcosyl, allows discrimination between amorphous and amyloid aggregates. Here, we used this property of amyloids to study the interdependence of their formation in yeast. We observed that SDS-resistant polymers of proteins with extended polyglutamine domains caused the appearance of SDS or sarcosyl-insoluble polymers of three tested chromosomally-encoded Q/N-rich proteins, Sup35, Rnq1 and Pub1. These polymers were non-heritable, since they could not propagate in the absence of polyglutamine polymers. Sup35 prion polymers caused the appearance of non-heritable sarcosyl-resistant polymers of Pub1. Since eukaryotic genomes encode hundreds of proteins with long Q/N-rich regions, polymer interdependence suggests that conversion of a single protein into polymer form may significantly affect cell physiology by causing partial transfer of other Q/N-rich proteins into a non-functional polymer state.
Key words: amyloid, prion, [PSI+], huntingtin, polyglutamine, Saccharomyces cerevisiae, Sup35/eRF3
Introduction
Amyloids are fibrous protein aggregates resulting from non-covalent polymerization of normally soluble proteins, accompanied by their deep conformational rearrangement and formation of a specific cross-β structure. Though interest in amyloids was initially caused by their relation to human and animal pathologies, this phenomenon may have broader significance, since a number of amyloids are functional (reviewed refs. 1–3).
Saccharomyces cerevisiae is a very convenient experimental model for studying the amyloid phenomenon, due to similarity of mechanisms of aggregate formation in higher organisms and in yeast and the fact that both mammals and yeast may contain self-propagating infectious amyloids, called prions.4,5 In contrast to mammalian prions, which are responsible for incurable neurodegenerative diseases, yeast prions manifest themselves as heritable elements with unusual genetic properties. The increasing number of discovered yeast prions suggests that this phenomenon is widespread.6–11
The most characterized yeast prion, [PSI+], relates to the prion state of the translation termination factor Sup35 (eRF3).6 The nonsense suppressor phenotype of [PSI+] reflects increased levels of nonsense codon readthrough due to partial aggregation and inactivation of Sup35.12,13 Generation of [PSI+] de novo requires transient Sup35 overproduction and the presence of another yeast prion, [PIN+], which is based on the Rnq1 protein.7,14 Prion polymers of Rnq1 serve as imperfect templates for polymerization of prionogenic proteins, thus facilitating appearance of other yeast prions.15,16 Remarkably, among the Sup35 amyloid polymers seeded by Rnq1 only a small portion has prion properties, while the vast majority belongs to a non-prion type.16
In addition to [PIN+], [PSI+] generation by overproduced Sup35 can be induced by overproduction of Rnq1, Ure2, Swi1 and eight other proteins,14 as well as by overproduction of proteins with the expanded polyglutamine (polyQ) domain of huntingtin.15 All of the identified yeast proteins possess glutamine and/or asparagine (Q/N)-rich regions, a trait characteristic of known prionogenic proteins in yeast, and it is likely that, similarly to polyQ proteins, overproduction of these Q/N-rich proteins leads to their polymerization in an amyloid form. However, of these only Ure2, Rnq1, Swi1 and Cyc8 were shown to exist in a prion state.6–8,11 This suggests that the other proteins aggregate stably only when they are overproduced, i.e., represent non-prion amyloids which can seed Sup35 prion polymerization as the [PIN+] prion does. Taken together, these data show that yeast prions and non-prion amyloids can promote the appearance of each other. Most probably, this effect relies on co-polymerization of Q/N-rich proteins.
Both in yeast and humans, the phenotypic effects of amyloids can manifest themselves via two mechanisms: either reduction of function of the aggregating protein or the interference of amyloids with cellular functions. The former mechanism applies to yeast prions and to some amyloid diseases, e.g., cystic fibrosis. In Alzheimer, Parkinson and polyQ disorders, misfolded proteins disrupt proper cellular function and cause cytotoxicity. For example, in Huntington disease and, probably, other polyQ disorders, proteins with extended polyQ tracts induce disease because they interfere with the normal function of cellular proteins. Indeed, it was shown that pathological huntingtin impairs gene transcription either by intranuclear aggregate formation, or by sequestration of transcription factors (reviewed in refs. 17 and 18). Also, the accumulation of chaperones, proteasomes and ubiquitin in polyQ aggregates suggests that insufficient protein folding and degradation are also involved in polyQ disease pathogenesis.19,20 Among other amyloidoses, polyQ disorders and, particularly, Huntington’s disease are especially attractive for modeling in yeast, because, similarly to them, yeast prions rely on domains enriched with Q. Similarly to yeast prions and Sup35 non-prion amyloids, the formation of polyQ amyloids in yeast depends on [PIN+],21 and, as in humans, it increases with the polyQ length. PolyQ overproduction may be toxic in yeast, and this effect is modulated by a suite of genome-encoded Q/N-rich proteins.22 It was suggested that this modulation is due to direct interaction with the polyQ-expanded proteins. In line with this suggestion, it was shown that polyQ aggregates in yeast contain host proteins with Q/N-rich domains.23
Previously we have shown that in yeast proteins with polyQ domains form SDS-resistant polymers16,24 similar to those formed by the Sup35 and Rnq1 proteins.25,26 Here, we used polymer analysis to show that aggregation of polyQ proteins, including mutant huntingtin, causes polymerization of the yeast Q/N-rich proteins Sup35, Rnq1 and Pub1 produced at their normal levels. We also show that the [PSI+] prion stimulates the appearance of Pub1 polymers.
