

Abstract
To identify genetic susceptibility factors conferring increased risk of venous thrombosis (VT), we conducted a multistage study, following results of a previously published GWAS that failed to detect loci for developing VT. Using a collection of 5862 cases with VT and 7112 healthy controls, we identified the HIVEP1 locus on chromosome 6p24.1 as a susceptibility locus for VT. Indeed, the HIVEP1 rs169713C allele was associated with an increased risk for VT, with an odds ratio of 1.20 (95% confidence interval 1.13–1.27, p = 2.86 × 10−9). HIVEP1 codes for a protein that participates in the transcriptional regulation of inflammatory target genes by binding specific DNA sequences in their promoter and enhancer regions. The current results provide the identification of a locus involved in VT susceptibility that lies outside the traditional coagulation/fibrinolysis pathway.
Main Text
We recently reported results of the only GWAS performed to date on venous thrombosis (VT) including deep vein thrombosis (DVT) and pulmonary embolism (PE).1 In a final analysis of 419 VT patients and 1228 healthy individuals typed with the Illumina Sentrix HumanHap300 array for 291,872 SNPs, only five SNPs reached statistical significance at α = 3.47 × 10−7. These SNPs were located within two well-established susceptibility loci for VTE, F5 (MIM 612309) and ABO (MIM 110300).2 In the current report, we pursued, by standard TaqMan genotyping technology (Applied Biosystems), the follow-up of nine SNPs that did not reach genome-wide significance but were nevertheless associated with VT at p < 1 × 10−5, corresponding to a false discovery rate estimate of 16% (Table 1). These nine SNPs were investigated in the MARTHA study,1 composed of 1129 VT cases and 801 controls of European origin.
Table 1.
SNPs Associated at p < 10−5 with VT in the GWAS Scan and Their Replication in the Independent MARTHA and FARIVE Studies
| Chr. | Gene(s) | rsID | Position | Alleles |
GWAS |
MARTHA |
FARIVE |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
MAFa |
p Valueb |
MAFa |
p Valueb |
MAFa |
p Valueb | ||||||||
| Controls (n = 1228) | Cases (n = 419) | Controls (n = 801) | Cases (n = 1129) | Controls (n = 607) | Cases (n = 607) | ||||||||
| 1p34.2 | PTPRF | rs11210892 | 43 872 671 | A/G | 0.32 | 0.40 | 6.70 × 10−6 | 0.32 | 0.37 | 1.83 × 10−3 | 0.33 | 0.33 | 0.811 |
| 3q26.31 | TNIK | rs10936688 | 172 656 249 | A/G | 0.36 | 0.44 | 3.15 × 10−6 | 0.39 | 0.37 | 0.187 | |||
| 6p24.1 | HIVEP1 | rs169713 | 12 028 503 | T/C | 0.21 | 0.28 | 7.24 × 10−6 | 0.23 | 0.29 | 2.08 × 10−5 | 0.21 | 0.24 | 0.089 |
| rs9380643 | 12 038 473 | C/T | 0.22 | 0.30 | 2.44 × 10−6 | 0.24 | 0.31 | 1.98 × 10−6 | 0.24 | 0.26 | 0.227 | ||
| 7p15.3 | IL6/TOMM7 | rs10229457 | 22 767 325 | G/A | 0.29 | 0.38 | 1.05 × 10−6 | 0.34 | 0.36 | 0.128 | - | - | - |
| 8p21.3 | ATP6V1B2/LZTS1 | rs952148 | 20 135 799 | C/T | 0.11 | 0.06 | 1.35 × 10−6 | 0.10 | 0.11 | 0.459 | - | - | - |
| 12p12.1 | ST8SIA1 | rs2268861 | 22 280 741 | G/A | 0.32 | 0.24 | 1.82 × 10−6 | 0.31 | 0.29 | 0.231 | - | - | - |
| 16q22.1 | NFATC3 | rs12598 | 66 783 016 | G/A | 0.08 | 0.04 | 7.64 × 10−6 | 0.06 | 0.05 | 0.074 | - | - | - |
| 22q13.31 | Gene desert | rs4823511 | 46 823 885 | A/G | 0.24 | 0.16 | 5.45 × 10−6 | 0.22 | 0.21 | 0.421 | - | - | - |
SNPs that did not pass the genome-wide significance level of 1.71 × 10−7 but showed suggestive evidence of association (p < 10−5) were investigated in the MARTHA study. SNPs that passed the Bonferroni-corrected significance level (p < 5.6 × 10−3) in MARTHA were further tested for association with VT in FARIVE.