Results
Polymers of Q70-Sup35MC and QY76-Sup35MC cause polymerization of Sup35NM.
Earlier we have shown that Rnq1 prion polymers can efficiently seed non-heritable polymerization of overproduced Sup35.16 Here we examined if polymers of the Q70-Sup35MC and QY76-Sup35MC proteins (designated hereafter as Q70 and QY76; Fig. 1) can seed polymerization of Sup35 in Δrnq1 cells. In the Q70 and QY76 proteins the prion domain of Sup35 was replaced with either 70 sequential glutamine or 76 glutamines and tyrosines [(QQQYQ)25Q] residues. In contrast to Q70, QY76 polymers are fragmented very efficiently due to the presence of tyrosines.24
Figure 1.

Schematic representation of the proteins used in this work. N, M, C, Sup35 domains. 2HA, double hemagglutinin tag. F, FLAG tag. Grey bars, amyloidogenic regions; Q/N, Q/N-rich; Q, Q-rich. Black bars, RNA-binding domains of Pub1.
Q70 and QY76 polymers can propagate in the absence of [PIN+], but Q70 polymers require [PIN+] for their appearance.24 To obtain polymers of Q70 and QY76, the strain 74-D694ΔS35 [psi−][PIN+] carrying the plasmid encoding Sup35C was transformed with the plasmids encoding either Q70 or QY76, and then the resident plasmid was lost. The transformants contained polymers of Q70 and QY76, as shown by SDD-AGE (Fig. 2). Then, the [PIN+] prion was eliminated by deleting the RNQ1 gene, which did not affect the presence of Q70 and QY76 polymers (Fig. 2). The resulting strains were transformed with the multicopy YEplac181-Sup35NM-2HA plasmid. The level of Sup35NM-2HA exceeded the level of chromosomally encoded Sup35 only about 2-fold (data not shown). Analysis of the transformants showed that they contained SDS-resistant complexes of Sup35NM-2HA. An important question is whether these complexes represent polymers composed mainly of Sup35NM-2HA, or arise via SDS-resistant binding of a limited number of Sup35NM-2HA molecules to the Q70 and QY76 seeds. In support of the former opportunity, the Sup35NM-2HA polymers differed significantly in size from QY76 polymers, and their sizes almost did not overlap. This indicates that Sup35NM-2HA polymers contained very little of QY76. Such a conclusion is less evident for Q70 seeds. However, it is supported by the fact that Q70 generated more Sup35NM-2HA polymers than QY76 (Fig. 2). Considering that the QY76 polymers were more numerous, which follows from their significantly smaller size, this also shows their significantly lower seeding efficiency.
Figure 2.

Polymers of Q70 and QY76 cause polymerization of the plasmid-encoded Sup35NM-2HAprotein in Δrnq1 cells. (A) The plasmid pRS315-SUP35C of the strain 74-D694ΔS35 [psi−][PIN+] was shuffled for either pQ70-Sup35MC or pQY76-Sup35MC. Then, the RNQ1 gene was deleted. Polymers of Q70 and QY76 were revealed using SDD-AGE and western blotting with polyclonal anti-Sup35NM antibody. (B) These strains were transformed with a multicopy plasmid, encoding Sup35NM-2HA. Appearance of Sup35NM-2HApolymers was tested using SDD-AGE and western blotting with monoclonal anti-HA antibody. PolyQ, the Q70 or QY76 protein; −70, −76, transformants that have lost the plasmid encoding indicated polyQ protein. Sup35NM-2HA, − or +, absence or presence of the plasmid encoding this protein. RNQ1, +, chromosomal wild type allele, Δ, disruption allele.
Replacement of the Q70- and QY76-encoding plasmids with the pRS315-SUP35C plasmid caused disappearance of the Sup35NM-2HA polymers. This shows that these polymers were of a non-prion nature and confirms that they were seeded solely by the polyQ/QY polymers (Fig. 2). The aggregation of Sup35NM, induced by Q70 and QY76 polymers in Δrnq1 cells, was also observed microscopically by fluorescence of Sup35NM-GFP (Fig. 3).
Figure 3.
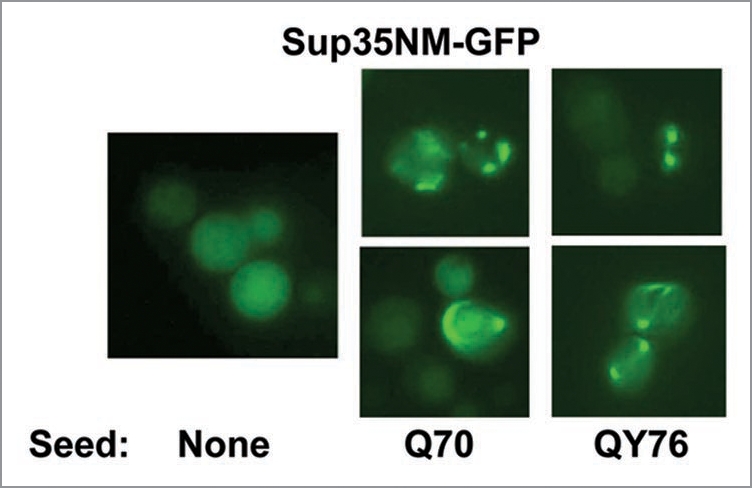
Polymers of Q70 and QY76 cause aggregation of Sup35NM-GFP in cells lacking [PIN+]. Multicopy plasmids encoding Q70 or QY76 were introduced into 74-D694 [psi−][PIN+] cells and then RNQ1 was deleted to remove [PIN+]. After this, the plasmid encoding Sup35NM-GFP was introduced. About 25% of transformed cells contained fluorescent dots. The same strain, but without the pQ70-Sup35MCand pQY76-Sup35MCplasmids was used as a control.
Polymerization of the mutant 103Q huntingtin causes depletion of soluble Sup35.
Similarly to the Q70 protein, the aggregation of pathological huntingtin is driven by its extended polyQ stretch. However, in contrast to Q70, overproduction of mutant 103Q huntingtin can be toxic for yeast and is routinely used for studying toxicity. The toxicity of 103Q may be related to a high GAL1-drived expression, as well as to the presence of the charged FLAG tag at its N-terminus.31 We tested the ability of pathological huntingtin exon I 103Q protein to cause conversion of the yeast chromosomally-encoded Sup35 protein into SDS-resistant polymers. The plasmid p103Q-GFP, encoding the 103Q-GFP chimera (Fig. 1) produced under the control of an inducible GAL1 promoter, was introduced into 74-D694 [psi−][PIN+] cells. The amount of 103Q-GFP SDS-resistant polymers was very low upon cell growth in glucose-containing medium, and increased greatly after transfer to raffinose- and then to galactose-containing media. As expected, the amount of Sup35 polymers also increased upon exchange of the carbon source, however it remained constant during growth on galactose-containing medium (Fig. 4A). The latter could be related to some decrease of the amount of total Sup35. The level of soluble Sup35 gradually decreased to about 10% of the original value (Fig. 4C), which should noticeably affect the efficiency of translation termination and cell viability.32 The Sup35 polymers observed here disappeared after loss of the p103Q-GFP plasmid (Fig. 4A and B) and thus were of a non-prion type.
Figure 4.

Time-dependent appearance of the 103Q-GFP and Sup35 polymers upon induction of 103Q-GFP synthesis. Cells of the strain 74-D694 [psi−][PIN+] with the multicopy p103Q-GFP plasmid were grown in glucose-containing medium (Gl), then in raffinose-containing medium (R), and then transferred to galactose-containing medium (Gal) and incubated for 1, 1.5, 2 and 3 h. After this, the 103Q-GFP-encoding plasmid was lost (−). (A) Polymers of 103Q-GFP and (B) Sup35 revealed by SDD-AGE. (C) The levels of polymer, monomer and total Sup35, SDS-PAGE analysis. The samples in the upper panel were not boiled before loading onto the gel. In the middle of the run, the gel was taken out, boiled to dissolve Sup35 polymers and allowed to run further. Blots were stained with anti-GFP (A) and anti-Sup35NM (B and C) antibodies.
Polymerization of Q70 causes appearance of Rnq1 and Pub1 polymers.
Next we studied the ability of Q70 to cause polymerization of the Rnq1 and Pub1 proteins. Both proteins have long Q/N-rich fragments (Fig. 1), which allows them to form amyloids.10,26 However, Rnq1 can adopt a prion form, which was not shown for Pub1.
In the strain 74-D694ΔS35 [psi−][pin−], the pRS315-SUP35C plasmid was replaced with pQ70-SUP35MC. Earlier we have shown that Q70 forms SDS-resistant polymers in [pin−] cells in a time-dependent manner: polymers were not observed after 30 cell generations with Q70 production, but appeared after 100 generations.24 Here, the polymers of Rnq1 and Pub1 appeared simultaneously with the Q70 polymers after 100 generations (Fig. 5). Replacement of the Q70-encoding plasmid with pRS315-SUP35C resulted in disappearance of the SDS-resistant Rnq1 and Pub1 polymers. This demonstrated that the Rnq1 and Pub1 polymers seeded by Q70 were of a non-prion type, because they could not propagate independently.