MAF, minor allele frequency.
p values of the Cochran-Armitage trend test.
Of these nine SNPs, three showed significant association with VT after Bonferroni correction for the number of tested SNPs (Table 1). Two of them, rs169713 (p = 2.08 × 10−5) and rs9380643 (p = 1.98 × 10−6), are located on chromosome 6p24.1, in the vicinity of the HIVEP1 (MIM 194540) locus, whereas the third one, rs11210892 (p = 1.83 × 10−3), maps between the PTPRF (MIM 179590) and JMJD2A (MIM 609764) genes on chromosome 1p34.1. Consequently, these three SNPs were further examined for support of association with VT in a third independent sample, the FARIVE study,1 composed of 561 cases with VT and 564 controls of French origin. As a result, none of these SNPs was significantly associated with VT in FARIVE (Table 1), although the rs169713-C allele was more frequent in cases compared to controls (0.24 versus 0.21, p = 0.089), as also observed in the GWAS and MARTHA samples (Figure 1). Because no evidence for heterogeneity across the three studies (p = 0.250) was detected by use of the Mantel-Haenszel method, and because the FARIVE study had a power of only ∼40% (calculated by the Cats software3) to detect the observed allele-frequency differences at a significance level of 0.05, rs169713 was further investigated in the MEGA study,4 composed of 3753 VT patients and 4519 controls of Dutch origin. In MEGA, the rs169713-C allele was significantly more frequent in cases than in controls (0.22 versus 0.20, p = 0.014) (Figure 1, Table S1 [available online]), even if the association was slightly weaker as compared to the other studies.
Figure 1.
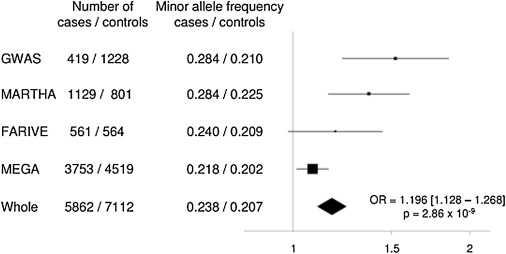
Forest Plot of the Association between rs169713 and VT Risk
Squares represent odds ratios with their 95% confidence interval under the assumption of additive allele effects. The whole odds ratio estimate was obtained from a combined analysis of all individual data sets after checking for homogeneity by use of the Mantel-Haenszel method.
With the combination of the four genotyped data sets, totalling 5862 cases and 7112 controls, the rs169713-C allele was significantly more frequent in cases than in controls (0.24 versus 0.21) and was associated with a combined odds ratio of 1.20 (95% confidence interval 1.13–1.27, p = 2.86 × 10−9) for VT. This effect was more pronounced (p = 0.001) in the GWAS and the MARTHA study (OR = 1.419 [1.266–1.591], p = 1.85 × 10−9) as compared to the FARIVE and MEGA studies (OR = 1.110 [1.035–1.190], p = 3.38 × 10−3). This was due to the fact that the frequency of the rs169713-C allele was higher in GWAS and MARTHA cases compared to FARIVE and MEGA cases, whereas its frequency was very homogeneous in the four control groups (Figure 1). Cases from the GWAS and MARTHA are patients referred to thrombophilia centers, whereas patients from FARIVE and MEGA were recruited from the general population. The former are often considered to be enriched in familial forms,5 a likely explanation for the observed higher frequency of the VT-disease-associated allele in this group. The rs169713-C allele association did not show any evidence for heterogeneity according to the age at onset, VT status (idiopathic versus nonidiopathic, DVT versus PE), or known risk factors for VT, such as sex, oral contraceptives, FV Leiden mutation, and ABO blood group (Tables S1–S8).