Figure 5.
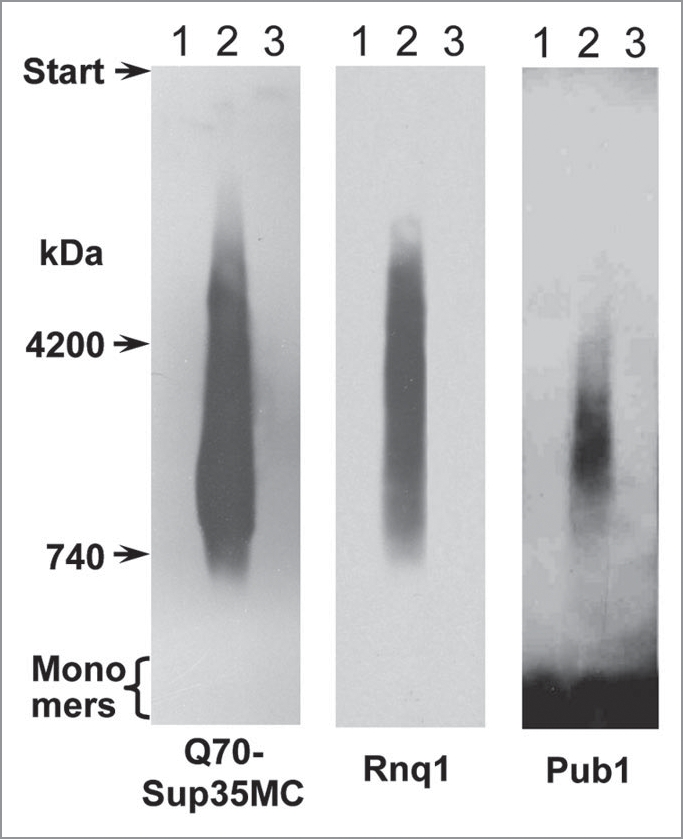
Co-appearance of SDS-resistant polymers of Q70, Rnq1 and Pub1. Cells of the 74-D694ΔS35 [psi−][pin−-] strain were transformed with the pQ70-SUP35MC plasmid. Samples were taken at 30 (lane 1) and 100 (lane 2) cell generations after transformation. Then the pQ70-SUP35MC plasmid was replaced with pRS315-SUP35C (lane 3). The samples were analyzed by SDD-AGE and western blotting. The proteins indicated below were revealed using antibodies to Sup35NM, Rnq1 and Pub1, respectively.
Pub1 polymerization depends on [PSI+].
PolyQ polymers were able to cause Pub1 polymerization, but Q70 and QY76 are not natural components of yeast cells. It was of interest, whether yeast prions can govern polymerization of this Q/N-rich protein. To answer this, we studied the dependence of Pub1 polymerization on the [PSI+] and [PIN+] prions. Examination of the 74-D694 [PSI+][PIN+] cells with Pub1 produced at normal and increased levels did not reveal SDS-resistant polymers of this protein (data not shown). However, it is known that some amyloids dissolve in cold SDS, but withstand a milder detergent, sarcosyl.33 Therefore, we modified the SDD-AGE procedure by replacing SDS with sarcosyl. This allowed us to observe Pub1 polymers in the [PSI+][PIN+] and [PSI+] ΔRNQ1, but not in the [psi-][PIN+] and [psi−][pin−] cells. Overproduction of Pub1 in cells containing [PSI+] strongly increased the level of its polymer form (Fig. 6). Thus, the presence of [PSI+], but not of [PIN+], caused Pub1 polymerization. Elimination of [PSI+] by Hsp104 overproduction caused disappearance of Pub1 detergent-resistant polymers. The inability of [PIN+] to cause noticeable Pub1 polymerization could be related to Rnq1 abundance, which is about 10-fold lower compared to Sup35.34 Expression from multicopy plasmid should yield Rnq1 levels comparable to Sup35, but this caused only weak Pub1 polymerization. Finally, multicopy expression of RNQ1 from the strong MET17 promoter caused more efficient polymerization of Pub1 than [PSI+] (Fig. 6). Thus, polymers of Rnq1 are less efficient than that of Sup35 in seeding Pub1 polymerization.
Figure 6.

Pub1 polymers depend on the [PSI+] determinant. (A) Polymers of Sup35 and Pub1 in [PSI+][PIN+] cells, (B) Absence of Pub1 polymers in [psi−][PIN+] cells, (C) Disappearance of Pub1 polymers upon [PSI+] elimination, (D) Pub1 can polymerize in [psi−][PIN+] cells overproducing Rnq1. + and −, presence or absence of a prion determinant; −*, [PSI+] was eliminated by Hsp104 overproduction; wt and ↑, wild type or increased levels of Pub1; +↑ and +M, Rnq1 was overproduced in [PIN+] cells from multicopy plasmids with either the native or MET17 promoter. Note: Pub1 monomers occupy a large area at the bottom of a gel. This is likely to be due to a high monomer proportion for Pub1 and low resolution of agarose gel for monomeric proteins aggravated by overloading of this area of the gel, which accommodates most of the cellular proteins.
Discussion
Sequestration of chaperones and other proteins by aggregates of mutant huntingtin can be involved in Huntington’s disease pathogenesis in humans17–20 and may be the reason for its toxicity in yeast.23,35 In a similar way, the toxicity of overproduced Sup35 in yeast [PSI+] cells is related to sequestration of Sup45 (eRF1) translation termination factor by Sup35 aggregates.36 The co-aggregating proteins may be divided into two groups. The first group consists of proteins, which bind aggregates of huntingtin. In yeast this includes chaperones, some glycolytic enzymes and other proteins.23 The amount of bound proteins should not significantly exceed the amount of aggregated huntingtin and their binding should be SDS-sensitive. The second group constitutes Q-rich proteins, which are presumed to polymerize as amyloids, being seeded by the huntingtin amyloid.15 The transition of these proteins into an amyloid form is not restricted quantitatively. For example, in the [PIN+] cells overproducing Sup35 the proportion of polymerized Sup35 to the Rnq1 protein constituting prion seeds exceeded 100-fold.16
Proteins of the latter group are of special interest. Genes were identified for several such proteins, whose deletion reduced or eliminated the toxicity of the otherwise toxic huntingtin exon I protein, whereas overproduction of other Q-rich proteins converted nontoxic huntingtin exon I proteins into toxic species.22 These proteins co-localized with 103Q aggregates, which implies that polyQ toxicity in yeast results from induced aggregation of many host Q-rich proteins. However, the co-localization of these proteins with 103Q does not reveal the nature of their aggregates. Here, we show that the aggregates of three tested yeast Q/N-rich proteins, Sup35, Rnq1 and Pub1, appearing in the presence of polymers of polyQ proteins, including mutant 103Q exon I huntingtin, represent SDS-resistant polymers, which strongly supports their amyloid nature. Most probably, the polymers of Sup35, Rnq1 and Pub1 appeared due to seeding by polyQ polymers, as it was shown previously for the seeding of Sup35 polymerization by Rnq1 polymers.15,16 Having appeared, such polymers could elongate independently of the Q70/QY76 seeds. This was evident in at least one case. The polymers of Sup35NM-2HA were significantly larger than the seeding polymers of QY76, and thus contained very little of QY76. Such a result was to be expected, considering the different fragmentation rate for QY76 and Sup35NM-2HA polymers. The latter grew without fragmentation and became large, while the growing QY76 polymers were frequently fragmented, and thus retained small size. Fragmentation also acted to remove QY76 seeds from the Sup35NM-2HA polymers.
Previously, we observed that the majority of Sup35 polymers seeded by the prion form of Rnq1 are non-heritable.16 Here, we observed again that the prionogenic proteins Sup35, Sup35NM and Rnq1 formed non-heritable polymers when seeded by other polymers. This suggests that, in general, all prionogenic proteins would preferentially form polymers of a non-prion type. Such polymers are infrequently fragmented by Hsp104, presumably because they are poorly recognized by this chaperone due to lack of solvent-exposed hydrophobic residues.24 The predominance of this kind of folding may be due to its thermodynamic preference or to the action of chaperones, which would bind to the emerging prion chaperon-recognizable folds and greatly complicate their appearance. The latter opportunity appears more likely, since in vitro the prion folds of Sup35 appear with no significant handicap, as is evident from their ability to transform yeast cells to the [PSI+] prion state.37
The polymerization of yeast Q/N-rich proteins may be seeded not only by polyQ/QY polymers. [PSI+]-dependent polymerization of the Pub1 protein is of special interest, since this protein plays an important role in posttranscriptional regulation of the expression of numerous yeast genes38,39 and in stress granule formation.40 Intriguingly, the Pub polymers were SDS-resistant when seeded by the polyQ polymers, but sensitive to SDS, though resistant to sarcosyl, when seeded by the prion form of Sup35, which suggests a structural difference between these Pub1 polymers. Pub1 polymers could not propagate on their own, being found only in the presence of the polyQ polymers or Sup35 prion polymers.