rs169713 maps to the pseudogene LOC100129761 and is 92 kb distant in 5′ orientation from the HIVEP1 gene. In the initial GWAS scan, the SNP with the highest significance was rs9380643, also strongly associated with VT in MARTHA. However, this SNP did not show any trend for association in FARIVE, and linkage disequilibrium (LD) analysis in MARTHA revealed that its effect was probably due to its strong LD with rs169713 (D′ = +0.87, r2 = 0.66). rs169713 was the second lead SNP at the HIVEP1 locus in the GWAS, and several other HIVEP1 SNPs showed suggestive evidence of association with VT (p < 10−3) (Table S8). According to public databases, there are many other SNPs at the HIVEP1 gene locus, including several insertion/deletion polymorphisms, which may not be well characterized by the DNA array used in the initial scan. LD analysis of the HapMap database via the SNAP software6 revealed that none of the SNPs located in the coding, intronic, and 3′ untranslated regions of the gene showed pairwise r2 > 0.32 with the rs169713, suggesting that the HIVEP1 promoter would probably be the candidate region harboring the functional variant(s). In silico analyses with the MatInspector7 and Patch tools both suggested that the rs169713-C allele potentially creates a binding site for AHR/ARNT transcription factors (factors activated by exposure to dioxin); however, additional fine-mapping studies and in-depth molecular functional studies are warranted for the detection of functional variant(s) causally related to VT phenotypes.
HIVEP1 belongs to the HIVEP gene family encoding very large sequence-specific DNA-binding proteins containing multiple zinc fingers.8,9 These genes have been reported to directly participate in the transcriptional regulation of a variety of genes by binding to their promoter NF-κB consensus sequences. Closely related sequences are found in the promoter enhancer elements of class I MHC, interleukin-2 receptor, and interferon-β genes, the latter being also linked to the inflammatory Toll-like receptor signaling pathway. HIVEP1 itself regulates the transcription of genes such as HIV, H-2K, and IFN-B.9 As part of an ongoing genome-wide expression analysis in monocytes of 1490 healthy individuals (unpublished data), an ontology analysis using the PANTHER software revealed that genes whose expression were strongly correlated to HIVEP1 gene expression were particularly enriched in those belonging to the interleukin signaling pathway. These data, if confirmed, would point to a possible role of the HIVEP1 locus in inflammation, a biological process whose role in thrombosis-related mechanisms is increasingly advocated.10–12 In addition, links between NF-κB and VT through regulation of the production of microparticles from endothelial cells and platelet activation emerged from recent works.13,14
According to databases of expressed sequence tag patterns, HIVEP1 is expressed in a variety of tissues. In that respect, we observed HIVEP1 expression in endothelial cells, platelets, monocytes, and macrophages from healthy donors human blood (Figure S1) as well as in human carotid atheroma plaques (Figure S2).
Given the the latter results and the key role of inflammation in atherosclerosis and its cardiovascular complications, additional investigations are warranted to assess whether the HIVEP1 locus could also contribute to the genetic susceptibility of cardiovascular disease phenotypes, linking VT to arterial thrombosis.15
Using a multistage strategy, we obtained strong evidence in favor of the implication of the HIVEP1 locus as a candidate for VT risk. To our knowledge, this study is the first to provide evidence of the role of a genetic variant on the risk of VT outside the traditional coagulation/fibrinolysis cascade.
Acknowledgments
All participants provided written informed consent, and the protocol was approved by the ethics committee of each participating institution. The FARIVE study was supported by grants from the Fondation pour la Recherche Médicale, the Program Hospitalier de Recherche Clinique, the Fondation de France, and the Leducq Foundation. The MARTHA study was supported by a grant from the Program Hospitalier de la Recherche Clinique. M.G. is supported by a grant funded by the Agence Nationale pour la Recherche (project ANR-07-MRAR-021). SM.B.-H. is supported by a grant from the EU-Project Network of Excellence, FP6-2005-LIFESCIHEALTH-6, Integrating Genomics, Clinical Research and Care in Hypertension, InGenious HyperCare proposal no. 037093 to Information and Communication Technologies (ICT), FP7-ICT-2007-2, project number 224635, Virtual Pathological Heart of the Virtual Physiological Human (VPH2). The Gutenberg Heart Study is funded through the government of Rheinland-Pfalz (“Stiftung Rheinland Pfalz für Innovation,” contract no. AZ 961-386261/733), the research programs “Wissen schafft Zukunft” and “Schwerpunkt Vaskuläre Prävention” of the Johannes Gutenberg-University of Mainz, and its contract with Boehringer Ingelheim and PHILIPS Medical Systems, including an unrestricted grant for the Gutenberg Heart Study. Specifically, the research reported in this article was supported by the National Genome Network “NGFNplus” (contract project no. A3 01GS0833) by the Federal Ministry of Education and Research, Germany.