Depending on the genetic background, [PSI+] exerts diverse phenotypic effects.41 While some of these effects were shown to result from the [PSI+]-related increase of nonsense codon readthrough,42 some others could be attributed to the ability of Sup35 prion aggregates to bind host proteins with important functions, for example, chaperones,43 or to cause conversion of polymerization-prone Q/N-rich proteins into an amyloid form. In these cases the strength of such [PSI+] manifestations should correlate with the proportion of the proteins that are inactivated by co-aggregation or polymerization. In our experiments, significant depletion of the studied Q-rich proteins was mainly achieved upon overproduction of the amyloid seeds. However, the overproduction of amyloids in rapidly dividing yeast cells models the accumulation of high amounts of amyloid aggregates in non-dividing neurons. The seeding efficiency varied depending on both the seed and polymerizing protein. Indeed, Sup35 prion polymers were more efficient than those of Rnq1 in seeding Pub1 polymerization, and Q70 polymers seeded polymerization of Sup35NM significantly more efficiently than QY76 ones. Considering the large number of proteins with long Q/N-rich stretches in both human and yeast cells,10,44 it is likely that at least some of them would be efficiently depleted in response to the accumulation of polyQ amyloids, which could broaden the influence of amyloid appearance on cell physiology.
Materials and Methods
Plasmids, strains and genetic methods.
In this work we used the strain 74-D694 [psi−][pin−] and its [PSI+] and [PIN+] derivatives,2727 as well as 74-D694ΔS35 [psi−] [PIN+] and [psi−][pin−] with SUP35 disrupted by the insertion of TRP1.24 The plasmids used in this study are described in Table 1. The 2.5 kb DNA fragment containing the PUB1 gene was amplified by PCR with primers 5′-ACG ACC ACA AAG GAT CCA GGG-3′ (−882 upstream of ORF) and 5′-GAC GAA GGA AGG AAA TAA GAC-3′ (+254 downstream of ORF), using genomic DNA of the strain 74-D694 as a template. The PCR product was inserted into the SmaI site of the URA3 YEplac195 plasmid,28 thus generating YEplac195-PUB1. The plasmid YEplac181-Sup35NM-2HA encodes Sup35NM-2HA with double HA (hemagglutinin) tag after the Sup35 amino acid 252. The plasmid YEplac181-HSP104 contains the HSP104 gene with 566 bp of upstream and 364 bp of downstream flanking sequences. To construct multicopy plasmids encoding Rnq1, the RNQ1 gene, including 487 bp (XhoI site) upstream and 186 bp downstream of the coding region, was amplified from yeast genomic DNA and inserted into YEplac181,28 by SalI and EclI36II sites, resulting in YEplac181-RNQ1. In this plasmid, the RNQ1 promoter fragment BsaBI-BglII (positions −236 to −9) was replaced for the PCR-amplified MET17 promoter (positions −429 to +15). This added amino acids sequence MPSHFGGSE to the N-terminus of the Rnq1 protein.
Table 1.
Plasmids
| Plasmid | Characteristics | Source |
| YEplac181-Sup35NM-2HA | Multicopy LEU2 plasmid encoding Sup35NM with double hemagglutinin tag at the C-terminus | This work |
| YEplac181-SUP35NM-GFP | Multicopy LEU2 plasmid encoding Sup35NM-GFP | 16 |
| pQ70-SUP35MC | Multicopy URA3 plasmid encoding Q70-Sup35MC | 24 |
| pQY76-Sup35MC | Multicopy URA3 plasmid encoding QY76-Sup35MC | 24 |
| p103Q-GFP | Multicopy URA3 plasmid, encoding 103Q protein fused with GFP under the control of GAL1 promoter | 45 |
| YEplac195-PUB1 | Multicopy URA3 plasmid encoding Pub1 | This work |
| YEplac181-HSP104 | Multicopy LEU2 plasmid encoding Hsp104 | This work |
| YEplac181-Met17-RNQ1 | Multicopy LEU2 plasmid encoding Rnq1 under the control of MET17 promoter | This work |
| pRS315-SUP35C | Centromeric LEU2 plasmid encoding Sup35C | 46 |
The RNQ1 gene disruption with HIS3 in the 74-D694 [PSI+][PIN+] strain was described previously.16 Yeast were grown at 30°C in rich (YPD, 1% yeast extract, 2% peptone, 2% glucose; or YPD-red, 1% yeast extract, 2% peptone, 4% glucose) or synthetic (SC, 0.67% yeast nitrogen base, 2% glucose supplemented with the required amino acids) media. To induce the synthesis of 103Q-GFP chimeric protein, the strain 74-D694 [psi−][PIN+] with the plasmid p103Q-GFP was incubated in liquid SC-Ura with 2% raffinose as a sole carbon source to OD600 of 1.0. Then, culture was diluted with equal volume of the same medium with galactose instead of raffinose. The final concentration of galactose in the medium was 2%. Cells of the strain 74-D694 [PSI+][PIN+] form white colonies which can grow on adenine-less medium due to suppression of the ade1-14 UGA mutation in the presence of [PSI+], while the loss of [PSI+] results in adenine auxotrophy and causes accumulation of red pigment related to impaired adenine biosynthesis. [PSI+] determinant of 74-D694 [PSI+][PIN+] was eliminated by overproduction of Hsp104. For this, the strain was transformed with the multicopy plasmid YEplac181-HSP104. One of the obtained transformants was streaked on YPD medium and red colonies were tested for the presence of Sup35 and Rnq1 SDS-resistant polymers. The clone containing only Rnq1 polymers was taken for further analysis.
Cells for fluorescent microscopy were grown overnight on medium selective for the YEplac181-SUP35NM-GFP plasmid along with either pQ70-SUP35MC or pQY76-SUP35MC. Cells were photographed using the OptonIII microscope (Carl Zeiss, Germany) with a 63 X objective.
Preparation of yeast cell lysates.
Yeast cultures were grown in liquid media to OD600 of 1.5. The cells were harvested, washed in water and lyzed by glass beads in buffer A: 30 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM dithiothreitol and 1% Triton X-100. To prevent proteolytic degradation, 10 mM phenylmethylsulfonyl fluoride and Complete⊥M protease inhibitor cocktail (Roche Applied Science) were added. Cell debris was removed by centrifugation at 1,500 g for 4 min.
Electrophoresis and blotting.
These were performed as described previously.25,29,30 Protein loads were equalized for each gel. For the analysis of SDS-resistant polymers we used horizontal 1.8% agarose gels in Tris-Acetate-EDTA (TAE) buffer with 0.1% SDS. Lysates were incubated in the sample buffer (0.5x TAE, 2% SDS, 5% glycerol and 0.05% Bromophenol Blue) for 5 min at 37°C. After electrophoresis, proteins were transferred from gels to Immobilon-P PVDF sheets (Millipore, USA) by vacuum blotting overnight (agarose gels), or electrophoretically (polyacrylamide gels). Bound antibody was detected using the ECL system (GE Healthcare). It should be noted that detergents (SDS or sarcosyl) in the non-boiled samples increase degradation of the Sup35 and Rnq1 monomers. This can result in the absence of Sup35 and Rnq1 monomer bands in SDD-AGE gels. All SDD-AGE experiments were repeated three times and typical pictures were presented. Rabbit polyclonal antibodies against Sup35, Rnq1 and Pub1 were used. Anti-GFP monoclonal antibody was obtained from Rusbiolink (Russia) and anti-HA monoclonal antibody—from Roche Applied Science.
Acknowledgements
The work was supported by the grants from Russian Foundation for Basic Research (# 08-04-0062) and the Wellcome Trust. Authors are grateful to M. Sherman for providing the plasmid p103Q-GFP and to O. Mitkevich for the help in obtaining antibody against Pub1. V.N.U. and A.B.V. contributed equally to this work.
Abbreviations
- polyQ
polyglutamine
- Q70
the Q70-Sup35MC protein
- QY76
the QY76-Sup35MC protein
- HA
hemagglutinin
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/11074
References
- 1.Gebbink MF, Claessen D, Bouma B, Dijkhuizen L, Wösten HA. Amyloids—a functional coat for microorganisms. Nat Rev Microbiol. 2005;3:333–341. doi: 10.1038/nrmicro1127. [DOI] [PubMed] [Google Scholar]
- 2.Inge-Vechtomov SG, Zhouravleva GA, Chernoff YO. Biological roles of prion domains. Prion. 2007;1:228–235. doi: 10.4161/pri.1.4.5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mironova LN, Goginashvili AI, Ter-Avanesyan MD. Biological functions of amyloids: facts and hypotheses. Mol Biol. 2008;42:710–719. [PubMed] [Google Scholar]
- 4.Vishnevskaya AB, Kushnirov VV, Ter-Avanesyan MD. Neurodegenerative amyloidoses: yeast model. Mol Biol. 2007;41:346–354. [PubMed] [Google Scholar]
- 5.Kushnirov VV, Vishnevskaya AB, Alexandrov IM, Ter-Avanesyan MD. Prion and nonprion amyloids: a comparison inspired by the yeast Sup35 protein. Prion. 2007;1:179–184. doi: 10.4161/pri.1.3.4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wickner RB. [URE3] as an altered Ure2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 7.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 8.Du Z, Park KW, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nature Genet. 2008;40:460–465. doi: 10.1038/ng.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nemecek J, Nakayashiki T, Wickner RB. A prion of yeast metacaspase homolog (Mca1p) detected by a genetic screen. Proc Natl Acad Sci USA. 2009;106:1892–1896. doi: 10.1073/pnas.0812470106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel BK, Gavin-Smyth J, Liebman SW. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nature Cell Biol. 2009;11:344–349. doi: 10.1038/ncb1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 13.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 14.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN+] Cell. 2001;106:171–182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 15.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci USA. 2004;101:12934–12939. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salnikova AB, Kryndushkin DS, Smirnov VN, Kushnirov VV, Ter-Avanesyan MD. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its non-heritable amyloids. J Biol Chem. 2005;280:8808–8812. doi: 10.1074/jbc.M410150200. [DOI] [PubMed] [Google Scholar]