The authors thank Marie Billerey for her excellent technical assistance in the MARTHA project.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Patch program, http://www.gene-regulation.com
Protein ANalysis THrough Evolutionary Relationships (PANTHER) software, http://www.pantherdb.org
References
- 1.Trégouët D.A., Heath S., Saut N., Biron-Andreani C., Schved J.F., Pernod G., Galan P., Drouet L., Zelenika D., Juhan-Vague I. Common susceptibility alleles are unlikely to contribute as strongly as the FV and ABO loci to VTE risk: results from a GWAS approach. Blood. 2009;113:5298–5303. doi: 10.1182/blood-2008-11-190389. [DOI] [PubMed] [Google Scholar]
- 2.Morange P.E., Tregouet D.A. Deciphering the molecular basis of venous thromboembolism: Where are we and where should we go? Br J Haematol. 2009;148:495–506. doi: 10.1111/j.1365-2141.2009.07975.x. [DOI] [PubMed] [Google Scholar]
- 3.Skol A.D., Scott L.J., Abecasis G.R., Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 4.Blom J.W., Doggen C.J., Osanto S., Rosendaal F.R. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293:715–722. doi: 10.1001/jama.293.6.715. [DOI] [PubMed] [Google Scholar]
- 5.Majerus P.W. Human genetics. Bad blood by mutation. Nature. 1994;369:14–15. doi: 10.1038/369014a0. [DOI] [PubMed] [Google Scholar]
- 6.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O'Donnell C.J., de Bakker P.I. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cartharius K., Frech K., Grote K., Klocke B., Haltmeier M., Klingenhoff A., Frisch M., Bayerlein M., Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin A.S., Jr., LeClair K.P., Singh H., Sharp P.A. A large protein containing zinc finger domains binds to related sequence elements in the enhancers of the class I major histocompatibility complex and kappa immunoglobulin genes. Mol. Cell. Biol. 1990;10:1406–1414. doi: 10.1128/mcb.10.4.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan C.M., Maniatis T. A DNA-binding protein containing two widely separated zinc finger motifs that recognize the same DNA sequence. Genes Dev. 1990;4:29–42. doi: 10.1101/gad.4.1.29. [DOI] [PubMed] [Google Scholar]
- 10.Esmon C.T. Inflammation and thrombosis. J. Thromb. Haemost. 2003;1:1343–1348. doi: 10.1046/j.1538-7836.2003.00261.x. [DOI] [PubMed] [Google Scholar]
- 11.Antoniades C., Bakogiannis C., Tousoulis D., Antonopoulos A.S., Stefanadis C. The CD40/CD40 ligand system: linking inflammation with atherothrombosis. J. Am. Coll. Cardiol. 2009;54:669–677. doi: 10.1016/j.jacc.2009.03.076. [DOI] [PubMed] [Google Scholar]
- 12.Ridker P.M., Silvertown J.D. Inflammation, C-reactive protein, and atherothrombosis. J. Periodontol. 2008;79(8, Suppl):1544–1551. doi: 10.1902/jop.2008.080249. [DOI] [PubMed] [Google Scholar]
- 13.Simoncini S., Njock M.S., Robert S., Camoin-Jau L., Sampol J., Harlé J.R., Nguyen C., Dignat-George F., Anfosso F. TRAIL/Apo2L mediates the release of procoagulant endothelial microparticles induced by thrombin in vitro: a potential mechanism linking inflammation and coagulation. Circ. Res. 2009;104:943–951. doi: 10.1161/CIRCRESAHA.108.183285. [DOI] [PubMed] [Google Scholar]
- 14.Malaver E., Romaniuk M.A., D'Atri L.P., Pozner R.G., Negrotto S., Benzadón R., Schattner M. NF-kappaB inhibitors impair platelet activation responses. J. Thromb. Haemost. 2009;7:1333–1343. doi: 10.1111/j.1538-7836.2009.03492.x. [DOI] [PubMed] [Google Scholar]
- 15.Prandoni P., Bilora F., Marchiori A., Bernardi E., Petrobelli F., Lensing A.W., Prins M.H., Girolami A. An association between atherosclerosis and venous thrombosis. N. Engl. J. Med. 2003;348:1435–1441. doi: 10.1056/NEJMoa022157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.