- 17.Cha JH. Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. 2000;23:387–392. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- 18.Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 19.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 20.Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci USA. 2002;99:16412–16418. doi: 10.1073/pnas.182426899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntingtin toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. Proc Natl Acad Sci USA. 2006;103:11051–11056. doi: 10.1073/pnas.0604548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Meriin AB, Costello CE, Sherman MY. Characterization of proteins associated with polyglutamine aggregates. Prion. 2007;1:128–135. doi: 10.4161/pri.1.2.4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrov IM, Vishnevskaya AB, Ter-Avanesyan MD, Kushnirov VV. Appearance and propagation of polyglutamine-based amyloids in yeast: Tyrosine residues enable polymer fragmentation. J Biol Chem. 2008;283:15185–15192. doi: 10.1074/jbc.M802071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 26.Vitrenko YA, Gracheva EO, Richmond JR, Liebman SW. Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM. J Biol Chem. 2007;282:1779–1787. doi: 10.1074/jbc.M609269200. [DOI] [PubMed] [Google Scholar]
- 27.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 28.Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 29.Shkundina IS, Kushnirov VV, Tuite MF, Ter-Avanesyan MD. The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics. 2006;172:827–835. doi: 10.1534/genetics.105.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kushnirov VV, Alexandrov IM, Mitkevich OV, Shkundina IS, Ter-Avanesyan MD. Purification and analysis of prion and amyloid aggregates. Methods. 2006;39:50–55. doi: 10.1016/j.ymeth.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci USA. 2006;103:11045–11050. doi: 10.1073/pnas.0604547103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valouev IA, Kushnirov VV, Ter-Avanesyan MD. Yeast polypeptide chain release factors eRF1 and eRF3 are involved in cytoskeleton organization and cell cycle regulation. Cell Motyl Cytoskeleton. 2002;52:161–173. doi: 10.1002/cm.10040. [DOI] [PubMed] [Google Scholar]
- 33.Taneja V, Maddelein ML, Talarek N, Saupe SJ, Liebman SW. A non-Q/N-rich prion domain of a foreign prion, [Het-s], can propagate as a prion in yeast. Mol Cell. 2007;27:67–77. doi: 10.1016/j.molcel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello E, Sherman MY. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 2009;23:451–463. doi: 10.1096/fj.08-117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vishveshwara N, Bradley ME, Liebman SW. Sequestration of essential proteins causes prion associated toxicity in yeast. Mol Microbiol. 2009;73:1101–1114. doi: 10.1111/j.1365-2958.2009.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 38.Ruiz-Echevarria MJ, Peltz SW. The RNA binding protein Pub1 modulates the stability of transcripts containing upstream open reading frames. Cell. 2000;101:741–751. doi: 10.1016/s0092-8674(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 39.Duttagupta R, Tian B, Wilusz CJ, Khounh DT, Soteropoulos P, Ouyang M, et al. Global analysis of Pub1p targets reveals a coordinate control of gene expression through modulation of binding and stability. Mol Cell Biol. 2005;25:5499–5513. doi: 10.1128/MCB.25.13.5499-5513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchan JR, Muhlard D, Parker R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol. 2008;183:441–455. doi: 10.1083/jcb.200807043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- 42.True HL, Berlin I, Lindquist SL. Epigenetic regulation reveals hidden genetic variation to produce complex traits. Nature. 2004;431:184–187. doi: 10.1038/nature02885. [DOI] [PubMed] [Google Scholar]
- 43.Bagriantsev SN, Gracheva EO, Richmond JE, Liebman SW. Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol Biol Cell. 2008;19:2433–2443. doi: 10.1091/mbc.E08-01-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitchelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci USA. 2000;97:11910–11915. doi: 10.1073/pnas.97.22.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gokhale KC, Newnam GP, Sherman MY, Chernoff YO. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J Biol Chem. 2005;280:22809–22818. doi: 10.1074/jbc.M500390200. [DOI] [PubMed] [Google Scholar]
- 46.Urakov VN, Valouev IA, Kochneva-Pervukhova NV, Packeiser AN, Vishnevsky AY, Glebov OO, et al. N-terminal region of Saccharomyces cerevisiae eRF3 is essential for the functioning of the eRF1/eRF3 complex beyond translation termination. BMC Mol Biol. 2006;7:34–46. doi: 10.1186/1471-2199-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]